

Abstract
Although prospective logistic regression is the standard method of analysis for case-control data, it has been recently noted that in genetic epidemiologic studies one can use the “retrospective” likelihood to gain major power by incorporating various population genetics model assumptions such as Hardy-Weinberg-Equilibrium (HWE), gene-gene and gene-environment independence. In this article, we review these modern methods and contrast them with the more classical approaches through two types of applications (i) association tests for typed and untyped single nucleotide polymorphisms (SNPs) and (ii) estimation of haplotype effects and haplotype-environment interactions in the presence of haplotype-phase ambiguity. We provide novel insights to existing methods by construction of various score-tests and pseudo-likelihoods. In addition, we describe a novel two-stage method for analysis of untyped SNPs that can use any flexible external algorithm for genotype imputation followed by a powerful association test based on the retrospective likelihood. We illustrate applications of the methods using simulated and real data.
Keywords: Case-control studies, Empirical-Bayes, Genetic epidemiology, Haplotypes, Model averaging, Model robustness, Model selection, Retrospective studies, Shrinkage
1 Introduction
Case-control study designs are now widely used to study the role of genetic susceptibility in the etiology of rare complex diseases. Typically, a case-control study involves recruiting all or a large fraction of the diseased subjects (cases) that arise in an underlying study base and then sampling a comparable number of healthy subjects (controls), ideally from the exact same study base, and possibly matched with the cases by some socio-demographic characteristics such as race, age and gender. Biological samples and questionnaire data collected on the sampled subjects are then used to determine their genetic susceptibility, such as SNP genotypes and history of some non-genetic (environmental) exposures. For rare diseases such as cancers, case-control studies are cost efficient compared to a cross-sectional or prospective cohort studies because they dramatically reduce the number of non-diseased subjects to study.
In general, the standard method for analysis of case-control data is the prospective logistic regression ignoring the retrospective nature of the underlying design. The validity of this approach relies on the classic results by Cornfield (1956) who showed the equivalence of prospective- and retrospective odds-ratios. The efficiency of the approach was established in two other classic papers by Andersen (1970) and Prentice and Pyke (1979), who showed that the prospective analysis of case-control data yields the proper maximum-likelihood estimates of the odds ratio parameters of the logistic model under a “semiparametric” setup that allows the distribution of the underlying covariates to remain completely unrestricted. More recently, it has been shown that even in the presence of missing data and measurement error in covariates, the “prospective” treatment of case-control data can yield proper maximum-likelihood estimates as long as the distribution of the underlying covariates is allowed to remain unrestricted (Roeder et al., 1996).
A special feature for studies in genetic epidemiology is that it is often reasonable to assume certain models for the population distribution of the genetic and environmental covariates of interest. The Hardy-Weinberg-Equilibrium (HWE) law, for example, which implies a simple relationship between allele and genotype frequencies at a given chromosomal locus, is a natural model for a random mating, large, stable population in the absence of new genetic mutations, inbreeding and selective survivorship among genotypes (see Hartl and Clark, 2007, Chapter 3). Genes which are physically apart and hence are not expected to be in linkage disequilibrium (LD) are also expected to be independently distributed in a homogeneous population. It is often also natural to assume a subject’s genetic susceptibility, a factor which is determined at birth, is independent of his/her subsequent environmental exposures. A pertinent question then is what is the most appropriate method for analysis of case-control data in genetic epidemiology where some natural model assumptions exist for the distribution of genetic and environmental factors in the underlying population.
We will assume data on some genetic (G) and environmental (E) exposures are collected in a case-control study involving N0 controls (D = 0) and N1 cases (D = 1). If one ignores the retrospective nature of the case-control design, one can conduct inference based on the prospective-likelihood
| (1) |
where N = N1 + N0. The fundamental likelihood for case-control data, however, known as the “retrospective” likelihood, is given by
| (2) |
In the absence of any missing data, it is evident from the classical theory that the prospective-likelihood (1) provides a valid way of testing and estimation of the odds-ratio association parameters of the underlying logistic regression model. In fact, the prospective-likelihood yields the same maximum-likelihood estimates for the odds-ratio association parameters that could be obtained by maximization of the proper retrospective likelihood (2) while allowing pr(G, E), the joint distribution of G and E, to remain completely non-parametric. Under constraints on pr(G, E), however, the retrospective likelihood would not yield the same maximum-likelihood estimator as that from the prospective likelihood. More importantly, the retrospective-likelihood can exploit various population genetics model assumptions such as HWE, gene-gene and gene-environment independence to gain major efficiency over the prospective-likelihood for inference on various association and interaction parameters. At the same time, if the underlying model assumptions are violated, then the use of the retrospective likelihood can lead to serious bias for both testing and estimation procedures. In the presence of missing data, a further complication is that the use of the prospective likelihood may not be even strictly valid in certain settings, such as that described in Section 4 for estimation of haplotype effects, where for the purpose of identifiability LP also requires some modeling assumptions, thus destroying its equivalence with LR that is known to hold under unspecified covariate distribution. Thus, to date, a debate remains about the most appropriate method of analysis of case-control studies in genetic epidemiology.
In this article, we will review some modern developments for analysis of case-control studies in genetic epidemiology using the prospective- and retrospective-likelihoods. We will describe the methods primarily through two different types of applications: (a) association testing for genotyped and imputed single nucleotide polymorphisms (SNP) (Section 2 and Section 3) and (b) estimation of haplotype effects and haplotype-environment interactions in the presence of phase ambiguity (Section 4). In each section, we aim to provide new intuitive insights into the alternative methods by constructions of various score tests (Section 2 and 3) and pseudo-likelihoods (Section 4). As a byproduct, in Section 3, we also propose a novel “retrospective” method for association testing for untyped SNPs which can easily use any external algorithm for imputation of genotypes. In each section, we will use numerical examples to illustrate the bias and efficiency trade-off between the alternative methods. We will conclude the article with a discussion and recommendations for practical data analysis.
2 Association Analysis for Single Nucleotide Polymorphisms (SNPs)
2.1 The Prospective Approach
The genotype information for an individual SNP in a case-control study can be represented by the 2 × 3 contingency table defined by cross-tabulation of case-control and genotype status. Let D be the indicator of case (D = 1) or control (D = 0) status and let G be the number of minor alleles carried by an individual (G = 0, 1, 2). Let ndg denote the number of subjects with genotype G = g and disease status D = d observed in the case-control sample. Suppose we are interested in testing the association of the disease outcome with a SNP-genotype using a population logistic regression model of the form
| (3) |
where the function m(·) is chosen in a suitable way to reflect an assumed mode of genetic effect. If, for example, G denotes the count for the minor allele at a SNP locus, then one can chose m(G) = G, m(G) = I(G ≥ 1) or m(G) = I(G = 2) to model the effect of the minor allele as additive (in the logistic scale), dominant or recessive. One can also consider a saturated model by allowing m(G) to be a vector of two dummy variables associated with heterozygous (G = 1) and homozygous variant (G = 2) genotypes and β to be the corresponding log-odds-ratios. The prospective analysis of case-control data yields an asymptotically unbiased estimate for the genotype-odds-ratio parameters β, but not for the intercept parameter α.
