

Abstract
Kennedy Disease/Spinal Bulbar Muscular Atrophy (KD/SBMA) is a progressive neurodegenerative disease caused by genetic polyglutamine expansion of the androgen receptor. We have recently found that overexpression of wildtype androgen receptor in skeletal muscle of transgenic mice results in a KD/SBMA phenotype. This surprising result challenges the orthodox view that KD/SBMA requires expression of polyglutamine expanded androgen receptor within motoneurons. Theories relating to the etiology of this disease drawn from studies of human patients, cellular and mouse models are considered with a special emphasis on potential myogenic contributions to as well as the molecular etiology of KD/SBMA.
Keywords: Neuromuscular systems, Sexual dimorphism, Testosterone, Axonopathy, Myopathy, Polyglutamine disease
Role of androgen receptor in neuromuscular systems
Androgens are generally considered to have anabolic actions on neuromuscular systems. Sexually dimorphic neuromuscular systems provide excellent models for understanding how androgens exert these anabolic actions, and studies of these model systems have generally concluded that androgenic anabolic actions are largely, although not exclusively, mediated via androgen receptors (AR). Several reviews on this topic exist, including an article in this issue (Forger and Sengelaub), and so we will only briefly comment on these studies in order to introduce our experiments relating to androgens and neuromuscular pathology.
The importance of AR to sexual differentiation of rodent copulatory neuromuscular systems is illustrated by testicular feminization mutation (Tfm) mutant rats, in which the normal function of androgen in promoting greater levator ani (LA) muscle size and spinal nucleus of the bulbocavernosus (SNB) motoneuron number and size is disabled by a loss of function mutation in the ligand binding domain of the AR gene (Breedlove and Arnold, 1981; Forger et al., 1993; Sengelaub et al., 1989; Yarbrough et al., 1990). Tfm mutants firmly establish the necessity of AR in neuromuscular anabolism, but do not speak to which cellular targets of androgen mediate these effects. It is equally plausible that AR in muscle fibers, motoneurons, and/or those in other cell populations are responsible for sexual differentiation and maintenance of neuromuscular systems and rather elaborate experiments are required to address this site of action question (Johansen et al., 2004; Morris et al., 2004).
Available evidence supports dissociable roles for motoneuron and muscle AR in mediating androgenic action on the SNB system. Some effects of androgen on motoneurons (i.e. those that maintain motoneuron size and gene expression), appear to rely on AR in motoneurons, and are therefore said to be direct, or cell autonomous (Monks et al., 2001, 1999; Watson et al., 2001). Other effects, such as the developmental rescue of both muscle and motoneurons and maintenance of adult motoneuron dendrites, affect motoneurons cell non-autonomously (i.e. indirectly) via actions on target muscles (Freeman et al., 1996; Rand and Breedlove, 1995). Nonetheless, the evidence that implicates AR in muscle in these effects does not resolve which cells in muscle mediate actions on the development and maintenance of neuromuscular systems. It is to answer precisely this question of site of action of androgen in the SNB system that we embarked on a series of studies characterizing the role of AR in skeletal muscle.
We initially addressed the cellular and molecular basis of the unusual androgen sensitivity of the LA muscle and found that along with unusually high AR protein in this muscle in relation to the extensor digitorum longus control muscle (Monks et al., 2006), the LA also has relatively more AR in LA myonuclei but not fibroblast nuclei, two prominent cell types in skeletal muscle that express AR, and which are therefore potential targets of androgen in this tissue (Monks et al., 2004). These studies suggested to us that it would be valuable to generate transgenic (Tg) mice in which AR is overexpressed solely in muscle fibers. We will further describe the generation of these mice and their phenotype below. The ultimate goal of these studies was to cross these Tg mice with testicular feminization (Tfm) mice, which have no functional AR in any tissue. Using this cross, a male mouse in which AR is expressed only in skeletal muscle fibers can be obtained, allowing for a test whether AR in muscle fibers is sufficient to masculinise of the SNB system.
One incidental prediction we had of Tg mice that overexpress AR in muscle fibers is that they would have an exaggerated neuromuscular response to androgens, and to some extent this has been borne out, although the nature of this response is surprising. In pursuing these studies, we discovered that overexpression of AR in skeletal muscle fibers results in a neuromuscular phenotype that recapitulates many of the key features of a human disease named both Kennedy disease and spinal bulbar muscular atrophy (KD/SBMA), which is caused by a characteristic mutation of the AR gene.
KD/SBMA overview and description
A polymorphic number of glutamine (Q) repeats exists in the N-terminal transactivation domain of the AR gene. This polymorphism is thought to contribute to individual differences in androgen sensitivity, as shorter repeat lengths increase AR transactivation, and longer repeat lengths decrease AR transactivation. At the population level, lower repeat lengths are associated with increased incidence of a number of androgen-mediated disorders including benign prostate hyperplasia and prostate cancer, whereas longer repeat lengths, up to about 36, are associated with male infertility (Casella et al., 2003; Mifsud et al., 2001). Q repeat expansion beyond this normal range is associated not only with androgen insensitivity affecting primary and secondary sexual characteristics (e.g., gynecomastia, hypogonadism, progressive loss of libido and erectile function, oligospermia and azoospermia) but also with the progressive neuromuscular atrophy known clinically as KD/SBMA (Kennedy et al., 1968; Walcott and Merry, 2002). KD/SBMA is relatively rare, with prevalence being reported as approximately 1/400,000 (Fischbeck, 1997). In humans, the disorder is characterized by late onset muscular atrophy beginning in the hips, then shoulders and progressing to muscles innervated by the brainstem (bulbar muscles), resulting in difficulty with walking, speech and swallowing (Atsuta et al., 2006; Fischbeck, 1997; Kennedy et al., 1968). The most common cause of death appears to be pneumonia associated with respiratory difficulties (Atsuta et al., 2006). One puzzling aspect of KD/SBMA is that some cells (i.e. specific neuromuscular systems) seem particularly susceptible to pathology, despite AR being expressed in many cell types.
The genetic basis of KD/SBMA was uncovered gradually. This disease shows an X-linked pattern of inheritance, which subsequently localized the allele to the long arm of the X-chromosome through polymorphism linkage analysis (Fischbeck et al., 1986). Molecular genetic studies of the AR gene subsequently revealed that KD/SBMA is specifically associated with increased number of CAG nucleotide repeats (which code for Q) in exon 1 of the AR gene (La Spada et al., 1991). Furthermore, the number of repeats in this polyQ tract is predictive of age of onset and of disease severity, with more repeats resulting in earlier onset and more severe symptomology (Atsuta et al., 2006; La Spada et al., 1992). Most experimental models support a causal role for expansions of this polyQ tract in many pathological features of KD/SBMA as will be described below. Toxicity resulting from polyQ expansion is also observed with several other proteins, and forms a class of neurodegenerative disorders, including Huntington’s disease and the spinal and cerebellar ataxias (Zoghbi and Orr, 2000).
Studies of spinal cord and skeletal muscles from clinical samples have indicated several histopathological hallmarks of KD/SBMA. The most famous of these is AR-immunoreactive inclusions, which are proteinaceous aggregates visible at the light microscope level. Inclusions are observed in diverse neural and non-neural tissues (Adachi et al., 2005). In the spinal cord, motoneurons are typically atrophic, and decreased ventral root size is also observed, reflecting motoneuron loss (Sobue et al., 1989). Dorsal root ganglion pathology has also been reported, corresponding with mild sensory disturbance (Sobue et al., 1989). In muscle, several atrophic features are typically described, including notably clumps of small, angulated muscle fibers (Katsuno et al., 2006b). Inclusions are not reported in skeletal muscle samples from KD/SBMA patients (Adachi et al., 2005). These histopathological changes in muscle have generally been considered to be secondary to denervation resulting from motoneuron pathology, in other words to be neurogenic in origin (Kennedy et al., 1968; Nagashima et al., 1988). More recent studies have interpreted some histopathology in muscle as myogenic in origin (Katsuno et al., 2006b; Yu et al., 2006a).
There is currently no treatment for KD/SBMA, although mouse models have suggested several viable therapeutic approaches. Notably, we now know that the mechanism of polyQ AR pathology in these models is androgen dependent, and this has prompted clinical trials in which androgens are reduced in KD/SBMA patients. A clinical trial of leuprorelin, which reduces testicular production of androgen, indicates that this treatment reduces inclusion formation in scrotal skin of patients, although motor function is not significantly affected (Banno et al., 2006). Additionally, one case has recently been reported in which androgen administration to a KD/SBMA patient resulted in an acceleration of symptoms, which was reversed with cessation of androgen treatment (Kinirons and Rouleau, 2008). Finally, clinical trials organized by the neurogenetics branch of NINDS are currently underway to evaluate the efficacy of Dutasteride, a 5-α-reductase inhibitor, in treating KD/SBMA (trial number NCT00303446).
