

Abstract
The DNA of most vertebrates is depleted in CpG dinucleotide: a C followed by a G in the 5′ to 3′ direction. CpGs are the target for DNA methylation, a chemical modification of cytosine (C) heritable during cell division and the most well-characterized epigenetic mechanism. The remaining CpGs tend to cluster in regions referred to as CpG islands (CGI). Knowing CGI locations is important because they mark functionally relevant epigenetic loci in development and disease. For various mammals, including human, a readily available and widely used list of CGI is available from the UCSC Genome Browser. This list was derived using algorithms that search for regions satisfying a definition of CGI proposed by Gardiner-Garden and Frommer more than 20 years ago. Recent findings, enabled by advances in technology that permit direct measurement of epigenetic endpoints at a whole-genome scale, motivate the need to adapt the current CGI definition. In this paper, we propose a procedure, guided by hidden Markov models, that permits an extensible approach to detecting CGI. The main advantage of our approach over others is that it summarizes the evidence for CGI status as probability scores. This provides flexibility in the definition of a CGI and facilitates the creation of CGI lists for other species. The utility of this approach is demonstrated by generating the first CGI lists for invertebrates, and the fact that we can create CGI lists that substantially increases overlap with recently discovered epigenetic marks. A CGI list and the probability scores, as a function of genome location, for each species are available at http://www.rafalab.org.
Keywords: CpG island, Epigenetics, Hidden Markov model, Sequence analysis
1. INTRODUCTION
DNA methylation is a type of chemical modification of DNA that can be inherited without changing the DNA sequence. This type of heritable mechanism is referred to as “epigenetic inheritance.” DNA methylation involves the addition of a methyl group to DNA and typically occurs at a C followed, in the 5′ to 3′ direction, by a G. Biologists refer to this dinucleotide as a CpG, where the p implies the 5′ to 3′ direction. Figure 1(a) is a simplified illustration of how DNA methylation is maintained during cell division. DNA methylation is of particular interest because it is involved in gene regulation. It affects the transcription of genes in 2 ways. First, the methylation of DNA can impede the binding of transcriptional proteins to the gene, thus blocking transcription. Second, methylated DNA may be bound by proteins that start a series of chemical events that result in the formation of compact DNA that renders it inactive. Note that although 2 cell types in an organism have the same genome, their methylation pattern can be different (Figure 1(b)). The fact that DNA methylation is heritable makes it the most prominent mechanism used by differentiated cells to pass tissue-specific transcription patterns to daughter cells in cell division. Therefore, DNA methylation is regarded as the “fifth base” of the genome and is of great interest to biologists.
Fig. 1.

Cartoon illustrating how DNA methylation is inherited in cell division on how it could be involved in tissue differentiation. (a) The fact that the complement of a CpG is also a CpG facilitates the inheritance mechanism. The cartoon illustrates how, during mitotic cell division, DNA methylation is inherited. (b) This cartoon illustrates how 2 cells can have the same genomic sequence but a different methylation pattern.
The DNA of most vertebrates is depleted in CpG dinucleotides. The remaining CpGs tend to cluster in regions referred to as CpG islands (CGI) (Figure 2). Interest in CGI grew when it was demonstrated that, in vertebrates, they are enriched in regions of the genome involved in gene transcription referred to as “promoters” (Bird, 1986). Furthermore, many investigators have observed altered DNA methylation of CGI in development and cancer (Feinberg, 2007). Irizarry and others (2008) recently demonstrated that “CGI shores,” defined as regions within 2000 (bp) but not inside CGI, are useful predictors for genomic locations that are differentially methylated across different tissues and between cancer and normal samples.
Fig. 2.

A genomic region of 40 000 bases from chromosome 1 is shown. The ticks on the x-axis represent CpG locations. The points represent CpG rates in segments of length 256 bases The curve is the result of a kernel smoother of the points. Approximately 20% of the genome are Cs and 20% are Gs. Thus, we expect about 4% of dinucleotides to be CpG. However, most points are well below rates 4% with 2 clusters well above 4%. The latter are CGI.
A specific example of the need for knowledge of CGI locations is their use in the construction of high-throughput assays. The traditional technique for measuring DNA methylation, bisulfite modification-based sequencing, is labor intensive and not suitable for genome-wide studies. New molecular biology techniques, along with the use of microarrays or second-generation sequencing technologies, has made high-throughput unbiased methylation profiling feasible. However, whole-genome assays are too costly for most research groups. Knowledge of CGI locations provide manufacturers a way to construct cost-effective products that focus on regions known to be associated with important epigenetic events (Agilent, 2008; Meissner and others, 2008).
Although existing CGI lists have been widely used, comprehensive measurements of methylation, enabled by recent advances in technology, demonstrate that the current definition needs to be improved. Furthermore, the current definition was developed for humans and interest in measuring DNA methylation in other organisms motivates the need for a more general definition. In Section 2, we describe existing approaches to detecting CGI and point out their limitations. In Section 3, we motivate the need for a new approach and the statistical model that we use to redefine the concept of a CGI. In Section 4, we present the model. In Section 5, we describe improvements over existing approaches obtained from fitting our model. Finally, in Section 6 we summarize our findings.