The score-function for β under the prospective-likelihood (1) can be written as
Under the null hypothesis, β = 0, we can estimate p = pr(D = 1|Gi) as p̂ = N1/(N1 + N0) since under that hypothesis, G does not influence D. Then the score function can be written as
which is proportional to the difference between the empirical mean of m(G) in the cases (D = 1) and in the controls (D = 0). We suppose without loss of generality that the indices for the cases are {i = 1, …, N1} and those for the controls are {i = N1 + 1, …, N1 + N0}. If, for example, we assume m(G) = G, i.e. the additive effect, then corresponds to the numerator of the Cochran-Armitage trend test (?, Chapter 7) that is widely used for single-SNP association testing. More generally, a “prospective” score-test can be constructed under any genetic model based on and its variance under the null hypothesis of no association that be estimated by
where Vm(G) the pooled-sample variance of m(Gi).
2.2 Retrospective Approach
The retrospective likelihood, LR, for the genotype data of a single-SNP can be written as the product of two sets of multinomial probabilities:
where pdg = pr(G = g|D = d), d = 0 and 1, denotes the population genotype frequencies for the controls and the cases, respectively. Given the genotype probabilities for the controls, we can characterize the genotype probabilities for the cases according to the formula (Satten and Kupper, 1993)
| (4) |
where ψg(β) denotes the odds-ratio associated with the genotype G = g as specified by the logistic model (3). Thus, the retrospective likelihood can be parameterized in terms of the genotype probabilities of the controls and the disease-odds-ratio parameters β. The maximization of the retrospective likelihood LR, without imposing any further constraints on the genotype probabilities for the controls, will lead to the same estimator for β that would be obtained by maximization of LP (Prentice and Pyke, 1979). In fact, it can be shown that the retrospective- and prospective-profile likelihoods of β become identical after maximization of the corresponding likelihoods with respect to the associated nuisance parameters (Roeder et al., 1996). Thus, the associated tests, including score-, Wald- and likelihood-ratio tests, are identical under the retrospective and prospective likelihoods.
Now suppose we are willing to assume that HWE holds in the underlying population and that the disease is rare so that HWE also holds approximately in the control population. In the retrospective likelihood LR, we can write the genotype probabilities for the controls as a function of the frequency, f, of the minor allele as
It is easy to show that the score-function for β associated with the retrospective likelihood can be written as
which under the null hypothesis of no association reduces to
| (5) |
Moreover, under the null hypothesis, the allele frequency f can be substituted for by its maximum-likelihood estimate
| (6) |
where n+g denotes the frequency for genotype G = g in the pooled sample of cases and controls. Thus, corresponds to the difference between the empirical mean of the function m(G) in cases and its expected value under HWE and the null hypothesis of no association. In contrast, note that, corresponds to the difference between the empirical mean of the function m(G) in cases and the empirical mean for the same function in the controls. If the expectation in the retrospective score-function (5) is estimated empirically without assuming HWE then, as expected, it can be easily shown that the retrospective and prospective scores are the same. If, however, we assume HWE to evaluate the retrospective score function, then it would have smaller variance than that for the prospective score. In particular, this can be seen from the estimate of the variance estimate given by
where
By the Cauchy-Schwartz inequality, asymptotically, implying that the retrospective score test is asymptotically more powerful than its prospective counterpart when the assumption of HWE is valid.
Chen and Chatterjee (2007) compared the performance of 2 d.f. Wald-tests of association based on the retrospective and prospective likelihoods. They observed major gains in power for the test based on the retrospective-likelihood for the detection of non-multiplicative effects, e.g., recessive effects. Notice that if we assume an additive model, i.e. m(G) = G, then the prospective and retrospective score-functions and become identical because in this case . The larger the departure of the effect of a SNP from the additive form, the greater the gain in efficiency for the retrospective method. Application of retrospective methods for association testing, however, requires caution because of their sensitivity to the underlying model assumption. In particular, it can be seen from the formula of that the unbiasedness of that score function crucially depends on the assumption of HWE being correct for the underlying population. Satten and Epstein (2004) and Chen and Chatterjee (2007) have noted that even modest violation of HWE can cause serious inflation in Type-I error in association tests based on the retrospective likelihood.
2.3 Empirical-Bayes Methods
Luo et al. (2009) considered an empirical-Bayes type shrinkage estimation approach to develop a 2 d.f. single-SNP association test that can gain power by exploiting the model assumptions of HWE for the underlying population and yet is resistant to bias when the model assumptions are violated. The method involves estimation of genotype-specific disease odds ratio parameters by data-adaptive “shrinkage” of a “prospective” model-free estimator that does not require the HWE assumption towards a “retrospective” model-based estimator that directly exploits the HWE constraints. The amount of “shrinkage” is sample-size and data-adaptive, so that in large samples the method has no bias whether the assumption of HWE holds or not and yet the method can gain efficiency by shrinking the analysis towards HWE, but only to the extent that the data validate the assumptions. In what follows, we provide some insight into the empirical-Bayes method through the construction of a score-test. For numerical illustration, however, we will focus on the Wald test as originally developed in Luo et al. (2009).
Let and denote the sample mean and variance for the function m(G), respectively. Further, let τ̂ = m̄(G) − Ef̂,HWEm(G) denote the difference between the empirical and expected means of m(G) when the latter quantity is computed assuming HWE and under the estimate of allele frequency f̂ given in (6). Intuitively, τ̂ can be viewed as an estimate of the bias in estimation of the population mean of m(G) under the assumption of HWE. An empirical-Bayes type score function can be now defined as
| (7) |
where EEB {m(G)} is the empirical-Bayes estimate for the mean of the function m(G) underH0, given by
Thus, EEB {m(G)} corresponds to a weighted average of the empirical mean of m(G) and its expected mean under HWE, with the weights defined by an estimate of the bias for the estimate of the population mean of m(G) under HWE and an estimate of the variance of the empirical mean of m(G). As τ̂2 decreases, i.e. the evidence of bias due to the violation of HWE becomes smaller, EEB {m(G)} gives more weight to the more precise HWE-based estimator of the population mean of m(G). Conversely, as decreases, i.e the sample mean of m(G) becomes more precise, then EEB {m(G)} puts more weight to the robust model-free estimator m̄(G). The original perspective for constructing such weighted combinations of model-based and model free estimators from an empirical-Bayes point of view can be found in Mukherjee and Chatterjee (2008). Simple methods for variance estimation for such estimators have been also described in that article.