Experimental models of KD/SBMA
Initial attempts to model KD/SBMA used transfection of polyQ expanded AR in cells of neural (Brooks et al., 1997; Darrington et al., 2002; Kobayashi et al., 2000; Nakajima et al., 1997) and non-neural (Avila et al., 2003; Becker et al., 2000; Mandrusiak et al., 2003; Merry et al., 1998; Takeshita et al., 2006) origin. In embryonic spinal neurons, transfection of AR with 65 Q repeats (AR65Q) did not obviously reduce viability or the ability of AR to translocate (Brooks et al., 1997). However, N-terminal truncated AR constructs with a greater number of repeats (AR97Q and AR112Q) did reduce viability of transfected cells in a Q repeat length-dependent fashion (Merry et al., 1998). AR-immunoreactive inclusions occur in several cellular models, although polyQ expansion AR isn’t always necessary or sufficient for inclusions or toxicity, as will be discussed below.
Several Tg mouse models of KD/SBMA have been developed, all involving an expansion of Q repeats, either as a pure Q tract, or within a truncated or full length AR gene (Table 1). Notably, Tg mice produced by several different labs expressing full length AR gene containing an expansion of the polyQ tract, show many features in common with KD/SBMA in humans. Initial models expressed polyQ AR with cytomegalovirus (CMV) or prion protein (PrP) promoters, which are expressed in many cell types, these models are informative, but are somewhat limited in being forced overexpression constructs.
Table 1.
Mouse models of KD/SBMA
| Original reference | Construct promoter- protein (#Q) | Phenotype | Onset | Androgen dependent |
|---|---|---|---|---|
| Bingham et al. (1995) | Mx-AR(45Q) NSE-AR(45Q) |
Very low transgene expression No phenotype |
N/A | N/A |
| La Spada et al. (1998) | AR-AR(45Q) | No phenotype reported, repeat instability | N/A | N/A |
|
Abel et al. (2001) Chevalier-Larsen et al. (2004) |
PrP-Δ AR(112Q) | CNS but not muscle inclusions, motor deficits, no obvious neuropathy or myopathy, some male infertility | 10–12 weeks | Y |
| Abel et al. (2001) | NFL-Δ AR(112Q) | Some CNS inclusions, motor deficits, no obvious neuropathy | 34 weeks | ? |
| Adachi et al. (2001) | AR-239Q | CNS inclusions, motor deficits, no neuropathy loss | 10–12 weeks | N |
| Katsuno et al. (2002) | CMV/β–actin-AR(97Q) | CNS, muscle inclusions, motor deficits motor deficits, neuropathy; Neurogenic and myogenic myopathy | 10–12 weeks | Y |
| McManamny et al. (2002) | CMV-AR(65Q) CMV-AR(120Q) |
Mild atrophy, not fully characterized No inclusions, motor deficits, neuropathy, testis atrophy, myopathy |
3 weeks | N |
| Sopher et al. (2004) | AR-AR(100Q) | CNS inclusions (but not motoneurons), neuropathy, myopathy, reduced VEGF | 32–48 weeks | ? |
| Yu et al. (2006a) | AR(113Q) KI | CNS and muscle inclusions, motor deficits neuropathy, testicular atrophy, myopathy transcriptional dysregulation, electrophysiological disturbance | Adult | Y |
| Monks et al. (2007) | HAS-AR(22Q) | No inclusions, motor deficits, axonopathy myopathy, transcriptional dysregulation, VEGF | Perinatal/10–12 weeks | Y |
ΔAR = Truncated AR, CMV = cytomegalovirus promoter, HSA = human skeletal actin promoter (muscle specific), KI = knock-in of polyQ tract, Mx = interferon-induced cellular resistance protein, NFL = neurofilament light chain (neuron specific), NSE = Neuron specific enolase, PrP = prion protein promoter.
Two additional models have been developed which include endogenous regulatory elements to address this concern. In the first, a Tg mouse has been generated using a yeast artificial chromosome (YAC) containing a 450 kb clone of the human AR gene modified to include 100 Q repeats. This model has the advantage of likely containing the normal regulatory elements of the AR gene (Sopher et al., 2004). A second mouse model has been created in which a polyQ tract of 113 repeats has been knocked-in to the endogenous AR allele (Yu et al., 2006a). This model has the advantage of having essentially all of the endogenous regulatory elements of the AR gene and additionally does not include potential interactions between the endogenous wildtype (WT) AR and a polyQ AR transgene.
Taken as a whole, these models indicate that several features of KD/SBMA can be reproduced in mice using polyQ alleles. The KD/SBMA phenotype presents clinically with several atrophic symptoms. These mice tend to lose weight and adopt postural abnormalities including an arched back (kyphosis). They also display motor dysfunction, including reduced spontaneous movement, and have pronounced weakness in standard tests such as the length of time which mice can hang suspended (hang test). Most models have onset of symptoms in early adulthood and exhibit evidence of androgen dependence, such as a sex bias in pathology, with males being more affected than females, castration improving affected males, and testosterone inducing symptoms in females (Table 1). A paradoxical reduction in normal androgen function is also observed in some models, which includes impaired male fertility associated with altered testicular morphology (McCampbell et al., 2000; Thomas et al., 2006b; Yu et al., 2006b). Histopathology in some models indicates progressive neurodegeneration of spinal motoneurons and myopathy (McManamny et al., 2002; Sopher et al., 2004; Yu et al., 2006b), and in some models inclusions containing AR are observed in motoneurons and/or muscle (Abel et al., 2001; Adachi et al., 2001; Katsuno et al., 2002; Yu et al., 2006a).
Although these models prove that several features of KD/SBMA can be induced with polyQ alleles, considerable variability exists in the extent to which these various features are present within different models. Notably there has been much discussion concerning the presence or absence of inclusions in affected cells of various mouse models of KD/SBMA and this has allowed for a reconsideration of the mechanistic role of this histopathological aspect of KD/SBMA. Before we consider various etiological mechanisms that might underlie KD/SBMA, we will describe Tg mice that overexpress AR with a WT number of Q repeats solely in skeletal muscle using the human skeletal actin (HSA) promoter. These HSA-AR Tg mice have a phenotype that is remarkably similar to that of polyQ AR Tg mice.
HSA-AR Tg Mice
The AR expressed by HSA-AR is a clone derived from WT laboratory rats (Tan et al., 1988), comprising 22 Q repeats, which is well within the normal range in humans and is typical of mice (Fig. 1A). We elected to use the HSA promoter to drive expression of AR because this promoter has been characterized by several groups (Brennan and Hardeman, 1993; Miniou et al., 1999) and consistently expresses strongly and exclusively in muscle fibers. The HSA promoter was cloned from the human ACTA1 gene, which is an actin isoform specific for muscle lineage cells. In our hands, this promoter does not appear to be expressed in other muscle types, including cardiac or smooth muscle (Monks et al., 2007). We verified HSA expression in two ways: we generated HSA-LacZ reporter mice, which use the same promoter to express β-galactosidase, an enzyme that is easily detected histochemically, and we measured AR protein in several tissues in HSA-AR Tg mice. We obtained very similar results when measuring expression using the HSA promoter to those reported by others (i.e. specific and uniform expression in skeletal muscle fibers). We detected no transgene expression in other tissues. Importantly, we could not detect any transgene expression in nervous tissue including spinal motoneurons (Monks et al., 2007).
Fig. 1.