2. PREVIOUS WORK
A formal definition of a CGI was provided by Gardiner-Garden and Frommer (1987). A CGI is defined as a region of at least 200 bp, with the proportion of Gs or Cs, referred to as “GC content,” greater than 50%, and observed to expected CpG ratio (O/E) greater than 0.6. The observed to expected ratio is calculated by dividing the proportion of CpG dinucleotides in the region by what is expected by chance when bases are assumed to be independent outcomes of a multinomial distribution. The formula currently used is
with N the number of bp in the segment under consideration. Various computer algorithms have been developed that efficiently scan the genome for regions satisfying the definition. The most widely used CGI list is based on this definition and is hosted by the UCSC Genome Browser (Kent and others, 2002). However, this definition is somewhat arbitrary because the choice of the cutoffs has a great influence on what is considered an island. The cutoff choice was likely derived from exploratory data analysis (Gardiner-Garden and Frommer, 1987 Figure 1) but neither a biological argument nor a formal statistical motivation was used.
Alternative algorithmic definitions have been proposed. For example, Takai and Jones (2002) considered slightly different cutoffs: a minimum length of 500 bp, a minimum GC content of 55% and a minimum O/E of 0.65. They demonstrated that by using the new cutoffs, the enrichment for promoter regions of genes was largely not affected, whereas most undesirable Alu-repetitive elements were excluded compared to the UCSC Genome Browser's CGI list. Repetitive elements are sequences that appear over and over again on the genome. As a specific type of repetitive element, the Alu sequence appears more than 1 000 000 times. They are not associated with epigenetic marks but satisfy the CGI definition, therefore biologists rather exclude them.
Glass and others (2007) described a completely different algorithm. For every CpG, they recorded the length of a segment needed to cover the nearest 27 CpGs. They then observed that, for certain species, a histogram of these lengths shows a bimodal distribution. The histogram was used to select a cutoff and regions associated with the first mode are defined as CpG “clusters” (their terminology for CGI). However, both these alternative definitions also depend on cutoffs based on a difficult to interpret scale.
Because we assume that the underlying structure of the genome includes unobserved states (CGI and baseline), which are presumed to be locally correlated along the genome (see, e.g. Figure 2), hidden Markov models (HMMs) are a natural method to consider. Churchill (1989) introduced the use of HMM to sequence analysis. More recently, HMMs have been effectively used to partition genomes into segments of similar stochastic structure (e.g. Muri, 1998; Nicolas and others, 2002; Boys and Henderson, 2004). In these approaches, the hidden state is assumed to be a homogeneous first-order Markov chain. The distribution of the observed base at location t, conditioned on the hidden state, is a heterogeneous first-order Markov chain. States are then inferred from the base-to-base transitions observed in the genome in question. In the examples cited above, this approach is effectively used to discover heterogeneities in the genome of bacteria (Nicolas and others, 2002) and to segment these genomes (Boys and Henderson, 2004).
In general, HMMs have been extensively used in sequence analysis to discover functional elements in various genomes. In a seminal book on the topic, Durbin and others (1998) proposed the use of HMMs for the task of detecting CGIs. Specifically, 8 states are assumed: the 4 nucleotides in each of the 2 states (CGI and baseline). Regions for which the state (CGI or baseline) is predetermined (using the current definition) are used to estimate the transition probabilities. With the transition probabilities in place and a sequence of dinucleotides, CGI and baseline states can be predicted by fitting an HMM.
3. MOTIVATION
3.1 Limitation of existing methods
Recent advances in technology have enabled high-throughput measurement of epigenetic events such as differentially methylated regions (DMRs) across tissue types. Newly available data have motivated the need for a more flexible CGI definition. For example, we examined data published by Irizarry and others (2008) and found many DMRs not associated with CGIs but that were nevertheless in the shores of CpG-enriched sequences. For example, one DMR reported by Irizarry and others (2008) was within 1000 bp of a CpG cluster not currently defined as a CGI (Figure 3(a)). Furthermore, this region coincides with a gene promoter. Despite coinciding with 2 functional elements associated with CGI, this region meets only 2 of the 3 criteria of the formal definition: O/E is only 0.5. Therefore, this region is not in the Genome Browser list of CGI. Visual inspection of the base composition around other DMRs not associated with CGI demonstrated that this was a general problem (data not shown).
Fig. 3.

The observed to expected ratio (green) and percentage of G + C (orange) are shown for 2 regions of the human genome. CpG clusters are denoted with bars along the bottom or top of the plot. (a) For a region covering the 5′ end of CLSTN3 a CpG dense region that is not in current CGI list is denoted by the lime green bar at the bottom. (b) The top (pink) bar denotes one of Glass and others (2007) CGI that engulfs 3 Genome Browser CGIs (denoted with purple bars at the bottom). The regions between the Genome Browser CGI have low observed to expected ratio.