2.4 The Cancer Genetics Markers of Susceptibility (CGEMS) Study
We illustrate the performance of alternative 2 d.f. single SNP association tests using data from the Cancer Genetics Markers of Susceptibility (CGEMS) study (Yeager et al., 2007; Hunter et al., 2007; Thomas et al., 2008), an NCI enterprize initiative to conduct multi-stage whole-genome association studies to identify susceptibility genes giving rise to increased risks of prostate and breast cancers. In this article, we will focus on data from the initial scan for the prostate cancer study, involving genotype data on about 550, 000 SNPs from 1, 172 cases and 1, 157 controls. The details of the CGEMS study design and the results from the initial scan and subsequent replication studies can be found at the web site https://caintegrator.nci.nih.gov/cgems/.
We consider 449, 698 SNPs from 22 non-sex chromosomes with minor allele frequencies larger than 0.05. Table 1 displays the empirical proportions of the number of SNPs that are found to be significant at different nominal significance levels using 2 d.f. tests based on three different methods: (a) prospective; (b) retrospective and (c) empirical-Bayes (see Luo et al. (2009) for more details). For a well-designed study and a robust analytic method, the empirical proportions are expected to be fairly close to the nominal significant levels, given that vast majority of the SNPs are likely to be not associated with the disease. In Table 1, we observe that the empirical proportions of significant SNPs found by the prospective method closely follows the nominal significance levels. In contrast, the corresponding proportions for the retrospective test deviate severely from the nominal values in the range of α ≤ 10−3, indicating significantly inflated type-I error due to the violation of HWE for many SNPs. The last column of Table 1 shows that the empirical-Bayes procedure essentially corrects for all the bias of the retrospective method due to the violation of the HWE assumption.
Table 1.
The empirical proportions of significant SNPs detected by different methods at different nominal significance levels in the CGEMS prostate cancer study.
| α | Prospective | Retrospective | Empirical-Bayes |
|---|---|---|---|
| 5e-2 | 5.01e-2 | 5.66e-2 | 4.49e-2 |
| 1e-2 | 0.98e-2 | 1.43e-2 | 0.87e-2 |
| 1e-3 | 1.05e-3 | 3.85e-3 | 1.00e-3 |
| 1e-4 | 1.27e-4 | 2.24e-3 | 1.31e-4 |
| 1e-5 | 2.67e-5 | 1.76e-3 | 3.34e-5 |
| 1e-6 | 2.22e-6 | 1.47e-3 | 4.45e-6 |
Next, we conducted a simulation study to investigate the performance of various tests in ranking a true susceptibility locus in a genome-wide association study (GWAS) that include hundreds of thousands of “null” SNPs. To generate realistic linkage disequilibrium patterns, we simulated GWAS data mimicking the CGEMS study itself. Given MAF among controls and the disease-genotype odds-ratio parameters for a chosen susceptibility locus, we simulate genotype data at that locus for the cases and controls separately from the corresponding multinomial distributions. Given the genotype data at the susceptibility locus for a case or a control, we simulate genotype data for the remainder of the SNPs by assigning the whole genotype profile for a randomly selected subject from the controls of the CGEMS study who have the same genotype data at the given susceptibility locus as the sampled subject in our simulation study. This algorithm, as originally described by Yu et al. (2009), assumes that given the genotypes for the susceptibility locus, the risk of the disease is independent of all the remaining SNPs. We simulated 50 data sets with approximately 550 cases and 550 controls. For each data set, we tested for association for each of the approximately 450,000 SNPs using the prospective, retrospective and empirical-Bayes methods. The rank of the disease-associated SNP is obtained by sorting all the p-values in ascending order.
Table 2 displays the median ranks obtained by three methods for a true disease-associated SNP that has a recessive effect with a log-odds-ratio of β = log(3). As expected, the ranks of all tests decrease as the MAF increases. Comparing the ranks of different tests at a specific MAF, we can see that the standard prospective method generally has the lowest power in the sense that it assigns much higher rank to the susceptibility SNP than the two other tests. When MAF=0.1, we observe that the pure retrospective method performs the best in the sense that it assigns the lowest rank to the susceptibility SNPs among all the methods. In contrast when MAF≥ 0.2, we observe that the empirical-Bayes procedure assigns considerable lower rank to the susceptibility SNP than the pure retrospective method. Intuitively, the results can be explained from the fact that the retrospective method yields low p-values for many null SNPs due to the violation of the HWE assumption (see Table 1) and thus dilutes the rank of the real susceptibility SNP.
Table 2.
Simulated median ranks of a true susceptibility SNP with a recessive effect and log-odds-ratio value of log(3) for alternative tests. The results are based on 50 simulated datasets, each of which has approximately 550 cases and 550 controls and 450,000 SNPs.