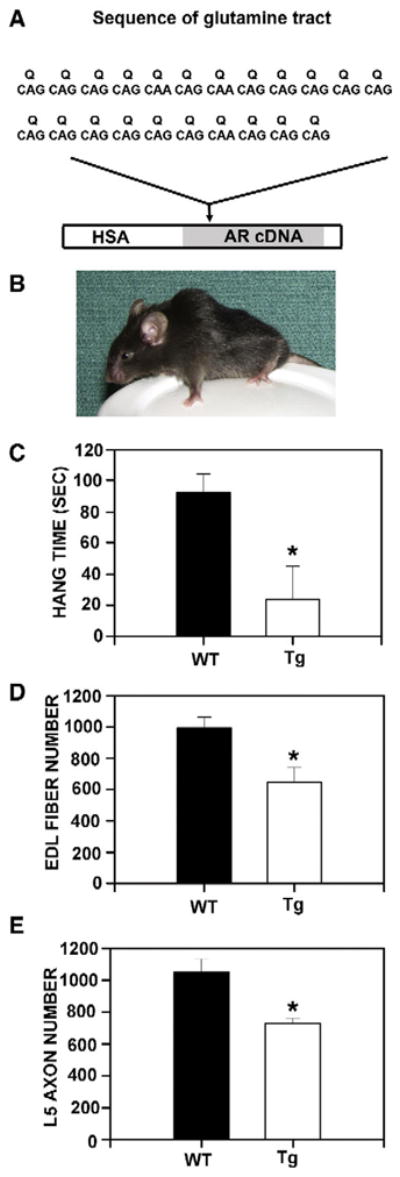
HSA-AR transgene and male phenotype. A) HSA-AR transgene comprises the human skeletal actin (HSA) promoter and a WT rat AR cDNA containing 22 glutamine (Q) repeats, coded for by the sequences indicated. This transgene expresses solely in skeletal muscle fiber lineage (Monks et al., 2007). B) An adult male HSA-AR Tg mouse from the 141 line. Note the small size and curvature of the spine (kyphosis) typical of polyQ AR mice. C) Latency to fall when held suspended from a cage lid (hang time) is significantly shorter for HSA-AR Tg males relative to WT brothers. The number of muscle fibers (D) from the extensor digitorum longus (EDL) muscle and motor axons (E) from the 5th lumbar ventral root (L5 axons) is significantly reduced in HSA-AR Tg males relative to WT brothers. Graphs represent mean±SEM and * indicates significantly different from WT as indicated by a t-test with p<0.05.
We obtained several HSA-AR founders from microinjection of the transgene, most of which were female, providing an initial clue that the transgene was exerting an androgen-dependent pathology. We subsequently determined that the male founders did not express the transgene detectably. After several rounds of breeding, we noticed that very few Tg males were found at weaning, an observation that has been borne out for many generations in two different institutions. Further investigation revealed that many Tg males die perinatally, likely as a result of androgen production by the Tg males themselves at this developmental stage. As adults, most HSA-AR males appear infertile, despite apparently normal testicular anatomy.
The males that survive are characterized by a neuromuscular phenotype that varies in severity between founding lines and according to transgene copy number. The most severe form associated with the highest transgene expression is morphologically and behaviourally obvious at weaning and is characterized by reduced body weight, altered posture (they appear kyphotic —Fig. 1B), decreased spontaneous locomotion, weakness and motor dysfunction (Fig. 1C) typical of the KD/SBMA phenotype of polyQ mice. Along with these motor deficits, males have marked myopathy, including a reduction in the number of muscle fibers overall (Fig. 1D). In addition to myopathy, these males have reduced motor axon number (Fig. 1E), although motoneuron number does not differ between WT and Tg males, suggestive of axonopathy without motoneurons loss. Because we know that the transgene is muscle specific, we may infer that this neuropathology is secondary to muscle pathology.
Motor dysfunction and reduced body weight is exhibited by males of 3 of the 7 founding lines with detectable transgene expression whereas unmanipulated females of all founding lines are behaviourally asymptomatic and have normal body weight. The remaining 4 founding lines also had reduced male viability, and surviving males were behaviorally asymptomatic. This sex limitation of the neuromuscular phenotype is suggestive of androgen dependence, and indeed, castrating HSA-AR Tg males results in dramatic improvement of motor function, including increased stride length (Fig. 2A), and hang time when held suspended (Fig. 2B).
Fig. 2.

Androgen dependence of motor dysfunction in HSA-AR Tg mice. Tg males exhibit impairments in motor tasks such as gait analysis and Hang test, which are somewhat normalized by castration. A) The HSA-AR male’s paw prints (blue for rear paws and red for front paws) are shown prior to castration (middle) and following castration (right). A WT brother’s prints are shown for comparison (left). Stride length, which is related to strength, for each mouse is indicated. B) Latency to fall when held suspended from a cage lid (hang time) is significantly shorter for HSA-AR Tg males (open bars) relative to WT brothers (filled bars) prior to castration (INTACT), but not following castration (CASTRATE). Androgen dependence is also indicated by androgen administration to HSA-AR Tg females (C, D). In this experiment, HSA-AR Tg females (triangles) are compared to WT sisters (circles), all animals are given either testosterone in a silastic capsule (T — empty symbols) or the vehicle capsule alone (VEH — closed symbols). Motor dysfunction is only observed in T treated Tg females (TG/T —open triangles) and begins 3 days following T treatment. Graphs represent mean ± SEM and * indicates significantly different from VEH as indicated by PLSD comparisons with p<0.05.
In further support of the androgen dependence of the HSA-AR phenotype, adult Tg females that are treated with testosterone in the male range exhibit loss of body weight/muscle mass and motor dysfunction (Figs. 2C, D) similar to their male counterparts (including weakness and kyphosis). These symptoms manifest within days of testosterone treatment. Although several myopathic features typical of Tg males were also present in testosterone-treated Tg females, no loss in muscle fiber number was observed in treated females. Interestingly, acute androgen treatment of female HSA-AR mice does not result in axon loss, even though it results in myopathy and motor dysfunction comparable to that observed in Tg males. This observation reinforces the notion that the transgene affects muscle primarily and that axon loss in males reflects more chronic, indirect, effects of androgen.
Of course, behavioural neuroendocrinologists are keenly aware of the potential contributions of aromatization of testosterone and estrogenic mediation of androgenic effects. To address this possible mechanism of androgen action, we additionally treated Tg females with pharmacological doses of estrogen or dihydrotesterone alone, and the results of this experiment strongly support AR-mediation, rather than estrogenic mediation. Females treated with either testosterone or dihydrotestosterone had reduced hang time and body weight, whereas treatment with estradiol was without effect (Fig. 3).
Fig. 3.

Androgens but not estrogens induce atrophy in HSA-AR Tg females. Female HSA-AR mice are asymptomatic unless treated with androgens. Seven days of treatment with silastic capsules containing testosterone (T), dihydrotestosterone (DHT), but not empty capsule vehicles (VEH), or capsules containing 17β estradiol (E2), results in inability to hang suspended (top), and a marked reduction in body weight (bottom). Graphs represent mean ± SEM and * indicates significantly different from VEH as indicated by a t-test with p<0.05.
To further explore potential molecular mechanisms, we determined hindlimb muscle mRNA expression of several genes dysregulated in polyQ models of SBMA (Fig. 4), including upregulation of several that are associated with denervation (MyoD, myogenin and acetylcholine receptor alpha), and downregulation of a candidate growth factor (vascular endothelial growth factor — VEGF). The results of these studies extend the similarity between HSA-AR mice and polyQ AR mouse models and suggest a model similar to that proposed for female-typical development of the SNB system, in which motoneurons retract their axons and are lost due to deprivation of target-derived growth factors (Johansen et al., 2004; Morris et al., 2004).
Fig. 4.

Gene expression is dysregulated in HSA-AR Tg mice. Alteration in gene expression in pooled hindlimb muscles are observed in HSA-AR Tg male mice, including decreased mRNA for vascular endothelial growth factor isoforms 164 and 188 (VEGF — A), and increased mRNA for Myogenin (B), acetylcholine receptor α subunit (AChR — C) and myogenic differentiation gene (MyoD — D). For A–D, fold change is relative to WT samples after normalizing to GAPDH expression. E) Pathology in HSA-AR mice is not due to simple toxicity due to overexpression, because testosterone treatment significantly reduces transgene expression while inducing pathology. The number of copies of AR mRNA in extensor digitorum longus muscle of HSA-AR Tg females treated with silastic capsules containing testosterone (T) or an empty silastic capsule vehicle (VEH) was measured by quantitative PCR normalized to known amounts of AR cDNA. Graphs represent mean± SEM and * indicates significantly different from B as indicated by a t-test with p<0.05.
Is this a simple case of non-specific toxicity?
One might ask whether HSA-AR mice tell us anything at all about KD/SBMA. Perhaps HSA-AR mice exhibit neuromuscular atrophy that results from non-specific toxicity of an overexpressed protein. Would any protein cause similar problems if sufficiently overexpressed? This particular possibility is unlikely for several reasons. Firstly, several strains of Tg mice using the HSA promoter do not have a similar neuromuscular phenotype, notably including HSA-LacZ mice that we generated. Additionally, the HSA-AR phenotype is androgen dependent, which indicates that it is mediated by AR function specifically, rather than non-specific toxicity. Finally, AR mRNA expression in skeletal muscle decreases following androgen treatment of HSA-AR females, and is lower in unmanipulated males than females, when pathology is present (Fig. 4E).