None of the existing competing algorithms solve this problem. By focusing only on promoters of known genes, we find that the definition proposed by Takai and Jones (2002), although successfully filtering out more undesirable repetitive regions, results in even less sensitivity for functional epigenetic elements. Furthermore, the Genome Browser list was filtered to remove repeats, which is a viable solution that does not involve changing to a more restrictive definition. The algorithm described by Glass and others (2007) has limitations as well. A specific problem is that several smaller clusters agglomerate into larger ones (Figure 3(b) shows an example). As a consequence, relatively long stretches of CpG depleted regions are included in the CGI. Furthermore, the 27 CpG requirement results in a list that leaves out many shorter CpG clusters that are associated with DMRs. For example, the CGI described above (Figure 3(a)), is excluded.
Similarly, more statistically based approaches have limitations. Although the model proposed by Durbin and others (1998) serves as an elegant illustration, implementing the approach has not yielded a practical method for genome-wide identification of CGI. To elaborate, note that the typical HMM approach in sequence analysis models the transitions between bases directly. When applied to CGIs, the fundamental difference between the 2 states must therefore be the transition from C to G, with islands having a bigger transition probability. However, below we demonstrate that for this approach to fit the data, we would have to include much more than 2 hidden states, due to the variability in base composition observed in most genomes. Moreover, in our experience, the level of complexity required by an HMM, applied to the individual bases, impedes the development of a procedure useful for the creation of CGI lists.
If CGIs are simply a cluster of CpGs, then a procedure that scans through the genome searching for regions with larger than expected CpG rates would suffice. However, the evolutionary theory for CGI motivates a more sophisticated approach. Briefly, the human genome is depleted of CpG because the mutation rate for this specific dinucleotide is higher than others (Lander and others, 2001). CGIs are believed to be the result of certain segments of the genome being somewhat protected from the mechanism that leads to this mutation. This is a possible explanation for the association of CGI and locations relevant to development. This evolutionary argument, based on differing mutation rates, suggest that the fundamental property that defines a CGI is not the CpG density per se but the CpG density conditioned on GC content. This is because regions that originally had high GC content had more CpG dinucleotides which, even unprotected, resulted in relatively high CpG counts. Gardiner-Garden and Frommer's definition, based on the observed to expected ratio as opposed to just the number of CpG, agrees with the above described theory. Our data exploration, described below, supports and builds on this approach.
3.2. Data exploration
We divided the human genome into nonoverlapping segments of length 256 bases after removing the Alu-repetitive elements. The histogram of the CpG rates of these segments (figure not shown) does not provide a clear cutoff for distinguishing CGI from baseline. However, if we stratify the segments by GC content (Figure 4), distinct bimodal distributions of CpG rates are observed. The 2 modes support the existence of 2 states: CGI and baseline. The fact that the center of the 2 modes increases with GC content implies that we should consider rates of CpG counts relative to the GC content of the segments. That is, we consider the number of CpGs relative to a quantity measuring the number of opportunities for CpGs, similar to considering the number of events is relative to the size of the risk set in survival analysis. Note that the O/E concept of Gardiner-Garden and Frommer is a clever and simple method for adhering to this principal.
Fig. 4.

Histogram of CpG rates in nonoverlapping genomic segments of length 256 bases, stratified by the GC contents in each segment. GC content strata is shown on top of each histogram. x-axis is the CpG rate. y-axis shows the percentage of segments belonging to each CpG rate category.
Our data exploration revealed another interesting characteristic of the human genome. Figure 5 shows GC content for a representative region of the genome (with no repetitive elements). There appears 2 states for GC content as well. In Section 4, we describe the relevance of this characteristic in our approach to defining islands.
Fig. 5.

GC content plots. A region with no Alu repeats was divided into nonoverlapping segments of length 256 bases. The points are the GC content of each segment. The curve is the result of a kernel smoother of the points.
Figures 2 and 4 support the claims that CpGs are clustered and that O/E can be satisfactorily modeled by 2 states. Therefore, a 2-state HMM is a natural method to consider. However, modeling the emission probability at a single location is complicated because GC content, needed to compute O/E, varies widely across the genome, as seen in Figure 5. Another complication is that the distribution of CpG counts at a single location is somewhat complicated because outcomes from consecutive locations are not independent. For example, CG dinucleotide cannot start at consecutive bases. In Section 4, we described a procedure, motivated by HMMs, that overcomes the described problems of existing approaches and the difficulties of modeling sequence data directly. By modeling CpG counts in small bins instead of base-to-base transitions, the complexity of the emission model is greatly reduced. The models are therefore relatively simple and can be fit without cutoff choices which facilitated the extension to species for which CGI have never been reported.