| MAF | Prospective | Retrospective | Empirical-Bayes |
|---|---|---|---|
| 0.1 | 112163 | 8117 | 44319 |
| 0.2 | 1888 | 203 | 52 |
| 0.3 | 656 | 210 | 27 |
| 0.4 | 15 | 82 | 2 |
3 Association Analysis for Imputed SNPs
The forms of the prospective- and retrospective-scores suggest how they can be modified easily for SNPs that may not have been directly genotyped, but can be “imputed” conditional on neighboring SNPs and estimates of linkage disequilibrium from HapMap or other similar data bases. Let 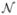 (G) denote the neighboring genotype information for an untyped SNP-locus with unobserved genotype G. The prospective score for such an untyped SNP can be defined by taking the conditional expectation of the “complete data” score-function
given the observed data, i.e the neighboring genotype information. More formally, the prospective score for an untyped SNP can be written as
(G) denote the neighboring genotype information for an untyped SNP-locus with unobserved genotype G. The prospective score for such an untyped SNP can be defined by taking the conditional expectation of the “complete data” score-function
given the observed data, i.e the neighboring genotype information. More formally, the prospective score for an untyped SNP can be written as
| (8) |
where the conditional expectations are taken with respect to a suitable imputation model such as those described by Nicolae (2006), Marchini et al. (2007) and others. The retrospective score for an untyped SNP can be similarly defined by the conditional expectation of the “complete data” retrospective score-function
given the observed data (G) in the form
| (9) |
Notice that in the retrospective score function, the contribution of the term EHWE,f {m(G)} is a constant term given the allele frequency f. The estimation of the allele frequency f for an untyped SNP, however, requires imputation. In particular, under the “complete data” model we can write the estimate of the allele frequency under the null hypothesis of no association as
Thus, given an imputation model, we can estimate the allele frequency f as
| (10) |
We further need the variances for and under the null hypothesis to obtain the corresponding score tests. The variance of can be estimated as
where VE{m(G)|(G)} is the pooled-sample variance of E{m(G)|(Gi)}. The prospective-score test is based on the test statistic given by
where the superscripts T and – denote transpose and generalized inverse, respectively. Asymptotically, this statistic follows a chi-squared distribution under the null hypothesis of β = 0, with the degrees of freedom given by the dimension of m(G). The variance of the retrospective score , after adjusting for the estimation of the allele frequency f by f̂ given by (10) and can be estimated by
where Q is pooled-sample covariance between E{m(G)|N(Gi)} and E{G|N(Gi)}. The variance of can also be alternatively estimated by the robust sandwich-type estimate given as
where the efficient score
The retrospective-score test is then based on the test statistic given by
which again follows a chi-squared distribution asymptotically under the null hypothesis, with the degrees of freedom given by the dimension of m(G). In both the prospective- and retrospective-sore tests given above, we obtain the conditional probability Pr{G|(Gi)} directly from some external reference database, e.g. HapMap, a strategy similar to the proposal of Nicolae (2006).
We now demonstrate the potential power advantages that might be achieved by imputing the untyped SNP, using numerical studies following two scenarios as in Tables 1 and 2 of Nicolae (2006). In Scenario 1, the untyped SNP can be perfectly predicted by the genotypes of the typed SNPs, namely the (see Stram et al., 2004, for definition), while in Scenario 2 the untyped SNP is moderately predicted by the genotypes of the typed SNPs with . The SNP profiles together with the haplotype frequencies estimated from HapMap CEU samples in the two scenarios are summarized in Tables 3 and 4. Also listed in Tables 3 and 4 are the haplotype frequencies we actually used to simulate the SNP data for the case-control sample, which moderately deviate from those seen in the HapMap CEU sample to reflect the potential discrepancy between the HapMap and study samples. The haplotype pair for each person is generated according to HWE.
Table 3.
The SNP profiles and haplotype frequencies for the region considered in Scenario 1, where the untyped SNP can be perfectly predicted by genotyped SNPs A1, …, A4 ( ). Also listed are the haplotype frequencies estimated from the CEU sample in HapMap. Part of the data are from Table 1 of Nicolae (2006).
| Haplotype of SNPs A1– T – A2– A3– A4 | Frequency | Frequency from HapMap |
|---|---|---|
| 1-0-0-0-0 | 0.158 | 0.058 |
| 0-1-0-1-0 | 0.400 | 0.300 |
| 1-1-0-1-0 | 0.050 | 0.050 |
| 1-1-1-0-1 | 0.358 | 0.558 |
| 0-1-1-0-1 | 0.022 | 0.017 |
| 1-1-0-0-1 | 0.012 | 0.017 |
Table 4.
The SNP profiles and haplotype frequencies for the region considered in Scenario 2, where the untyped SNP is moderately predicted by genotyped SNPs A1, …, A3 ( ). Also listed are the haplotype frequencies estimated from the CEU sample in HapMap. Part of the data are from Table 2 of Nicolae (2006).
| Haplotype of SNPs A1– T – A2– A3 | Frequency | Frequency from HapMap |
|---|---|---|
| 0-0-0-0 | 0.088 | 0.058 |
| 0-0-1-1 | 0.027 | 0.017 |
| 0-1-0-0 | 0.302 | 0.342 |
| 0-1-1-0 | 0.008 | 0.008 |
| 1-0-1-0 | 0.242 | 0.142 |
| 1-0-1-1 | 0.333 | 0.433 |
We simulated the case-control status by the logistic regression model (3), where the genetic determinant G is given by the minor allele count of the untyped SNP, and the function m(·) is given by the recessive, dominant, or additive genetic mode. The intercept α = −3.0, which yields an overall disease rate around 5%. Each analysis is based on a case-control sample with 1000 cases and 1000 controls. The simulation results are based on 1000 (3000) repetitions for evaluation of test power (size). All the tests are performed at a significance level of 0.01. The score tests are performed using the correct genetic model, and the retrospective-score tests are based on the robust sandwich-type variance estimates; results based on model-based variance estimates are quite similar and are omitted. When performing the prospective- and retrospective-score tests with imputed genotypes for the untyped SNP, we use thed haplotype frequency estimates from HapMap to obtain the conditional probabilities Pr{G|(Gi)}, even though the case-control sample is actually from a population with moderately different haplotype frequencies. To see the degree of recovery of missing information achieved by imputation, we also perform the prospective-and retrospective-score tests based on the true genotypes at the untyped SNP. In addition, we perform the multi-marker Hotelling’s T2 test based on genotypes at typed SNPs (Xiong et al., 2002; Chapman et al., 2003), which is equivalent to the prospective-score test derived from the logistic regression model (3) with the covariates m(G) given as the vector of genotypes for all the typed SNPs.
Results for this simulation study are presented in Tables 5 (Scenario 1) and 6 (Scenario 2). It is seen that the score tests with imputed genotypes have size matching reasonably well with the nominal value of 1%, even though the imputation is based on haplotype frequencies that are obtained from the HapMap data and are different from the true frequencies. From the results regarding power, we see that imputing the untyped SNP in either the prospective- or the retrospective-score test can achieve substantial power gains as compared with the Hotelling’s T2 test based only on genotyped SNPs. The relative power improvement gained by imputation can still be quite remarkable even when the accuracy for predicting the untyped SNP using the genotyped SNPs is only of a moderate level (Scenario 2, where ). On the other hand, the prediction accuracy does affect the degree of recovery of the missing information that may be achieved by imputation: in Scenario 1, with perfect prediction of the untyped SNP, the tests using imputed genotypes do attain the full power we would obtain if the tests were based on the true genotype of the untyped SNP. In Scenario 2, with moderate prediction of the untyped SNP, imputation of the untyped SNP can recover partial but not full power. It is worth remembering that, with exact data, the retrospective-score test is usually more powerful than the prospective-score under the dominant or recessive model, and the two tests are essentially equivalent under the additive model. Here we observe the same phenomena when the prospective- and retrospective-score tests are based on imputed genotypes.
Table 5.