Perhaps a more fruitful approach is to ask to what extent HSA-AR mice and polyQ mice share traits and etiological mechanisms. The extent of overlap in phenotype is already surprising from the perspective of a strong orthodox model in which a critical number of Q repeats is necessary for the KD/SBMA disease process. Of course, it is difficult to say what the critical number of Q repeats is, as vastly greater number of Q repeats (at least 2–3 times greater) are necessary for pathology in cellular and mouse models of KD/SBMA relative to the human case. Conversely, in some cellular models of KD/SBMA, no Q repeats at all are necessary for toxicity or inclusion formation (Stenoien et al., 1999), and AR constructs with WT polyQ repeat lengths can also form inclusions and/or be toxic (although at lower frequency than polyQ AR) when overexpressed in cellular models (Ellerby et al., 1999; Stenoien et al., 1999; Thomas et al., 2004). A similar conclusion has been reached in a Tg model of SCA1, a related polyQ disease, in which overexpression of WT Ataxin 1 is sufficient for polyQ-like neurodegeneration (Fernandez-Funez et al., 2000; Tsuda et al., 2005).
There is currently no information available as to whether AR is overexpressed in human cases of KD/SBMA, although it is unlikely that this possibility has been evaluated. Current genetic diagnosis of KD/SBMA is based solely on the number of Q repeats contained within exon 1 of the AR gene. Because of similarity in clinical presentation between KD/SBMA and amyotrophic sclerosis (ALS), it remains possible that duplications in AR or other mutations affecting AR expression levels in muscle might be identified in some individuals currently diagnosed with ALS, for example. Nonetheless, we are not arguing that current diagnostic criteria for KD/SBMA should be revised, but rather that because overexpression of AR and polyQ expansion of AR manifest similarly, they may converge on similar pathological mechanisms, as is observed in other polyQ disorders.
Despite the surprising similarity in clinical presentation, histopathology and gene expression described above, HSA-AR mice do have several points of contrast with polyQ models, aside from the obvious difference in number of Q repeats. Notably, HSA-AR mice have significant perinatal mortality, whereas other polyQ AR mice generally have a later onset of symptoms, typically exhibiting motor dysfunction in early adulthood (Table 1). Nonetheless, the onset of motor dysfunction in surviving HSA-AR mice is similar to that in other polyQ AR models. An additional difference is that HSA-AR mice do not exhibit inclusions in muscle or neural tissues. It is worth mentioning that both age of onset and the manifestation of inclusions varies considerably amongst polyQ AR mice (Table 1). In the final analysis, we believe that it is reasonable to treat HSA-AR mice as a model of KD/SBMA, albeit an unorthodox one. HSA-AR mice suggest that expression level and number of Q repeats both contribute to, and are sufficient for, converging pathological processes. We additionally propose that muscle is an important determinant of pathology in KD/SBMA.
Theories of KD/SBMA etiology
Site of action
Based on clinical and histopathological findings, KD/SBMA has been traditionally thought of as neurogenic. This explanation of neural pathology resulting from a direct mechanism within motoneurons is appealing as it is parsimonious. In further support of this idea, polyQ AR is sufficient to cause pathology in cultured motoneurons (Merry et al., 1998; Palazzolo et al., 2007), and inclusions are present in motoneurons of KD/SBMA patients (Adachi et al., 2005) and some polyQ AR Tg mouse models (Adachi et al., 2001; Chevalier-Larsen et al., 2004; Katsuno et al., 2002; Yu et al., 2006a). Nonetheless, emerging evidence is challenging this view. For example, polyQ AR is not obviously more prone to form inclusions in motoneurons than in other cell types, even though this population of cells is considered more vulnerable to degeneration. Inclusions are prominent in non-neural cells (including scrotal epithelia of KD/SBMA patients) and are generally widespread throughout the nervous system of polyQ mice rather than being restricted to motoneurons (Table 1).
Another obstacle to the proposal that neurodegeneration results from cell autonomous actions of polyQ AR in motoneurons is that neuron specific expression of polyQ AR has thus far been unsuccessful in producing motoneuron pathology (Abel et al., 2001; Bingham et al., 1995). Mouse models exhibiting evidence of motoneuron pathology have expressed AR using the AR promoter (which is expressed in muscle as well as motoneurons), ubiquitous promoters, or in our model, a muscle specific promoter (Table 1). It is likely that motor deficits in CNS-specific polyQ AR may represent neural dysfunction, perhaps via altered electrophysiological properties of motoneurons or motor nerves (Yu et al., 2006a), rather than frank regression of motoneurons such as motor axon loss or motoneuron loss. Therefore, the available evidence, limited though it is, actually suggests that neural degeneration is non-cell autonomous and originates in skeletal muscle. Supporting this idea, inclusions and muscle pathology precede motoneuron pathology in the one polyQ AR mouse where this possibility has been assessed (Yu et al., 2006a). Finally, HSA-AR mice demonstrate that myogenic pathology is sufficient to reproduce many features of KD/SBMA. Taken together, these results warrant further investigation into myogenic contributions to KD/SBMA.
Given that there is evidence implicating muscle contributions to KD/SBMA, how might muscle dysfunction induce motoneuron degeneration? One salient model is provided by muscle-nerve interactions in which retrograde factors (e.g. VEGF) produced by muscle provide trophic support for motoneurons. Reduction in trophic factor delivery to motoneurons because of myopathy provides a plausible mechanism whereby myogenic neurodegeneration might occur. Another more complicated scenario might involve a deficit in axonal transport, resulting in functional denervation, such as that suggested by studies of polyQ AR models (Katsuno et al., 2006a; Morfini et al., 2006).
Induction of inclusions
Inclusions are reliably associated with pathology in a number of models of KD/SBMA and are considered a hallmark feature of this and other polyQ diseases. Nonetheless, the role of inclusions in the etiology of KD/SBMA remains controversial. There is little definitive evidence to determine whether these inclusions are causal, protective or unrelated to pathology. In a mouse model of a related polyQ disorder (SCA7), inclusions are actually indicative of normal functioning of the ubiquitin-proteasome pathway in neurons, and less healthy neurons are less likely to exhibit inclusions (Bowman et al., 2005), suggesting that inclusions may represent an adaptive response. Inclusions are additionally immunoreactive for ubiquitin and ubiquitin-like proteins as well as for steroid hormone receptor cofactors such as SRC1 and CBP (McCampbell et al., 2000; Stenoien et al., 1999). These observations, along with evidence of atypical proteolysis of polyQ AR (Merry et al., 1998), has led to a hypothesis in which activated polyQ AR contributes to inclusions and becomes toxic via aberrant proteolysis of AR.
Experimentally introducing polyQ peptides with a nuclear localization signal is sufficient to induce apoptosis in cultured cos-7 and PC12 cells, whereas polyQ peptides restricted to the cytoplasm are non-toxic to these cells (Yang et al., 2002), suggesting that the polyQ is itself toxic when localized to the nucleus. In support of this idea, a Tg mouse expressing a polyQ tract under control of the AR promoter has several features of KD/SBMA, including nuclear inclusions (Adachi et al., 2001). Furthermore, ligand-induced nuclear translocation (Takeyama et al., 2002) per se, rather than AR transactivation appear to trigger pathology, since hydroxyflutamide (OHF), which induces nuclear translocation of AR while inhibiting AR transactivation, leads to neurodegeneration in cellular and fly models of KD/SBMA. It is unclear whether this result is generalizable to cell (Darrington et al., 2002) or mouse (Katsuno et al., 2002) models using full length polyQ AR, in which flutamide does not induce inclusions and/or pathology.
Whereas polyQ repeats can be sufficient for inclusion formation and toxicity, it is doubtful that they are necessary for inclusion formation for reasons described above. Nonetheless, in several cellular models, repeat length is correlated with inclusion formation (Merry et al., 1998; Stenoien et al., 1999). Inclusions can be quite prominent within cells and in some cases likely account for a significant proportion of cellular protein. Therefore, it may be reasonable to infer that the functions normally carried out by protein species sequestered in inclusions may be compromised or dysregulated. Most hypotheses relating to this have focused on the potential shortages of transcriptional cofactors and indeed, transcriptional dysregulation is observed in cellular and mouse models of KD/SBMA (Katsuno et al., 2006a; Lieberman et al., 2002; Sopher et al., 2004; Yu et al., 2006a).