4. MODEL
For any given genome, we assumed that each chromosome is divided into 3 states: Alu repetitive elements, baseline, and CGI. Because the locations of the Alu-repetitive elements are well characterized, they are inherently not of interest for the current statistical problem and therefore such regions were removed. Hence, we characterize the problem as that of a semi-HMM, with a known state for Alu repetitive elements. Our analysis then considers the 2-state chain conditional on being in a non-Alu repetitive state.
We followed the basic statistical concepts first used by Churchill (1989), described by Durbin and others (1998) and used by bioinformatic tools such as Multiple EM for Motif Elicitation (Bailey and others, 2006), Motif Alignment & Search Tool (Bailey, 1998), and Basic Local Alignment Search Tool (Altschul and others, 1990). The foundation of these tools is the stochastic modeling of bases in the genome. We denote as the base at genomic location t, the probability that for , and the probability of being CpG at location t. The depletion of CpG implies that the probability of a C at location t followed by a G is less likely than would be predicted by chance under independence: . We have argued that a useful model for detection of CGI needs 2 states to describe changes in and . However, we have specified 3 parameters for each genomic location t, resulting in an overdetermined system. Placing parsimonious modeling assumptions on the chain of bases that imply in a 2-state stochastic process for the chain of CpGs would result in undue complexity. Instead, we describe and motivate simple assumptions that permitted the derivation of a useful model from the general model described above.
We first divided the non-Alu regions into nonoverlapping segments of length L bp. For the results shown here, we used . This choice is justified in Section 4.2. We denoted , , and as the number of C, G, and CpG in segment s, and the hidden state for segment s with 2 states: as CGI and as baseline. We assume is a stationary first-order Markov chain.
As discussed in Section 3.2, the GC content and CpG count are not independent. In order to fit an HMM on , one has to evaluate the joint likelihood by a complicated numerical method such as Monte Carlo Markov chain. Given the size of genomes (3 billion bases for human), we opted for an intuitive and computational feasible approach: we modeled the data generating process with a hierarchical model that we subsequently fit using direct estimates in an iterative stepped approach. The most complex portion of the model involves a model for the GC content, . We require a model that adheres to the following: (i) it must account for jumps in GC content, (ii) slowly varying trends must also be accounted for, and (iii) fitting must be reasonably fast and able to accommodate the large size of the data.
We first defined a latent Markov process to be the hidden state for segment s with states: as high GC count regions and as baseline. We assumed that was a stationary first-order Markov chain with invariant probabilities , say, and transition matrix P. Let be the collection of segments defined by a constant latent state. That is, , where is the smallest index so that ; , where is the smallest index so that ; and so on. This process divided the segments into regions of low or high GC content.
The lowest level of the hierarchy characterized the unknown proportion of GC content in segment s and was denoted by . The model for must account for the fast variation in the chain of GC content as well as the slow variation within segments of similar type, as shown in Figure 5. We posited the following model on :
where represented smooth deviations, while the additive constant represents jumps in the GC contents. Conditioned on , the observed GC content follows Binomial distribution:
For this approach, we approximated the binomial distribution with the normal density. We did not force a binomial variance and estimated it from the data when fitting HMM. This gives us added flexibility, though it requires to lie away from the 0 and 1 boundaries for the distributional assumptions to be valid. However, this is well indicated by the data. Under the above model assumptions, the GC content subtracting the slow variation forms a first-order HMM:
Note that we originally tried using smoothed GC content instead of the proposed 2-state model. However, results showed that CGIs associated with low GC content are generally not associated with epigenetic marks and need to be filtered out. Furthermore, we observed sharp changes in GC content as shown in Figure 5. To avoid arbitrarily selecting a cutoff for GC content and account for the GC content jumps, we implemented the HMM approach.
Finally, conditioned on and , we assumed an HMM on with Poisson emission probabilities with conditional means
Here, we are making the parsimony assumption that 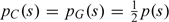 . This assumption, though perhaps aggressive if the bin sizes are small, is biologically well motivated. Further, the Poisson assumption is motivated in Section 4.1. Note that the parameters and can be interpreted as the O/E for the CGI and baseline regions, respectively.
. This assumption, though perhaps aggressive if the bin sizes are small, is biologically well motivated. Further, the Poisson assumption is motivated in Section 4.1. Note that the parameters and can be interpreted as the O/E for the CGI and baseline regions, respectively.
4.1. Motivation for Poisson model
An important model assumption is that the number of CpG occurrences in a segment of the genome approximately follows a Poisson distribution. Note that the counts are not binomial because CG dinucleotide cannot start at consecutive bases. We termed the distribution nonconsecutive binomial and proved that, asymptotically, we obtain the same results as if the counts were based on independent Bernoulli trials. A detailed proof can be found at Section 1 of the supplementary material available at Biostatistics online.
We examined the properties of a our random variable using simulations. Figure 6 shows the probability mass function of a nonconsecutive binomial and Poisson are similar for different L and p values.
Fig. 6.

Probability mass functions for (black circle) and (red triangle) for different values of L and p. The pmf functions are similar so is a good approximation of .