Size/Power (%) of the prospective- and retrospective-score tests (significance level=0.01) based on the imputed and true (in parenthesis) genotypes at the untyped causal SNP, using SNP data generated according to Table 3 (perfect prediction). Also shown are results for the Hotelling’s T2 test based only on genotypes at the typed SNPs. Results for power (size) are based on 1000 (3000) simulated data sets.
| β | Prospective Score imputed (true) | Retrospective Score imputed (true) | Hotelling’s T2 |
|---|---|---|---|
| Recessive model | |||
| 0 | 1.1 (1.1) | 1.1 (1.1) | 0.9 |
| 0.5 | 26.1 (26.1) | 33.7 (33.7) | 3.6 |
| 0.6 | 40.1 (40.1) | 55.3 (55.3) | 5.6 |
| Dominant model | |||
| 0 | 1.0 (1.3) | 1.0 (1.3) | 0.9 |
| 0.3 | 68.6 (68.6) | 72.9 (72.9) | 39.0 |
| 0.4 | 96.0 (96.0) | 96.7 (96.7) | 79.3 |
| Additive model | |||
| 0 | 1.2 (1.2) | 1.2 (1.2) | 0.9 |
| 0.2 | 43.0 (43.0) | 43.0 (43.0) | 24.2 |
| 0.3 | 86.4 (86.4) | 86.4 (86.4) | 65.5 |
As we noted earlier, when exact genotype data are available, the retrospective-score test is more sensitive to violation of the HWE assumption than the prospective-score test; i.e., the former is usually biased while the latter still remains unbiased when HWE does not hold. To assess the robustness properties for the prospective- and retrospective-score tests with imputed genotype data, we performed a further simulation study where the SNP haplotypes are still given as in Tables 3 and 4, but the haplotype pair Hdi = (ha, hb) for each person is given by the model with Pr{Hdi = (ha, hb)} = (1 − ζ)θa θb for ha ≠ hb and for ha = hb, where θa is the frequency for haplotype ha, and ζ, the fixation index quantifying the departure from HWE, is set to 0.05. We can see from the results listed in Table 7 that, with imputed genotype data, the prospective-score test, like its exact-data counterpart, still shows greater robustness in maintaining the type I error rates than the retrospective-score test. In particular, the retrospective-score test, based on the recessive or dominant model, may yield high type I error rates under violation of HWE, no matter whether exact or imputed genotype data are used. Thus, an empirical-Bayes type shrinkage method that can adapt between prospective and retrospective methods depending on bias-variance trade-off could be useful for analysis of both typed and untyped SNPs.
Table 7.
Size (%) of the prospective- and retrospective-score tests (significance level=0.01) based on the imputed and true (in parenthesis) genotypes at the untyped causal SNP, using SNP data generated according to scenarios 1 (Table 3) and 2 (Table 4) and a fixation index of 0.5 (violating HWE). Results are based on 3000 simulated data sets.
| Prospective Score imputed (true) | Retrospective Score imputed (true) | |
|---|---|---|
| Recessive model | ||
| scenario 1 | 0.8 (0.8) | 1.7 (1.7) |
| scenario 2 | 1.2 (1.2) | 5.9 (7.7) |
| Dominant model | ||
| scenario 1 | 0.9 (0.9) | 1.4 (1.4) |
| scenario 2 | 1.0 (0.8) | 3.2 (5.1) |
| Additive model | ||
| scenario 1 | 1.0 (1.0) | 1.0 (1.0) |
| scenario 2 | 0.7 (0.8) | 0.7 (0.8) |
We conclude this section with a discussion on the two types of association analyses recently developed for untyped SNPs: the full likelihood approach (Lin et al., 2008) and the two-stage approach (Nicolae, 2006; Marchini et al., 2007). The full likelihood approach uses a retrospective likelihood for the case-control sample and a likelihood for the external (such as HapMap) data, by which the imputation and association analysis are simultaneously performed in a one-stage manner. Conversely, the two-stage approach performs the imputation and association analysis separately: imputing missing genotypes in the first stage and then performing association analysis in the second stage. In the imputation stage of the two-stage approach, one can apply existing powerful external imputation algorithms such as Nicolae (2006) and Marchini et al. (2007), and hence the two-stage approach is convenient to implement. There has been some debate on the efficiency difference between the two approaches (Marchini and Howie, 2008; Lin and Hu, 2008). Our simulation results (Tables 5 and 6) suggest that the efficiency difference between the full likelihood and the two-stage approaches may be mostly due to the use of different likelihoods (prospective vs. retrospective) and not so much due to the use of one-stage versus two-stage analysis. In this section, we have shown that one can still use a retrospective likelihood even in a two-stage approach with powerful imputation performed at the first stage.
Table 6.
Size/Power (%) of the prospective- and retrospective-score tests (significance level=0.01) based on the imputed and true (in parenthesis) genotypes at the untyped causal SNP, using SNP data generated according to Table 4 (moderate prediction). Also shown are results for the Hotelling’s T2 test based only on genotypes at the typed SNPs. Results for power (size) are based on 1000 (3000) simulated data sets.
| β | Prospective Score imputed (true) | Retrospective Score imputed (true) | Hotelling’s T2 |
|---|---|---|---|
| Recessive model | |||
| 0 | 1.4 (1.2) | 1.2 (1.2) | 1.1 |
| 0.5 | 42.6 (92.2) | 47.0 (97.6) | 17.6 |
| 0.6 | 59.4 (99.1) | 66.4 (99.9) | 24.9 |
| Dominant model | |||
| 0 | 0.8 (1.1) | 0.9 (1.0) | 1.1 |
| 0.4 | 48.5 (95.6) | 54.3 (98.2) | 23.8 |
| 0.5 | 71.6 (99.6) | 77.2 (100) | 41.5 |
| Additive model | |||
| 0 | 1.0 (1.3) | 1.0 (1.3) | 1.1 |
| 0.3 | 60.2 (97.6) | 60.1 (97.6) | 40.6 |
| 0.4 | 92.5 (99.9) | 92.4 (99.9) | 77.4 |
4 Haplotypes
4.1 Definitions, Background and Missing Data
Although single-SNP association tests are often the primary methods for genome-wide association scans, many secondary or “downstream” analyses are often useful for detailed characterization of the risk of the disease associated with specific genomic regions of interest. One popular technique is haplotype-based association analysis, which involves studying the association of a disease with a genomic region in terms of the underlying “haplotypes”, the combination of alleles at multiple loci along individual homologous chromosomes. Originally, haplotype-based association analysis was considered a powerful technique for “indirect” association testing in situations where a causal SNP may not have been genotyped, but the haplotypes defined by multiple typed SNPs could serve as a good “surrogate” for the causal variant. With the advent of various imputation methods, although haplotype analysis has become less relevant for such indirect association testing, it remains a useful tool for parsimonious characterization of disease-risk associated with multiple, possibly interacting, loci within a given region. Moreover, it is conceivable that for some regions, the haplotypes, and not the individual SNPs, are functional units and thus for these regions stronger signals of associations could be detected by performing haplotype based regression analysis.