Transcriptional dysregulation
Several genes have been implicated as potential contributors to KD/SBMA based on observations of differences in gene expression between polyQ and WT AR. In one such study, transcriptional profiling of polyQ AR cells has been carried out, with the general conclusion that AR65Q has both a loss of normal AR function and a gain of function that is ligand independent (Lieberman et al., 2002).
It is generally unclear whether these changes in gene expression result from aberrant interactions of polyQ AR with the transcriptional machinery or whether they represent an indirect pathological response. In a related polyQ disorder (SCA1), results from a mouse model implicate deficiencies of polyQ Ataxin1 in the function of multiprotein complexes that Ataxin normally forms with a transcriptional repressor in the pathology (Bowman et al., 2007; Lam et al., 2006). These results suggest that polyQ diseases are not solely gain of function mutations, but that loss of function may also contribute to the pathology.
Another source of indirect evidence speaking to transcriptional dysregulation comes from studies of compounds which target cofactors. CREB binding protein (CBP) has been a popular target, as CBP is a histone acetyl-transferase (HAT) cofactor for AR in motoneurons, and is sequestered in inclusions (McCampbell et al., 2000; Minamiyama et al., 2004; Sopher et al., 2004). Additionally, dysregulation of at least one candidate gene (VEGF) is mediated via CBP (Sopher et al., 2004). Sodium butyrate, which broadly inhibits histone deacetylase (HDAC) activity, also reduces symptomology in polyQ AR mice (Minamiyama et al., 2004).
It appears that disruption of AR-cofactor interactions improves pathology via prevention of a toxic gain of function rather than by interfering with the normal function of AR. This conclusion has been reached from two lines of evidence. In the first, an AR-cofactor inhibitor (ASC-J9) improves atrophic symptoms but preserves fertility (Yang et al., 2007), establishing that these are dissociable. In the second, Tg polyQ AR mice crossed onto a Tfm background, thereby eliminating endogenous AR, have worse atrophic symptoms and only partially rescued Tfm phenotype (Thomas et al., 2006b).
This experiment suggests both that normal AR function can actually ameliorate KD/SBMA symptoms and that polyQ AR has greatly reduced WT AR function. It is unclear via what mechanism WT AR function inhibits polyQ AR function in this model. One possibility is that WT AR may promote beneficial gene expression. A further possibility suggested by studies of Ataxin1, is that WT AR may provide competitive antagonism of protein-protein interactions with polyQ AR. One caveat when interpreting the loss of function of polyQ expansion of AR is that polyQ AR is generally expressed at much lower levels than WT AR when driven by the same promoter, and so it may be underexpression of AR rather inherent deficiencies in polyQ AR that explain loss of function in cellular and mouse models (Brooks et al., 1997).
Aberrant interactions of AR with heat shock proteins
AR normally associates with several heat shock proteins (HSP), which are thought to aid in stability of unliganded AR and to facilitate cellular localization of AR, in part via interactions with cytoskeletal proteins. These functions are largely intact in polyQ AR models, most of which show correct nuclear translocation with ligand binding and limited toxicity and aggregates when unliganded. Nonetheless, the sequestration of HSPs and nuclear cofactors in inclusions (Stenoien et al., 1999; Walcott and Merry, 2002), suggests dysfunction in cellular trafficking and/or chaperone functions of HSPs when interacting with polyQ AR.
Experimentally altering HSP function can reduce pathological features in KD/SBMA models, providing support for a functional role for HSPs in mediating toxicity and inclusions formed by polyQ AR. For example, overexpression of HSP40 and/or HSP70 reduces inclusions and cell death in a cellular model (Kobayashi et al., 2000), likely via enhanced protein degradation via the ubiquitin-proteosome pathway (Bailey et al., 2002). Similarly, HSP105a and HSP90 can reduce inclusions and improve viability in cellular models (Ishihara et al., 2003; Thomas et al., 2006a). Furthermore, manipulation of HSP function via geldanamycin or related compounds improve KD/SBMA features in cellular and mouse models (Katsuno et al., 2005; Thomas et al., 2006a; Waza et al., 2005). Whereas the mechanism of geldanamycin/17AAG action is generally thought to occur via HSP40 and HSP70 induction, its protective effect is maintained even in cells that lack the intermediary HSP induction protein (Thomas et al., 2006a), suggesting that this compound likely mediates neuroprotection via HSP90 interactions, rather than induction of HSPs. Ongoing studies are evaluating the efficacy of these compounds in HSA-AR mice.
Aberrant proteolysis of polyQ AR
It appears that polyQ tracts are particularly difficult to protealize, potentially because of transglutaminase activity, in which extensive cross-linking occurs between glutamine and lysine. These transglutamine reactions appear important for polyQ AR toxicity in cultured cells and there is evidence of this type of cross-linking in nervous tissue samples of Tg polyQ AR mice (Mandrusiak et al., 2003). Several studies have proposed that proteolytic fragments containing the polyQ repeat mediate the pathological functions of polyQ AR. One study has implicated caspase-3 cleavage of a site in the N-terminal of AR in both the cytotoxic and inclusion forming aspects of polyQ AR (Ellerby et al., 1999). In a second line of evidence, aberrant proteolytic fragments have been described for greatly expanded glutamine tracts (Merry et al., 1998), which may relate to soluble oligomers formed by polyQ AR (Li et al., 2007). In at least one polyQ AR Tg model, the formation of these microaggregates, which include the polyQ tract, is androgen dependent and precedes KD/SBMA symptomology. These soluble aggregates may ultimately resolve the inconsistent relationship between frank aggregates and pathology which have been observed in mouse models of KD/SBMA, including HSA-AR mice (Table 1).
Summary
Explicit Tg mouse models of KD/SBMA, in which PolyQ AR is expressed either ubiquitously throughout the organism, specifically in the nervous system, or using the AR promoter, have generally resulted in a phenotype that is characterized by at least some of the following: sex limitation (males are affected but females are not), androgen dependence, reduced body weight, kyphosis, motor dysfunction, loss of motor axons, and muscular atrophy characterized by histopathological signs (Table 1). These pathological changes typical of KD/SBMA are generally ascribed to action of the polyQ AR on motoneurons, with muscle effects being a secondary consequence of denervation.
Nonetheless, all of these features are observed in HSA-AR Tg mice in which WT AR is overexpressed solely in muscle fibers and not in motoneurons. Because this phenotype runs contrary to the prevailing view that the symptoms of KD/SBMA result from a primary motoneuron pathology, we believe that characterizing this model will yield new insights into the neuromuscular atrophy typical of KD/SBMA as well as into the roles of AR in muscle fibers.
Footnotes
NINDS Grant support: NS051257 (DAM).
Trainee support: NIH National Research Service Award F31 NS54517-01 (to JAJ).