4.2. Choosing the segment length
The Poisson approximation, described in Section 4.1, requires L to be “large.” However, there is a trade-off in that smaller values of L provide better resolution for the edges of CGI. In this section, we present a simulation and data-motivated rationale for choosing this parameter.
Our simulations showed that the approximation was appropriate for length larger than (Figure 6). We further assessed the performance on real data by creating CGI lists as described in Section 5 for the human genome using segment lengths of and 32. The resulting lists were similar. Visual inspection revealed that there are various instances where smaller proximal CGIs were engulfed into a larger CGI, and using picked more short (less than 50 bp) CGIs. Finally, we created validation plots based on the association of CGI with epigenetic marks as described in Section 5 for each length; showed the best performance (Supplementary Figure S1 of the supplementary material available at Biostatistics online). Therefore, was used throughout this manuscript and we recommend its use in practice. However, we emphasize that for future applications, because of the computational shortcuts proposed, performing a similar sensitivity analysis on this parameter can be easily done.
4.3. Parameter estimation
We used an iterative stepped approach to fit the posited hierarchical model. The benefits of this strategy are many and most notably include the ability to use existing software for fitting, as well as making the computational problem of fitting the model feasible. Moreover, by fitting the model in stages, we thus obtain values based on the most direct evidence. This provides some robustness against model misspecification. However, this approach comes at the cost of theoretical continuity and perhaps leads to understating uncertainty in parameter estimates.
A complicated aspect of our approach was dealing with our assumption of a nonconstant conditional mean for . Typically, HMM algorithms presume a constant mean in each state. To solve this problem, we used an iterative algorithm starting with . Then the standard forward–backward algorithm, as described by Rabiner (1989), with an expectation-maximization (EM) algorithm (Dempster and others, 1977) was applied to . This algorithm provided estimates for the conditional means for each state, that is, and , the variance , as well as posterior probabilities for each state for each segment s. The posterior probabilities were thresholded to obtain a binary (0 or 1) estimate 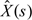 of . Then for each segment, we subtract the means from observed values to obtain the residuals:
of . Then for each segment, we subtract the means from observed values to obtain the residuals:
We then estimate by applying a smoother to . Specifically, we used a moving weighted average with weights obtained from Tukey's biweight kernel with a certain window size. To choose a proper smoothing window, we constructed CGI lists using different window sizes and evaluated their association with functional elements. A window size of 400 bp showed the best result (Supplementary Figure S2 of the supplementary material available at Biostatistics online) and therefore we used it throughout this article. We then iterated the process. Namely, we subtract the smooth estimate, say 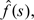 , from the observed GC content and apply the forward–backward algorithm to
, from the observed GC content and apply the forward–backward algorithm to 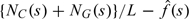 and repeat the above procedure until convergence.
and repeat the above procedure until convergence.
The use of HMMs and this iterated scheme, as opposed to a complete maximum likelihood solution, for example, is motivated by HMMs established applicability to sequence data, the availability of robust fitting algorithms and the satisfactory performance we have seen on the data. Moreover, as stated above, we have placed a high emphasis utilizing methods that can be easily implemented and use the most direct information available. Convergence is usually obtained quickly, in 5 iterations or so.
The result of this algorithm is a smoothed estimate of that accommodates change points from regions of high GC content and a slowly varying trend. By iterating these steps, we mirror a blocked maximization procedure, such as is common in backfitting and related procedures. At convergence. a smoothed estimate of f is obtained as well as estimates for the terms, which represent local constant increases or decreases in GC content.
With the estimate of in hand, estimating the HMM on is much simpler. Since, we assume
the HMM can be fit with standard forward–backward algorithm via an EM algorithm (Dempster and others, 1977). The result will give estimates for and and posterior probabilities for . Now we have state probabilities for the 2 latent Markov chains, one defining areas of high GC content, and one defining areas of high CpG content. Here, the areas of CpG content correctly accounts for the number of opportunities for CpG, rather than looking at the raw number in isolation. We estimate the posterior probabilities of being a CGI state, that is, , for each segment s. We also obtain the posterior probabilities of being in a high GC content state, that is, , for each segment s. Because the forward–backward algorithm calculates these quantities, they are readily available. We can then estimate the states for X and Y using these posteriors.
5. RESULTS
Our main motivation for the development of a new CGI definition was the fact that recently discovered epigenetic marks were not associated with CGI based on the current definition but were associated with CpG-enriched regions. Specifically, many DMRs not associated with existing CGI lists, were in CpG shores. Below we describe how CGI lists based on the results of fitting the HMMs, described in Section 4, improve coverage of these locations. As shown in Supplementary Figure S3 of the supplementary material available at Biostatistics online, the CGIs that reside in regions shown in Figure 3 can be correctly detected by thresholding posterior probabilities from 2 HMMs. To further evaluate the performance of the proposed model, we compare our list, which we refer to as the model-based CGI, to CGI lists provided by the UCSC Genome Browser (Kent and others, 2002), denoted as Genome Browser CGI, and the Glass and others (2007) CGI (Glass and others, 2007).