A technical problem for haplotype-based regression analysis is that typically the haplotype information for the study subjects is not directly observable. Instead, locus-specific genotype data are observed, which contain information on the pair of alleles a subject carries, but does not provide the “phase information”, that is which combinations of alleles appear across multiple loci along the individual chromosomes. In general, the genotype data of a subject will be phase-ambiguous whenever the subject is heterozygous at two or more loci. Statistically, the lack of phase information can be viewed as a special missing data problem.
For example, suppose A/a and B/b denote the major/minor alleles in two bi-allelic loci. A particular haplotype pair, called a diplotype, is the pair of alleles that are inherited from one’s parents. One such haplotype pair would be (AB) − (ab), and disease risk can be associated with the number of copies of particular haplotypes that one inherits. Unfortunately, the diplotypes are not observable directly, but instead we can observe only the unordered or combined genotypes, in this case (Aa) at the first locus and (Bb) at the second locus, i.e., (AaBb). However, when observing only the genotypes, the actual haplotype pair is unknown, or “phase ambiguous”, because the haplotype pair (Ab)−(aB) has the same set of unordered genotypes. Confronted with the unordered set of genotypes (AaBb), we know that the actual haplotype pair is either (AB) − (ab) or (Ab) − (aB), but we must use probability models to take into account the phase ambiguity when performing statistical inference.
In Section 2 we described “model-free” prospective and “model-based” efficient retrospective methods for analyzing SNP data, and we also described empirical-Bayes methods that data-adaptively move between the two. Just as in SNP data, for haplotype data there are also model-free and model-based methods, and accompanying empirical-Bayes methods.
A variety of methods have been developed for haplotype-based analysis of case-control data using the logistic regression model (Zhao et al., 2003; Lake et al., 2003; Epstein and Satten, 2003; Satten and Epstein, 2004; Spinka et al., 2005; Lin and Zeng, 2006; Chatterjee et al., 2006; Chen et al., 2009). Consider a general risk model similar to (3) but with the addition of environmental factors (E) and written in terms of the diplotypes, denoted as Hdi:
| (11) |
where the function m(·) is chosen in a suitable way to reflect an assumed mode of genetic effect. For example, suppose we are interested in the particular haplotype h*= (ab). A model that assumes an additive effect of this haplotype would have m(Hdi = hdi, E) linear in the number of copies of the haplotype h*.
4.2 Model-Based and Model-Free Methods
4.2.1 Identifiability
The data setup then is that we have observations on environmental exposure (E), genotypes G and cases and controls D. What is missing is the underlying diplotype Hdi. The retrospective likelihood is still (2), but the risk of disease depends on the diplotype Hdi and not otherwise on the genotype.
While models such as (11) seem straightforward enough for random samples, in retrospective samples a problem arises because of the phase ambiguity. In particular, all components of β may not be identifiable if the distribution of (Hdi, E) is left completely unrestricted (Epstein and Satten, 2003; Lin and Zeng, 2006). Thus, to make progress, some type of distributional assumptions are needed. Here we will distinguished between two approaches, both of them retrospective in nature but with different distributional assumptions. The first we call “model-free” in that very little is actually assumed about the haplotype distribution. If haplotypes were observable, this method reduces to ordinary prospective logistic regression, while in the rare disease case with phase ambiguity, the method reduces to that of Zhao et al. (2003). The second approach, which we call “model-based”, makes much stronger assumptions about the haplotype distribution, and reduces to the efficient retrospective approach of Chatterjee and Carroll (2005) if haplotypes were observable. The model-free method will thus be more robust but less efficient than the model-based method.
4.2.2 Model-Based Method
The model-based method (Spinka et al., 2005) has three aspects.
(A.1) Haplotypes and the environment are assumed independent in the population.
-
(A.2) The diplotypes are assumed to be in HWE in the population, so that
where θs denotes the population frequency for the haplotype hs.
(A.3) The distribution of the environmental variable E is left completely nonparametric.
The methodology Spinka et al. (2005) used to construct their profile likelihood was a nonparametric maximum likelihood estimator over the unknown distribution of E. However, there is an alternative derivation, one that is both more intuitive and much easier to work out. Indeed, it is a not sufficiently well known fact that for most purposes a case-control study can be viewed as a prospective study with missing data. Consider a sampling scenario where each subject from the underlying population is selected into the case-control study using a Bernoulli sampling scheme where the selection probability for a subject given his/her disease status D = d is proportional to Nd/pr(D = d). Inference with the actual case-control data can then be based on the pseudo-likelihood derived for such an alternative sampling scenario. Let δ = 1 denote that a subject is selected in the case-control sample under this Bernoulli sampling scheme and hence has been observed. Then in this alternative sampling scheme, and with the assumptions stated above, Spinka et al. (2005) compute pr(D = 1, G = g|E, δ = 1). This calculation is simple and in the rare disease case the resulting efficient model-based likelihood function reduces to
| (12) |
where pd = Nd/N, πd = pr(D = d), κ = α + log(p1/p0) − log(π1/π0), Ω = (β, θ, κ), and ℋG is the set of diplotypes consistent with the observed genotypes G.
4.2.3 Model-Free Method
The two important model assumptions in the model-based estimator are (A.1) and (A.2). Although because of identifiability some model assumptions must be made, they can be weakened tremendously, as follows (Chen, Chatterjee and Carroll, 2009):
(B.1) The haplotype and the environment are independent in the population given the genotype G.
(B.2) There population distribution for the diplotypes given the genotype G, called qfree(hdi|G, θ), can be derived assuming HWE.
Following the same alternative sampling scheme as described in Section ??, or by doing a nonparametric maximum likelihood analysis, we can compute pr(D = 1|G, E, δ = 1) under assumptions (B.1), (B.2) and (A.3) to be
| (13) |
To see why the likelihood Lfree requires far weaker assumptions than Lmodel, note that Lfree requires the haplotype-environment independence and HWE assumption only to specify the conditional distribution pr(Hdi|G, X) while Lmodel requires the same assumption to specify the entire joint distribution pr(Hdi, X). As a result, Lfree requires the haplotype-environment independence and HWE only to resolve the phase ambiguous genotypes. The likelihood contribution for the subjects with phase unambiguous genotypes, ie. G = Hdi, is the same as that for the standard prospective logistic regression. In contrast, Lmodel depends on the assumptions (A.1) and (A.2) irrespective a subject has missing phase or not.