References
- Abel A, Walcott J, Woods J, Duda J, Merry DE. Expression of expanded repeat androgen receptor produces neurologic disease in transgenic mice. Hum Mol Genet. 2001;10 (2):107–116. doi: 10.1093/hmg/10.2.107. [DOI] [PubMed] [Google Scholar]
- Adachi H, Kume A, Li M, Nakagomi Y, Niwa H, Do J, Sang C, Kobayashi Y, Doyu M, Sobue G. Transgenic mice with an expanded CAG repeat controlled by the human AR promoter show polyglutamine nuclear inclusions and neuronal dysfunction without neuronal cell death. Hum Mol Genet. 2001;10 (10):1039–1048. doi: 10.1093/hmg/10.10.1039. [DOI] [PubMed] [Google Scholar]
- Adachi H, Katsuno M, Minamiyama M, Waza M, Sang C, Nakagomi Y, Kobayashi Y, Tanaka F, Doyu M, Inukai A, Yoshida M, Hashizume Y, Sobue G. Widespread nuclear and cytoplasmic accumulation of mutant androgen receptor in SBMA patients. Brain. 2005;128 (Pt 3):659–670. doi: 10.1093/brain/awh381. [DOI] [PubMed] [Google Scholar]
- Atsuta N, Watanabe H, Ito M, Banno H, Suzuki K, Katsuno M, Tanaka F, Tamakoshi A, Sobue G. Natural history of spinal and bulbar muscular atrophy (SBMA): a study of 223 Japanese patients. Brain. 2006;129 (Pt 6):1446–1455. doi: 10.1093/brain/awl096. [DOI] [PubMed] [Google Scholar]
- Avila DM, Allman DR, Gallo JM, McPhaul MJ. Androgen receptors containing expanded polyglutamine tracts exhibit progressive toxicity when stably expressed in the neuroblastoma cell line, SH-SY 5Y. Exp Biol Med (Maywood) 2003;228 (8):982–990. doi: 10.1177/153537020322800815. [DOI] [PubMed] [Google Scholar]
- Bailey CK, Andriola IF, Kampinga HH, Merry DE. Molecular chaperones enhance the degradation of expanded polyglutamine repeat androgen receptor in a cellular model of spinal and bulbar muscular atrophy. Hum Mol Genet. 2002;11 (5):515–523. doi: 10.1093/hmg/11.5.515. [DOI] [PubMed] [Google Scholar]
- Banno H, Adachi H, Katsuno M, Suzuki K, Atsuta N, Watanabe H, Tanaka F, Doyu M, Sobue G. Mutant androgen receptor accumulation in spinal and bulbar muscular atrophy scrotal skin: a pathogenic marker. Ann Neurol. 2006;59 (3):520–526. doi: 10.1002/ana.20735. [DOI] [PubMed] [Google Scholar]
- Becker M, Martin E, Schneikert J, Krug HF, Cato AC. Cytoplasmic localization and the choice of ligand determine aggregate formation by androgen receptor with amplified polyglutamine stretch. J Cell Biol. 2000;149 (2):255–262. doi: 10.1083/jcb.149.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingham PM, Scott MO, Wang S, McPhaul MJ, Wilson EM, Garbern JY, Merry DE, Fischbeck KH. Stability of an expanded trinucleotide repeat in the androgen receptor gene in transgenic mice. Nat Genet. 1995;9 (2):191–196. doi: 10.1038/ng0295-191. [DOI] [PubMed] [Google Scholar]
- Bowman AB, Yoo SY, Dantuma NP, Zoghbi HY. Neuronal dysfunction in a polyglutamine disease model occurs in the absence of ubiquitin-proteasome system impairment and inversely correlates with the degree of nuclear inclusion formation. Hum Mol Genet. 2005;14 (5):679–691. doi: 10.1093/hmg/ddi064. [DOI] [PubMed] [Google Scholar]
- Bowman AB, Lam YC, Jafar-Nejad P, Chen HK, Richman R, Samaco RC, Fryer JD, Kahle JJ, Orr HT, Zoghbi HY. Duplication of Atxn1l suppresses SCA1 neuropathology by decreasing incorporation of polyglutamine-expanded ataxin-1 into native complexes. Nat Genet. 2007;39 (3):373–379. doi: 10.1038/ng1977. [DOI] [PubMed] [Google Scholar]
- Breedlove SM, Arnold AP. Sexually dimorphic motor nucleus in the rat lumbar spinal cord: response to adult hormone manipulation, absence in androgen-insensitive rats. Brain Res. 1981;225 (2):297–307. doi: 10.1016/0006-8993(81)90837-4. [DOI] [PubMed] [Google Scholar]
- Brennan KJ, Hardeman EC. Quantitative analysis of the human alpha-skeletal actin gene in transgenic mice. J Biol Chem. 1993;268 (1):719–725. [PubMed] [Google Scholar]
- Brooks BP, Paulson HL, Merry DE, Salazar-Grueso EF, Brinkmann AO, Wilson EM, Fischbeck KH. Characterization of an expanded glutamine repeat androgen receptor in a neuronal cell culture system. Neurobiol Dis. 1997;3 (4):313–323. doi: 10.1006/nbdi.1997.0126. [DOI] [PubMed] [Google Scholar]
- Casella R, Maduro MR, Misfud A, Lipshultz LI, Yong EL, Lamb DJ. Androgen receptor gene polyglutamine length is associated with testicular histology in infertile patients. J Urol. 2003;169 (1):224–227. doi: 10.1016/S0022-5347(05)64073-6. [DOI] [PubMed] [Google Scholar]
- Chevalier-Larsen ES, O’Brien CJ, Wang H, Jenkins SC, Holder L, Lieberman AP, Merry DE. Castration restores function and neurofilament alterations of aged symptomatic males in a transgenic mouse model of spinal and bulbar muscular atrophy. J Neurosci. 2004;24 (20):4778–4786. doi: 10.1523/JNEUROSCI.0808-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darrington RS, Butler R, Leigh PN, McPhaul MJ, Gallo JM. Ligand-dependent aggregation of polyglutamine-expanded androgen receptor in neuronal cells. Neuroreport. 2002;13 (16):2117–2120. doi: 10.1097/00001756-200211150-00025. [DOI] [PubMed] [Google Scholar]
- Ellerby LM, Hackam AS, Propp SS, Ellerby HM, Rabizadeh S, Cashman NR, Trifiro MA, Pinsky L, Wellington CL, Salvesen GS, Hayden MR, Bredesen DE. Kennedy’s disease: caspase cleavage of the androgen receptor is a crucial event in cytotoxicity. J Neurochem. 1999;72 (1):185–195. doi: 10.1046/j.1471-4159.1999.0720185.x. [DOI] [PubMed] [Google Scholar]
- Fernandez-Funez P, Nino-Rosales ML, de Gouyon B, She WC, Luchak JM, Martinez P, Turiegano E, Benito J, Capovilla M, Skinner PJ, McCall A, Canal I, Orr HT, Zoghbi HY, Botas J. Identification of genes that modify ataxin-1-induced neurodegeneration. Nature. 2000;408 (6808):101–106. doi: 10.1038/35040584. [DOI] [PubMed] [Google Scholar]
- Fischbeck KH. Kennedy disease. J Inherit Metab Dis. 1997;20 (2):152–158. doi: 10.1023/a:1005344403603. [DOI] [PubMed] [Google Scholar]
- Fischbeck KH, Ionasescu V, Ritter AW, Ionasescu R, Davies K, Ball S, Bosch P, Burns T, Hausmanowa-Petrusewicz I, Borkowska J, et al. Localization of the gene for X-linked spinal muscular atrophy. Neurology. 1986;36 (12):1595–1598. doi: 10.1212/wnl.36.12.1595. [DOI] [PubMed] [Google Scholar]
- Forger NG, Roberts SL, Wong V, Breedlove SM. Ciliary neurotrophic factor maintains motoneurons and their target muscles in developing rats. J Neurosci. 1993;13 (11):4720–4726. doi: 10.1523/JNEUROSCI.13-11-04720.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman LM, Watson NV, Breedlove SM. Androgen spares androgen-insensitive motoneurons from apoptosis in the spinal nucleus of the bulbocavernosus in rats. Horm Behav. 1996;30 (4):424–433. doi: 10.1006/hbeh.1996.0047. [DOI] [PubMed] [Google Scholar]
- Ishihara K, Yamagishi N, Saito Y, Adachi H, Kobayashi Y, Sobue G, Ohtsuka K, Hatayama T. Hsp105alpha suppresses the aggregation of truncated androgen receptor with expanded CAG repeats and cell toxicity. J Biol Chem. 2003;278 (27):25143–25150. doi: 10.1074/jbc.M302975200. [DOI] [PubMed] [Google Scholar]
- Johansen JA, Jordan CL, Breedlove SM. Steroid hormone masculinization of neural structure in rats: a tale of two nuclei. Physiol Behav. 2004;83 (2):271–277. doi: 10.1016/j.physbeh.2004.08.016. [DOI] [PubMed] [Google Scholar]