We created a CGI lists by considering regions of locations with posterior probability greater than 0.5. We also found that the CGIs that coincided with regions of baseline GC content were not associated with epigenetic marks (data not shown) and therefore we filtered these regions. Table 1 shows the joint distribution of the observed posteriors for X and Y. Note that the majority of locations with evidence of CGI state occur when the genome is in the high GC content state.
Table 1.
Joint distribution of posterior probabilities for X (GC content) and Y (CpG rate) on human hg18 genome. Numbers in each cell are the percentages of nonoverlapping 16 bp segments with posterior probabilities fall in a category. For example, there are 64.3% of the segments with both probabilities between 0 and 0.1
| Posterior probabilities for GC content | Posterior probabilities for CpG rate |
||||
| (0, 0.1] | (0.1, 0.5] | (0.5, 0.9] | (0.9, 1] | Total | |
| (0, 0.1] | 64.3 | 2.5 | 0.7 | 0.4 | 67.9 |
| (0.1, 0.5] | 1.6 | 0.1 | 0.0 | 0.0 | 1.7 |
| (0.5, 0.9] | 1.6 | 0.1 | 0.0 | 0.0 | 1.7 |
| (0.9, 1] | 23.0 | 1.9 | 1.2 | 2.6 | 28.7 |
| Total | 90.5 | 4.6 | 1.9 | 3.0 | 100 |
This CGI list is close to 93% of the DMRs reported by Irizarry and others (2008). This is a dramatic increase from 81% by the Genome Browser CGIs and 86% by the Glass and others (2007) CGIs. This improvement was made possible by the flexibility to control specificity. Note that the number of CGIs produced with a posterior probability cutoff of 0.50 was 118 710 and the number in the Genome Browser list is 28 226. To compare lists of similar specificity, we created model-based CGI lists with posterior probability cutoffs ranging from 0.50 to 0.9995 for the human and mouse genomes. We compared the association of each list with 2 functional elements: gene promoters and DMRs.
The Genome Browser CGIs are mostly annotated on the nonrepetitive region and the maximum percentage of repetitive bases for their CGIs is 35%. To make the results comparable, we filtered model-based CGIs and Glass and others (2007) CGIs and kept those with less than 35% repetitive bases. To assess sensitivity, we computed the percentage of human DMRs within 2000 bp of a CGI. We also performed comparisons similar to those previously used to assess CGI lists. Namely, we compared the percent of the transcriptional starting sites (TSS) of refseq genes (Pruitt and others, 2006) covered by CGIs for human and mouse, as done by Takai and Jones (2002) and Glass and others (2007), and the percent of mouse DMRs (Yagi and others, 2008) within 2000 bp of a CGI. To assess specificity in a comparable way for the 3 approaches, we computed the total number of bases covered by each CGI list. Figure 7 shows plots of sensitivity versus specificity.
Fig. 7.

Receiver operating characteristic-like plots showing the sensitivity versus total length for different CGI lists (used as a measure of specificity). The sensitivity is defined as the percentage of functional elements associated to CGI. The 3 figures are for different functional elements: (a) Human (hg18) TSS, (b) human (hg18) DMRs, (c) mouse (MM8) TSS, and (d) mouse (MM8) DMR, respectively.
Glass and others (2007) CGIs overlap with a larger percentage of human TSS than Genome Browser CGIs (64.5% versus 55.9%). However, to achieve this gain in sensitivity, twice as many bases are used. The ability to control specificity with the model-based CGIs demonstrates that small improvements in covering human TSS over the Genome Browser and Glass and others (2007) CGIs are possible at the same specificity level (Figure 7(a)). A more substantial improvement was achieved by the model-based approach in the overlap with the human DMRs (Figure 7(b)). Using a probability cutoff of 0.995, the total lengths of the model-based CGIs (22.7 Mbp) was comparable to the total length of the Genome Browser CGIs (21.1 Mbp), but the overlap with DMR increased from 81% to 86%. A cutoff of 0.9 made the model-based CGIs (37.2 Mbp) a little smaller in size to the Glass and others (2007) CGIs (41.1 Mbp) but the the overlap with DMR increased from 86% to 91%. We observed the same improvements for mouse genome as shown in Figures 7(c) and (d). Different biological applications may have difference sensitivity and specificity requirements. Our approach is flexible in this regard. We provide the CGI list that uses a cutoff value of 0.99 as the “canonical” list because it is associated with the inflection point of the receiver operating characteristic curve (Figure 7).