Note that Lfree(D, G, E, Ω) will contain little information on θ since it conditions on G. Thus, when implementing methods based on this likelihood, Chen et al. (2009) proposed to replace the score function for θ by the estimating function for θ based on the genotype data from the controls and assuming that the haplotypes are in HWE in the population.
4.3 Empirical-Bayes
In Section 4.2.2, we constructed a profile likelihood under strong assumptions leading to an efficient method that will not be robust to violations of the two major assumptions. Conversely, in Section 4.2.3 we computed a profile likelihood leading to much more robust inference, but at the cost of a steep loss of efficiency. Similarly to Section 2.3, here we briefly review a fully sample size- and data-adaptive empirical-Bayes method that Chen et al. (2009) described for gaining efficiency when warranted but is still robust.
Let β̂free and β̂model be the model-free and model-based estimates, with jth components βj,free and β̂j,model. Let V be the covariance matrix of ψ̂ = β̂free − β̂model, with the jth diagonal element of V being vi: a sandwich estimator vi can be computed, although a nonparametric bootstrap can also be used. Then one can define the empirical-Bayes estimator
| (14) |
The intuition behind (14) is that if the model fails, (β̂j,model − β̂j,free) will be large relative to vi, which as a variance is proportional to N−1, hence Wj ≈ 0, and hence the empirical-Bayes method will effectively become the model-free estimator. If however the model assumption holds, then vi and (β̂j,free − β̂j,model)2 are proportional to one another, so that Wj > 0 and the empirical-Bayes estimate goes part way towards the model-based estimator, and hence gains efficiency over the model-free estimate. Chen et al. (2009) describe the limiting distribution of (14) and how to compute an estimate of its variance.
Chen et al. (2009) illustrate application of the different methods in two case-control data examples. The examples were chosen in such a way that from a priori biologic grounds one would expect the gene-environment independence assumption to hold in one case, but not in the other. The two examples together illustrate how the different shrinkage estimators adapt to alternative scenarios of gene-environment distribution.
5 Discussion
Researchers now increasingly use the Cochran-Armitage trend test as the primary method for single-SNP association testing in the GWAS. The test is known to have robust power for the detection of effect of susceptibility SNPs under a range of realistic modes of inheritance that give rise to some sort of monotone relationship between disease-risk and allele count. As noted in Section 2, the retrospective and prospective methods have very similar, if not identical, power under the trend model and thus either could be used as the primary method for analysis of GWAS data. The trend test, however, can perform very poorly for the detection of SNPs for which the minor allele has a recessive effect. Thus, it is often recommended that a test under the recessive mode of inheritance be conducted as a secondary step to detect SNPs with recessive effects that may be missed by the primary trend test of association. The use of the retrospective method can be potentially beneficial at this stage. One, however, has to be cautious about creation of false positive results due to the violation of the HWE assumption. We recommend that if a retrospective method is to be used for potential power gain, then it should be used in conjunction with the empirical-Bayes type shrinkage estimation. Our numerical investigations suggest that such a method can indeed be more powerful than the conventional “prospective” methods without creating excess false positives, see Tables 1 and 2.
In this article, although we focus on association tests involving bi-allelic SNPs, the same issues are relevant for genetic association tests involving loci with more than two alleles. In particular, one can gain efficiency for analysis of case-control data by assuming HWE or other natural population-genetic models (Satten and Epstein, 2004; Lin and Zeng, 2006) to specify multi-allelic genotype frequency for the underlying population. The sensitivity of the methods to underlying model assumption can be reduced by appropriate shrinkage estimation techniques.
The difference between prospective and retrospective methods become more relevant for studies of gene-gene and gene-environment interactions, a topic that we have not directly addressed in this article. In particular, retrospective methods, such as the case-only analysis (Piegorsch et al., 1994), which assumes gene-gene or/and gene-environment independence for the underlying population, can gain dramatic power for testing and estimation of odds-ratio interaction parameters in the logistic regression model. Given that standard case-control analyses often have poor power for detection of multiplicative interactions due to small numbers of cases or controls in cells of crossing exposures, practitioners often find it is tempting to use the more powerful retrospective methods. The assumption of gene-environment independence, however, can be violated, either due to direct casual association between gene and environment or indirect association due to effects of family history and hidden population stratification. The assumption of gene-gene independence between physically distant genes can also be violated due to population stratification. Thus, we believe the development of shrinkage (Mukherjee and Chatterjee, 2008; Chen, Chatterjee and Carroll, 2009) and other type of data-adaptive techniques (Li and Conti, 2009) has been valuable for robust inference in case-control studies of genetic epidemiology.
Acknowledgments
Chatterjee’s research was supported by a gene-environment initiative grant from the National Heart Lung and Blood Institute (RO1HL091172-01) and by the Intramural Research Program of the National Cancer Institute. Chen’s research was supported by the National Science Council of ROC (NSC 95-2118-M-001-022-MY3). Carroll’s research was supported by a grant from the National Cancer Institute (CA57030) and by Award Number KUS-CI-016-04, made by King Abdullah University of Science and Technology (KAUST).
Contributor Information
NILANJAN CHATTERJEE, Email: chattern@mail.nih.gov, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, DHHS. Rockville MD 20852, U.S.A.
YI-HAU CHEN, Email: yhchen@stat.sinica.edu.tw, Institute of Statistical Science, Academia Sinica, Taipei 11529, Taiwan R.O.C.
SHENG LUO, Email: Sheng.T.Luo@uth.tmc.edu, Division of Biostatistics, University of Texas School of Public Health, Houston, TX 77030.