- Katsuno M, Adachi H, Kume A, Li M, Nakagomi Y, Niwa H, Sang C, Kobayashi Y, Doyu M, Sobue G. Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron. 2002;35 (5):843–854. doi: 10.1016/s0896-6273(02)00834-6. [DOI] [PubMed] [Google Scholar]
- Katsuno M, Sang C, Adachi H, Minamiyama M, Waza M, Tanaka F, Doyu M, Sobue G. Pharmacological induction of heat-shock proteins alleviates polyglutamine-mediated motor neuron disease. Proc Natl Acad Sci U S A. 2005;102 (46):16801–16806. doi: 10.1073/pnas.0506249102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuno M, Adachi H, Minamiyama M, Waza M, Tokui K, Banno H, Suzuki K, Onoda Y, Tanaka F, Doyu M, Sobue G. Reversible disruption of dynactin 1-mediated retrograde axonal transport in polyglutamine-induced motor neuron degeneration. J Neurosci. 2006a;26 (47):12106–12117. doi: 10.1523/JNEUROSCI.3032-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuno M, Adachi H, Waza M, Banno H, Suzuki K, Tanaka F, Doyu M, Sobue G. Pathogenesis, animal models and therapeutics in spinal and bulbar muscular atrophy (SBMA) Exp Neurol. 2006b;200 (1):8–18. doi: 10.1016/j.expneurol.2006.01.021. [DOI] [PubMed] [Google Scholar]
- Kennedy WR, Alter M, Sung JH. Progressive proximal spinal and bulbar muscular atrophy of late onset. A sex-linked recessive trait Neurology. 1968;18 (7):671–680. doi: 10.1212/wnl.18.7.671. [DOI] [PubMed] [Google Scholar]
- Kinirons P, Rouleau GA. Administration of testosterone results in reversible deterioration in Kennedy’s disease. J Neurol Neurosurg Psychiatry. 2008;79 (1):106–107. doi: 10.1136/jnnp.2006.101899. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y, Kume A, Li M, Doyu M, Hata M, Ohtsuka K, Sobue G. Chaperones Hsp70 and Hsp40 suppress aggregate formation and apoptosis in cultured neuronal cells expressing truncated androgen receptor protein with expanded polyglutamine tract. J Biol Chem. 2000;275 (12):8772–8778. doi: 10.1074/jbc.275.12.8772. [DOI] [PubMed] [Google Scholar]
- La Spada AR, Roling DB, Harding AE, Warner CL, Spiegel R, Hausmanowa-Petrusewicz I, Yee WC, Fischbeck KH. Meiotic stability and genotype-phenotype correlation of the trinucleotide repeat in X-linked spinal and bulbar muscular atrophy. Nat Genet. 1992;2 (4):301–304. doi: 10.1038/ng1292-301. [DOI] [PubMed] [Google Scholar]
- La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991;352 (6330):77–79. doi: 10.1038/352077a0. [DOI] [PubMed] [Google Scholar]
- La Spada AR, Peterson KR, Meadows SA, McClain ME, Jeng G, Chmelar RS, Haugen HA, Chen K, Singer MJ, Moore D, Trask BJ, Fischbeck KH, Clegg CH, McKnight GS. Androgen receptor YAC transgenic mice carrying CAG 45 alleles show trinucleotide repeat instability. Hum Mol Genet. 1998;7 (6):959–967. doi: 10.1093/hmg/7.6.959. [DOI] [PubMed] [Google Scholar]
- Lam YC, Bowman AB, Jafar-Nejad P, Lim J, Richman R, Fryer JD, Hyun ED, Duvick LA, Orr HT, Botas J, Zoghbi HY. ATAXIN-1 interacts with the repressor Capicua in its native complex to cause SCA1 neuropathology. Cell. 2006;127 (7):1335–1347. doi: 10.1016/j.cell.2006.11.038. [DOI] [PubMed] [Google Scholar]
- Li M, Chevalier-Larsen ES, Merry DE, Diamond MI. Soluble androgen receptor oligomers underlie pathology in a mouse model of spinobulbar muscular atrophy. J Biol Chem. 2007;282 (5):3157–3164. doi: 10.1074/jbc.M609972200. [DOI] [PubMed] [Google Scholar]
- Lieberman AP, Harmison G, Strand AD, Olson JM, Fischbeck KH. Altered transcriptional regulation in cells expressing the expanded polyglutamine androgen receptor. Hum Mol Genet. 2002;11 (17):1967–1976. doi: 10.1093/hmg/11.17.1967. [DOI] [PubMed] [Google Scholar]
- Mandrusiak LM, Beitel LK, Wang X, Scanlon TC, Chevalier-Larsen E, Merry DE, Trifiro MA. Transglutaminase potentiates ligand-dependent proteasome dysfunction induced by polyglutamine-expanded androgen receptor. Hum Mol Genet. 2003;12 (13):1497–1506. doi: 10.1093/hmg/ddg161. [DOI] [PubMed] [Google Scholar]
- McCampbell A, Taylor JP, Taye AA, Robitschek J, Li M, Walcott J, Merry D, Chai Y, Paulson H, Sobue G, Fischbeck KH. CREB-binding protein sequestration by expanded polyglutamine. Hum Mol Genet. 2000;9 (14):2197–2202. doi: 10.1093/hmg/9.14.2197. [DOI] [PubMed] [Google Scholar]
- McManamny P, Chy HS, Finkelstein DI, Craythorn RG, Crack PJ, Kola I, Cheema SS, Horne MK, Wreford NG, O’Bryan MK, De Kretser DM, Morrison JR. A mouse model of spinal and bulbar muscular atrophy. Hum Mol Genet. 2002;11 (18):2103–2111. doi: 10.1093/hmg/11.18.2103. [DOI] [PubMed] [Google Scholar]
- Merry DE, Kobayashi Y, Bailey CK, Taye AA, Fischbeck KH. Cleavage, aggregation and toxicity of the expanded androgen receptor in spinal and bulbar muscular atrophy. Hum Mol Genet. 1998;7 (4):693–701. doi: 10.1093/hmg/7.4.693. [DOI] [PubMed] [Google Scholar]
- Mifsud A, Sim CK, Boettger-Tong H, Moreira S, Lamb DJ, Lipshultz LI, Yong EL. Trinucleotide (CAG) repeat polymorphisms in the androgen receptor gene: molecular markers of risk for male infertility. Fertil Steril. 2001;75 (2):275–281. doi: 10.1016/s0015-0282(00)01693-9. [DOI] [PubMed] [Google Scholar]
- Minamiyama M, Katsuno M, Adachi H, Waza M, Sang C, Kobayashi Y, Tanaka F, Doyu M, Inukai A, Sobue G. Sodium butyrate ameliorates phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Hum Mol Genet. 2004;13 (11):1183–1192. doi: 10.1093/hmg/ddh131. [DOI] [PubMed] [Google Scholar]
- Miniou P, Tiziano D, Frugier T, Roblot N, Le Meur M, Melki J. Gene targeting restricted to mouse striated muscle lineage. Nucleic Acids Res. 1999;27 (19):e27. doi: 10.1093/nar/27.19.e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monks DA, Vanston CM, Watson NV. Direct androgenic regulation of calcitonin gene-related peptide expression in motoneurons of rats with mosaic androgen insensitivity. J Neurosci. 1999;19 (13):5597–5601. doi: 10.1523/JNEUROSCI.19-13-05597.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monks DA, Getsios S, MacCalman CD, Watson NV. N-cadherin is regulated by gonadal steroids in adult sexually dimorphic spinal motoneurons. J Neurobiol. 2001;47 (4):255–264. doi: 10.1002/neu.1033. [DOI] [PubMed] [Google Scholar]
- Monks DA, O’Bryant EL, Jordan CL. Androgen receptor immunoreactivity in skeletal muscle: enrichment at the neuromuscular junction. J Comp Neurol. 2004;473 (1):59–72. doi: 10.1002/cne.20088. [DOI] [PubMed] [Google Scholar]
- Monks DA, Kopachik W, Breedlove SM, Jordan CL. Anabolic responsiveness of skeletal muscles correlates with androgen receptor protein but not mRNA. Can J Physiol Pharm. 2006;84 (2):273–277. doi: 10.1139/y05-157. [DOI] [PubMed] [Google Scholar]
- Monks DA, Johansen JA, Mo K, Rao PR, Eagleson B, Yu Z, Lieberman AP, Breedlove SM, Jordan CL, Lieberman AP, Breedlove SM, Jordan CL. Overexpression of wildtype androgen receptor in muscle recapitulates polyglutamine disease. Proc Natl Acad Sci U S A. 2007;104 (46):18259–18264. doi: 10.1073/pnas.0705501104. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini G, Pigino G, Szebenyi G, You Y, Pollema S, Brady ST. JNK mediates pathogenic effects of polyglutamine-expanded androgen receptor on fast axonal transport. Nat Neurosci. 2006;9 (7):907–916. doi: 10.1038/nn1717. [DOI] [PubMed] [Google Scholar]
- Morris JA, Jordan CL, Breedlove SM. Sexual differentiation of the vertebrate nervous system. Nat Neurosci. 2004;7 (10):1034–1039. doi: 10.1038/nn1325. [DOI] [PubMed] [Google Scholar]