Another advantage of our approach is that we can easily fit the HMMs to genomes of other species. We fitted the model to 12 species: Homo sapiens (human), Pan troglodytes (chimpanzee), Mus musculus (mouse), Bos taurus (cow), Canis lupus familiaris (dog), Gallus gallus (chicken), Apis mellifera (bee), Drosophila melanogaster (fruit fly), Caenorhabditis elegans (worm), Arabidopsis thaliana (Arabidopsis), Escherichia Coli and Saccharomyces cerevisiae (yeast). CGIs have only been reported for vertebrates. We therefore tested for the presence of CGI by computing a likelihood ratio comparing a model with 2 states to a model with 1 state. Of the 12 species we tested, only the unicellular organisms, that is, yeast and E. Coli, did not have significant evidence in favor of the presence of CGI. We are therefore reporting the first CGI lists for bee, worm, and fruit fly. Previous approaches were not successful because the required cutoffs for these species are very different than for humans. This is demonstrated by examining the fitted and parameters as shown in Table 2. Note that these can be interpreted as the average O/E in the baseline and CGI regions, respectively.
Table 2.
In the HMM, the parameters a0 and a1 represent the average observed to expected ratios in the baseline and island regions. The table below shows the estimated parameters in 12 species. The numbers in the parenthesis are the estimated standard errors
| a0 | a1 | |
| Human | 0.16 (0.001) | 0.56 (0.005) |
| Chimp | 0.16 (0.001) | 0.55 (0.005) |
| Mouse | 0.14 (0.001) | 0.46 (0.006) |
| Cow | 0.17 (0.003) | 0.50 (0.005) |
| Dog | 0.16 (0.002) | 0.64 (0.004) |
| Chicken | 0.18 (0.003) | 0.70 (0.003) |
| Bee | 0.89 (0.028) | 1.55 (0.008) |
| Fruit fly | 0.82 (0.007) | 1.00 (0.024) |
| Worm | 0.84 (0.007) | 1.26 (0.034) |
| Arabidopsis | 0.50 (0.006) | 0.95 (0.004) |
| Yeast | 0.78 (0.011) | 0.80 (0.016) |
| E. coli | 1.12 (0.003) | 1.12 (0.003) |
6. DISCUSSION
We have proposed a procedure for building CGI lists based on HMMs. The main motivation for the development of a new approach was the observation that many DMRs were near regions of high CpG density that did not meet the current definition or any of the alternative definitions. Our new approach improved the overlap with known TSS and DMRs in both human and mouse. The improvements achieved with our approach was mainly due to the data-driven nature of the procedure. Many of the CpG dense regions were left out by algorithmic approaches because they did not satisfy a predetermined rule. Rerunning these algorithms with different cutoffs is no easy task. However, generating CGI lists with different cutoff for the HMM-generated posterior probabilities is trivial. Furthermore, our model has been used to define CGI for 30 species (Irizarry and others, 2009). Note that for some of these species, we are providing the very first list of CGIs.
Figure 8 shows GC content versus O/E for the model-based human CGI list. The red horizontal and vertical lines are from Gardiner-Garden and Frommer CGI definition (GC content > 50%, O/E > 0.6). Based on the current definition only the points above the horizontal line and to the right of the vertical line are CGIs. Various of the model-based CGI do not satisfy the original definition. A histogram of the lengths of model-based CGIs shows many model-based islands are smaller than the formal definition's requirement of 200 bases (Figure 8(b)). These figures demonstrate how the added flexibility permits shorter regions with slightly lower O/E.
Fig. 8.

Statistical characteristics of model-based CGI list for human (hg18). (a) GC content versus O/E. The red vertical and horizontal lines represent the cutoffs used by the Gardiner-Garden and Frommer definition: O/E > 0.6, GC content > 0.5. (b) Histogram of CGI lengths. The vertical line is at the minimum length requirement of Gardiner-Garden and Frommer CGI definition (200 bp).
Our probability-based estimates have units that are interpretable across species. Thus, in a sense, we have transformed the problem onto a standardized scale which will facilitate discussion of thresholding definitions. Because of this, fitting the model to the genomes of other species was simple—no additional user input or algorithmic tweaking was required. To demonstrate this, we fitted the model to the genome of 12 species.
In addition to providing CGI information for these species in isolation, it led to some interesting scientific findings when compared across specifies within taxonomic and evolutionary classes. Strong evidence for the presence of CGI was found for all multicellular organisms. The estimated model parameters confirmed that vertebrates are CpG depleted in their baseline level. Invertebrates were not CpG depleted in their baseline levels but showed higher than expected levels in the CGI. Arabidopsis was somewhere in between. Evidence of methylation has been reported for species for which we found evidence of CGI. The fruit fly had the weakest evidence for the presence of CGI. Interestingly, only small amounts of methylation are detected for this organism (Lyko and others, 2000).
A promising application of the newly defined CGIs is the creation of efficient DNA methylation arrays or enrichment schemes for second generation sequencing. For example, we can construct microarrays that tile only CGI shores. For that purpose using the current Genome Browser definition will miss out on a substantial number of DMRs. It would also be possible to construct this array for any species for which the genome has been sequenced. Furthermore, the ability to control specificity will permit us to deal with different array densities. Note that using a low cutoff value for posterior probabilities produces many new CGIs as compared to the Genome Browser list. As our biological knowledge advances we will be able to check how many of the new ones are functional.