RAYMOND J. CARROLL, Email: carroll@stat.tamu.edu, Department of Statistics, Texas A&M University, College Station, TX 77843-3143, U.S.A
References
- Andersen EB. Asymptotic properties of conditional maximum-likelihood estimators. Journal of the Royal Statistical Society, Series B. 1970;32:283–301. [Google Scholar]
- Chapman JM, Cooper JD, Todd JA, Clayton DG. Detecting disease associations due to linkage disequilibrium using haplotype tags: a class of tests and the determinants of statistical power. Human Heredity. 2003;56:18–31. doi: 10.1159/000073729. [DOI] [PubMed] [Google Scholar]
- Chatterjee N, Carroll RJ. Semiparametric maximum likelihood estimation in case-control studies of gene-environment interactions. Biometrika. 2005;92:399–418. [Google Scholar]
- Chatterjee N, Chen J, Spinka C, Carroll RJ. Comment on the paper likelihood based inference on haplotype effects in genetic association studies by d y lin and d zeng. Journal of American Statistical Association. 2006;101:108–110. [Google Scholar]
- Chen J, Chatterjee N. Exploiting hardy-weinberg equilibrium for efficient screening of single snp associations from case-control studies. Human Heredity. 2007;63:196–204. doi: 10.1159/000099996. [DOI] [PubMed] [Google Scholar]
- Chen YH, Chatterjee N, Carroll RJ. Shrinkage estimators for robust and efficient inference in haplotype-based case-control studies. Journal of American Statistical Association. 2009 doi: 10.1198/jasa.2009.0104. page To appear. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornfield J. A statistical problem arising from retrospective studies. Proceedings of the Third Berkeley Symposium on Mathematical Statistics and Probability.1956. [Google Scholar]
- Epstein MP, Satten GA. Inference on haplotype effects in case-control studies using unphased genotype data. American Journal of Human Genetics. 2003;73:1316–1329. doi: 10.1086/380204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl DL, Clark AG. Principles of Population Genetics. 4 Sinauer Associates; 2007. [Google Scholar]
- Hunter DJ, Kraft P, Jacobs KB, Cox DG, Yeager M, Hankinson SE, Wacholder S, Wang Z, Welch R, Hutchinson A, Wang J, Yu K, Chatterjee N, et al. A genome-wide association study identifies alleles in fgfr2 associated with risk of sporadic postmenopausal breast cancer. Nature Genetics. 2007;39:870–874. doi: 10.1038/ng2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake SL, Lyon H, Tantisira K, Silverman EK, Weiss ST, Laird NM, Schaid DJ. Estimation and tests of haplotype-environment interaction when linkage phase is ambiguous. Human Heredity. 2003;55:56–65. doi: 10.1159/000071811. [DOI] [PubMed] [Google Scholar]
- Li D, Conti DV. Detecting gene-environment interactions using a combined case-only and case-control approach. American Journal of Epidemiology. 2009;169:497–504. doi: 10.1093/aje/kwn339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DY, Hu Y. Reply to marchini and howie. American Journal of Human Genetics. 2008;83:539–540. doi: 10.1016/j.ajhg.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DY, Hu Y, Huang BE. Simple and efficient analysis of disease association with missing genotype data. American Journal of Human Genetics. 2008;82:444–45. doi: 10.1016/j.ajhg.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DY, Zeng D. Likelihood-based inference on haplotype effects in genetic association studies. Journal of the American Statistical Association. 2006;101(473):89–104. [Google Scholar]
- Luo S, Mukherjee B, Chen J, Chatterjee N. Shrinkage estimation for robust and efficient screening of single-snp sssociation from case-control genome-wide association studies. Genetic Epidemiology. 2009 doi: 10.1002/gepi.20428. To apprear. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchini J, Howie B. Comparing algorithms for genotype imputation. American Journal of Human Genetics. 2008;83:535–539. doi: 10.1016/j.ajhg.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchini J, Howie B, Myers S, GM, PD A new multipoint method for genome-wide association studies by imputation of genotypes. Nature Genetics. 2007;39:906–913. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- Mukherjee B, Chatterjee N. Exploiting gene-environment independence for analysis of case-control studies: an empirical bayes approach to trade off between bias and efficiency. Biometrics. 2008;64:685–694. doi: 10.1111/j.1541-0420.2007.00953.x. [DOI] [PubMed] [Google Scholar]
- Nicolae DL. Testing untyped alleles (tuna)-applications to genome-wide association studies. Genetic Epidemiology. 2006;30:718–727. doi: 10.1002/gepi.20182. [DOI] [PubMed] [Google Scholar]
- Piegorsch WW, Weinberg CR, Taylor JA. Non-hierarchical logistic models and case-only designs for assessing susceptibility in population-based case-control studies. Statistics in Medicine. 1994;13:153–162. doi: 10.1002/sim.4780130206. [DOI] [PubMed] [Google Scholar]
- Prentice RL, Pyke R. Logistic disease incidence models and case-control studies. Biometrika. 1979;66:403–412. [Google Scholar]
- Roeder K, Carroll RJ, Lindsay BG. A semiparametric mixture approach to case-control studies with errors in covariables. Journal of the American Statistical Association. 1996;91:722–732. [Google Scholar]
- Satten GA, Epstein MP. Comparison of prospective and retrospective methods for haplotype inference in case-control studies. Genetic Epidemiology. 2004;27(3):192–201. doi: 10.1002/gepi.20020. [DOI] [PubMed] [Google Scholar]
- Satten GA, Kupper LL. Conditional regression analysis of the exposure- disease odds ratio using known probability-of-exposure values. Biometrics. 1993;49:429–440. [PubMed] [Google Scholar]
- Spinka C, Carroll RJ, Chatterjee N. Analysis of case-control studies of genetic and environmental factors with missing genetic information and haplotype-phase ambiguity. Genetic Epidemiology. 2005;29:108–127. doi: 10.1002/gepi.20085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas G, Jacobs KB, Yeager M, Kraft P, Wacholder S, Orr N, Yu K, Chatterjee N, Welch R, Hutchinson A, et al. Multiple novel loci identified in a genome-wide association study of prostate cancer. Nature Genetics. 2008;40:310–315. doi: 10.1038/ng.91. [DOI] [PubMed] [Google Scholar]
- Xiong M, Zhao J, Berwinkle E. Generalized t2 test for genome association studies. American Journal of Human Genetics. 2002;70:1257–1268. doi: 10.1086/340392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeager M, Orr N, Hayes RB, Jacobs KB, Kraft P, Wacholder S, Minichiello MJ, Fearnhead P, Yu K, Chatterjee N, et al. Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nature Genetics. 2007;39:645–649. doi: 10.1038/ng2022. [DOI] [PubMed] [Google Scholar]
- Yu K, Li QWBA, Pfeiffer R, Rosenberg P, Caporaso N, Kraft P, Chatterjee N. Pathway analysis by adaptive combination of p-values. Genetic Epidemiology. 2009 doi: 10.1002/gepi.20422. To appear. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao LP, Li SS, Khalid NA. Method for the assessment of disease associations with single-nucleotide polymorphism haplotypes and environmental variables in case-control studies. American Journal of Human Genetics. 2003;72:1231–1250. doi: 10.1086/375140. [DOI] [PMC free article] [PubMed] [Google Scholar]