- Nagashima T, Seko K, Hirose K, Mannen T, Yoshimura S, Arima R, Nagashima K, Morimatsu Y. Familial bulbo-spinal muscular atrophy associated with testicular atrophy and sensory neuropathy (Kennedy–Alter–Sung syndrome). Autopsy case report of two brothers. J Neurol Sci. 1988;87 (2–3):141–152. doi: 10.1016/0022-510x(88)90240-7. [DOI] [PubMed] [Google Scholar]
- Nakajima H, Kimura F, Nakagawa T, Ikemoto T, Furutama D, Shinoda K, Kato S, Shimizu A, Ohsawa N. Effects of androgen receptor polyglutamine tract expansion on proliferation of NG108-15 cells. Neurosci Lett. 1997;222 (2):83–86. doi: 10.1016/s0304-3940(97)13348-1. [DOI] [PubMed] [Google Scholar]
- Palazzolo I, Burnett BG, Young JE, Brenne PL, La Spada AR, Fischbeck KH, Howell BW, Pennuto M. Akt blocks ligand binding and protects against expanded polyglutamine androgen receptor toxicity. Hum Mol Genet. 2007;16 (13):1593–1603. doi: 10.1093/hmg/ddm109. [DOI] [PubMed] [Google Scholar]
- Rand MN, Breedlove SM. Androgen alters the dendritic arbors of SNB motoneurons by acting upon their target muscles. J Neurosci. 1995;15 (6):4408–4416. doi: 10.1523/JNEUROSCI.15-06-04408.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengelaub DR, Jordan CL, Kurz EM, Arnold AP. Hormonal control of neuron number in sexually dimorphic spinal nuclei of the rat: II. Development of the spinal nucleus of the bulbocavernosus in androgen-insensitive (Tfm) rats. J Comp Neurol. 1989;280 (4):630–636. doi: 10.1002/cne.902800412. [DOI] [PubMed] [Google Scholar]
- Sobue G, Hashizume Y, Mukai E, Hirayama M, Mitsuma T, Takahashi A. X-linked recessive bulbospinal neuronopathy. A clinicopathological study. Brain. 1989;112 (Pt 1):209–232. doi: 10.1093/brain/112.1.209. [DOI] [PubMed] [Google Scholar]
- Sopher BL, Thomas PS, Jr, LaFevre-Bernt MA, Holm IE, Wilke SA, Ware CB, Jin LW, Libby RT, Ellerby LM, La Spada AR. Androgen receptor YAC transgenic mice recapitulate SBMA motor neuronopathy and implicate VEGF164 in the motor neuron degeneration. Neuron. 2004;41 (5):687–699. doi: 10.1016/s0896-6273(04)00082-0. [DOI] [PubMed] [Google Scholar]
- Stenoien DL, Cummings CJ, Adams HP, Mancini MG, Patel K, DeMartino GN, Marcelli M, Weigel NL, Mancini MA. Polyglutamine-expanded androgen receptors form aggregates that sequester heat shock proteins, proteasome components and SRC-1, and are suppressed by the HDJ-2 chaperone. Hum Mol Genet. 1999;8 (5):731–741. doi: 10.1093/hmg/8.5.731. [DOI] [PubMed] [Google Scholar]
- Takeshita Y, Fujinaga R, Zhao C, Yanai A, Shinoda K. Huntingtin-associated protein 1 (HAP1) interacts with androgen receptor (AR) and suppresses SBMA-mutant-AR-induced apoptosis. Hum Mol Genet. 2006;15 (15):2298–2312. doi: 10.1093/hmg/ddl156. [DOI] [PubMed] [Google Scholar]
- Takeyama K, Ito S, Yamamoto A, Tanimoto H, Furutani T, Kanuka H, Miura M, Tabata T, Kato S. Androgen-dependent neurodegeneration by polyglutamine-expanded human androgen receptor in Drosophila. Neuron. 2002;35 (5):855–864. doi: 10.1016/s0896-6273(02)00875-9. [DOI] [PubMed] [Google Scholar]
- Tan JA, Joseph DR, Quarmby VE, Lubahn DB, Sar M, French FS, Wilson EM. The rat androgen receptor: primary structure, autoregulation of its messenger ribonucleic acid, and immunocytochemical localization of the receptor protein. Mol Endocrinol. 1988;2 (12):1276–1285. doi: 10.1210/mend-2-12-1276. [DOI] [PubMed] [Google Scholar]
- Thomas M, Dadgar N, Aphale A, Harrell JM, Kunkel R, Pratt WB, Lieberman AP. Androgen receptor acetylation site mutations cause trafficking defects, misfolding, and aggregation similar to expanded glutamine tracts. J Biol Chem. 2004;279 (9):8389–8395. doi: 10.1074/jbc.M311761200. [DOI] [PubMed] [Google Scholar]
- Thomas M, Harrell JM, Morishima Y, Peng HM, Pratt WB, Lieberman AP. Pharmacologic and genetic inhibition of hsp90-dependent trafficking reduces aggregation and promotes degradation of the expanded glutamine androgen receptor without stress protein induction. Hum Mol Genet. 2006a;15 (11):1876–1883. doi: 10.1093/hmg/ddl110. [DOI] [PubMed] [Google Scholar]
- Thomas PS, Jr, Fraley GS, Damian V, Woodke LB, Zapata F, Sopher BL, Plymate SR, La Spada AR. Loss of endogenous androgen receptor protein accelerates motor neuron degeneration and accentuates androgen insensitivity in a mouse model of X-linked spinal and bulbar muscular atrophy. Hum Mol Genet. 2006b;15 (14):2225–2238. doi: 10.1093/hmg/ddl148. [DOI] [PubMed] [Google Scholar]
- Tsuda H, Jafar-Nejad H, Patel AJ, Sun Y, Chen HK, Rose MF, Venken KJ, Botas J, Orr HT, Bellen HJ, Zoghbi HY. The AXH domain of Ataxin-1 mediates neurodegeneration through its interaction with Gfi-1/senseless proteins. Cell. 2005;122 (4):633–644. doi: 10.1016/j.cell.2005.06.012. [DOI] [PubMed] [Google Scholar]
- Walcott JL, Merry DE. Ligand promotes intranuclear inclusions in a novel cell model of spinal and bulbar muscular atrophy. J Biol Chem. 2002;277 (52):50855–50859. doi: 10.1074/jbc.M209466200. [DOI] [PubMed] [Google Scholar]
- Watson NV, Freeman LM, Breedlove SM. Neuronal size in the spinal nucleus of the bulbocavernosus: direct modulation by androgen in rats with mosaic androgen insensitivity. J Neurosci. 2001;21 (3):1062–1066. doi: 10.1523/JNEUROSCI.21-03-01062.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waza M, Adachi H, Katsuno M, Minamiyama M, Sang C, Tanaka F, Inukai A, Doyu M, Sobue G. 17-AAG, an Hsp90 inhibitor, ameliorates polyglutamine-mediated motor neuron degeneration. Nat Med. 2005;11 (10):1088–1095. doi: 10.1038/nm1298. [DOI] [PubMed] [Google Scholar]
- Yang W, Dunlap JR, Andrews RB, Wetzel R. Aggregated polyglutamine peptides delivered to nuclei are toxic to mammalian cells. Hum Mol Genet. 2002;11 (23):2905–2917. doi: 10.1093/hmg/11.23.2905. [DOI] [PubMed] [Google Scholar]
- Yang Z, Chang YJ, Yu IC, Yeh S, Wu CC, Miyamoto H, Merry DE, Sobue G, Chen LM, Chang SS, Chang C. ASC-J9 ameliorates spinal and bulbar muscular atrophy phenotype via degradation of androgen receptor. Nat Med. 2007;13 (3):348–353. doi: 10.1038/nm1547. [DOI] [PubMed] [Google Scholar]
- Yarbrough WG, Quarmby VE, Simental JA, Joseph DR, Sar M, Lubahn DB, Olsen KL, French FS, Wilson EM. A single base mutation in the androgen receptor gene causes androgen insensitivity in the testicular feminized rat. J Biol Chem. 1990;265 (15):8893–8900. [PubMed] [Google Scholar]
- Yu Z, Dadgar N, Albertelli M, Gruis K, Jordan C, Robins DM, Lieberman AP. Androgen-dependent pathology demonstrates myopathic contribution to the Kennedy disease phenotype in a mouse knock-in model. J Clin Invest. 2006a;116 (10):2663–2672. doi: 10.1172/JCI28773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Dadgar N, Albertelli M, Scheller A, Albin RL, Robins DM, Lieberman AP. Abnormalities of germ cell maturation and sertoli cell cytoskeleton in androgen receptor 113 CAG knock-in mice reveal toxic effects of the mutant protein. Am J Pathol. 2006b;168 (1):195–204. doi: 10.2353/ajpath.2006.050619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY, Orr HT. Glutamine repeats and neurodegeneration. Annu Rev Neurosci. 2000;23:217–247. doi: 10.1146/annurev.neuro.23.1.217. [DOI] [PubMed] [Google Scholar]