SUPPLEMENTARY MATERIAL
Supplementary material is available at http://biostatistics.oxfordjournals.org.
FUNDING
National Institutes of Health (1R01GM083084-01 to R.I., 1R01RR021967-01A2 to H.J., T32GM074906 to H.W., P50HG003233 to A.F.)
Supplementary Material
Acknowledgments
We thank Herve Pages and Robert Gentleman for help with the Bioconductor packages. Conflict of Interest: None declared.
References
- Agilent. 2008. http://www.chem.agilent.com/Scripts/PDS.asp?lPage=50884. [Google Scholar]
- Altschul S, Gish W, Miller W, Myers E, Lipman D. Basic local alignment search tool. Journal of Molecular Biology. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Bailey T. Combining evidence using p-values: application to sequence homology searches. Bioinformatics. 1998;14:48–54. doi: 10.1093/bioinformatics/14.1.48. [DOI] [PubMed] [Google Scholar]
- Bailey T, Williams N, Misleh C, Li W. MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Research. 2006 doi: 10.1093/nar/gkl198. 34 (Web Server issue), W369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird A. CpG-rich islands and the function of DNA methylation. Nature. 1986;321:209–213. doi: 10.1038/321209a0. [DOI] [PubMed] [Google Scholar]
- Boys R, Henderson D. A Bayesian approach to DNA sequence segmentation. Biometrics. 2004;60:573–581. doi: 10.1111/j.0006-341X.2004.00206.x. [DOI] [PubMed] [Google Scholar]
- Churchill GA. Stochastic models for heterogeneous DNA sequences. Bulletin of Mathematical Biology. 1989;51:79–94. doi: 10.1007/BF02458837. [DOI] [PubMed] [Google Scholar]
- Dempster A, Laird N, Rubin D. Maximum likelihood from incomplete data via the EM algorithm. Journal of the Royal Statistical Society, Series B (Methodological) 1977;39:1–38. [Google Scholar]
- Durbin R, Eddy S, Krogh A, Mitchison G. Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids. Cambridge: Cambridge University Press; 1998. [Google Scholar]
- Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447:433–440. doi: 10.1038/nature05919. [DOI] [PubMed] [Google Scholar]
- Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. Journal of Molecular Biology. 1987;196:261–282. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- Glass J, Thompson R, Khulan B, Figueroa M, Olivier E, Oakley E, van Zant G, Bouhassira E, Melnick A, Golden A and others. CG dinucleotide clustering is a species-specific property of the genome. Nucleic Acids Research. 2007;35:6798. doi: 10.1093/nar/gkm489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry R, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M and others. Genome-wide methylation analysis of human colon cancer reveals similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nature Genetics. 2008;41:178–186. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry R, Wu H, Feinberg A. A species-generalized probabilistic model-based definition of CpG islands. Mammalian Genome. 2009;20:674–680. doi: 10.1007/s00335-009-9222-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent W, Sugnet C, Furey T, Roskin K, Pringle T, Zahler A, Haussler D. The human genome browser at UCSC. Genome Research. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander E, Linton L, Birren B, Nusbaum C, Zody M, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W and others. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Lyko F, Ramsahoye B, Jaenisch R. Development: DNA methylation in Drosophila melanogaster. Nature. 2000;408:538–540. doi: 10.1038/35046205. [DOI] [PubMed] [Google Scholar]
- Meissner A, Mikkelsen T, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein B, Nusbaum C, Jaffe D and others. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–770. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muri F. Modelling bacterial genomes using hidden Markov models. In: Payne R, Green PJ, editors. COMPSTAT '98 Proceedings in Computational Statistics. Heidelberg: Physica; 1998. pp. 89–100. [Google Scholar]
- Nicolas P, Bize L, Muri F, Hoebeke M, Rodolphe F, Ehrlich SD, Prum B, Bessières P. Mining Bacillus subtilis chromosome heterogeneities using hidden Markov models. Nucleic Acids Research. 2002;30:1418–1426. doi: 10.1093/nar/30.6.1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt K, Tatusova T, Maglott D. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Research. 2006;33(Database issue):D501–4. doi: 10.1093/nar/gki025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabiner L. A tutorial on hidden Markov models and selected applications inspeech recognition. Proceedings of the IEEE. 1989;77:257–286. [Google Scholar]
- Takai D, Jones P. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:3740. doi: 10.1073/pnas.052410099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi S, Hirabayashi K, Sato S, Li W, Takahashi Y, Hirakawa T, Wu G, Hattori N, Hattori N, Ohgane J and others. DNA methylation profile of tissue-dependent and differentially methylated regions (T-DMRs) in mouse promoter regions demonstrating tissue-specific gene expression. Genome research. 2008;18:1969. doi: 10.1101/gr.074070.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.