

Abstract
Importance of the field
Pancreatic cancer (PC) is a deadly disease that is so far intractable to currently available treatment regimens. Although well described in different tumors types, the importance of apoptosis inducer Par-4 in PC has not been appreciated. PC is a oncogenic kras driven disease which is known to down-regulate Par-4. Therefore, this review highlights its significance and builds a strong case supporting the role of Par-4 as a possible therapeutic target in PC.
Areas covered in this review
This article covers literature based evidence spanning the last 15 years on Par-4 and its significance in PC.
What the reader will gain
This review provides comprehensive knowledge of the significance of Par-4 and its association with kras status in PC, along with the crosstalk with crucial resistance and survival molecules NF-kB and Bcl-2 that ultimately are responsible for the overall poor outcome of different therapeutic approaches in this disease.
Take home message
Par-4 holds promise as a potential therapeutic target that can be induced by chemopreventive agents and small molecule inhibitors either alone or in combination with standard chemotherapeutics leading to selective apoptosis in PC cells. It also acts as a chemosensitizer and therefore warrants further clinical investigations in this deadly disease.
Keywords: Par-4, Pancreatic cancer, Apoptosis
Pancreatic cancer is a dreadful disease that is poorly amenable to current treatment modalities. Last year the estimated deaths from this intractable malignancy was about 35000 making it the most elusive of all cancers [1]. Less than five percent survive 5 years post diagnosis and complete remission is rare. PC arises through the accumulation of inherited and acquired genetic alterations [2] and it is well established that kras mutations are one of the most common types of genetic abnormality in PC [3–5]. Point mutation in the kras gene occurs in 70–90% of cases and the vast majority occurs at codon 12 of the oncogene. Consequently, a considerable research effort has been made to define the function of kras in normal and neoplastic cells and to target kras for PC treatment. Various strategies have been developed to target kras for the treatment of human cancers since kras mutations occur very early and are the most frequent [6] in PC followed by mutation or silencing of p53 [7], p16 [8;9] and DPC4/smad4 [10–12]. Addressing the contributions of these genes in tumor cell survival after treatment may point to new ways for improving therapy for this disease. For pancreatic cancer, kras mutations have been reported as a negative prognostic factor after surgery and adjuvant chemo-radiation therapy [13]. Activated kras (mutant or oncogenic) are known to inactivate genes, which are directly involved in the regulation of growth inhibition and apoptosis. One such example is that oncogenic kras inhibits TGF-beta (TGF-β) signaling by down regulating TGF-β receptor II (RII) expression [14;15]. A majority of pancreatic tumors show loss of expression of RII and are resistant to exogenous TGF-β mediated growth inhibition [16;17]. In this context, oncogenic kras was also found to down regulate the pro-apoptotic gene, Par-4 [18].
Par-4 and its significance in PC
Prostate apoptosis response-4 (Par-4), also known as PRKC, apoptosis, WT1, regulator (PAWR) is a human gene coding for a tumor-suppressor protein that induces apoptosis in cancer cells, but not in normal cells [19;20]. Par-4 contains a leucine zipper domain that was first identified in prostate cancer cells undergoing apoptosis in response to an exogenous insult [21]. It is ubiquitously expressed in normal tissues and cell types and is typically found in the cytoplasm [22–25]. In contrast, Par-4 localizes both to the cytoplasm and the nucleus in many if not all cancer cells and clinical specimens [26]. Endogenous Par-4 expressed in normal and cancer cells does not, by itself, cause apoptosis. However, inhibition of endogenous Par-4 with antisense oligonucleotides, a dominant-negative leucine zipper domain, or RNA interference precludes apoptosis by exogenously applied agents (such as tumor necrosis factor–related apoptosis-inducing ligand, tumor necrosis factor, growth factor withdrawal, chemotherapeutic agents, or ionizing radiation), which highlights that Par-4 function is essential for apoptosis via diverse cell death pathways [27]. Consistent with this observation that endogenous Par-4 has apoptotic potential, Par-4 knockout mice spontaneously develop tumors of the liver, lung, and endometrium and exhibit prostatic intraepithelial neoplasia (PIN) [28].
Ectopic Par-4 over expression is sufficient to induce apoptosis in most cancer cells, but not in normal or immortalized cells, and this action of Par-4 does not require the leucine zipper domain [29]. This selectivity is certainly very important from a therapeutic point of view. To delineate of the mechanism of this differential activity one needs to understand the inherent differences between cancer and normal cells. In a important study, Gurmurthy et al., have shown that phosphorylation of T155 residue of PAR-4 is critical for apoptosis [27]. This T155 phosphorylation is governed by a protein kinase A (PKA) that is well known to be constitutively elevated in cancer cells [30]. In normal cells, basal PKA activity levels are relatively low and phosphorylation of T155 fails to occur, therefore, normal cells are resistant to apoptosis by ectopic Par-4. Elevation of PKA in normal cells by cAMP-doxorubicin or vincristine induces apoptosis by a mechanism that is dependent on PKA-mediated phosphorylation of the T155 residue of endogenous Par-4 [27]. Further, apoptosis by ectopic Par-4 involves activation of the Fas death receptor signaling pathway and concurrent NF-kB inhibition, which withdraws the anti-apoptotic roadblocks and allows the caspase cascade to proceed uninterrupted [31]. Neither p53 nor PTEN are required for apoptosis by ectopic Par-4. Importantly, over expression of Par-4 in prostate cancer xenografts by i.t. injections of adenoviral Par-4, either in s.c. tumors in nude mice or in orthotopic tumors in the prostatic milieu, results in apoptosis and tumor growth inhibition, therefore implying that Par-4 has therapeutic potential. Nuclear translocation of Par-4 is essential for apoptosis and is mediated by a nuclear localization signal [32]. It is apparent that endogenous Par-4 is in a nonfunctional state and unable to affect apoptosis in cancer cells unless translocated to the nucleus. Akt1 binding and phosphorylation of Par-4 results in 14-3-3–mediated cytoplasmic retention of Par-4 and abrogation of apoptosis [33]. Therefore, one the mechanism through which Akt targeting agents induce apoptosis could be through the involvement of Par-4.
The Par-4 gene has been mapped to chromosome 12q21 and interestingly. this region (12q21) is often deleted in pancreatic cancer [34]. Further, it has been suggested that loss of this chromosome arm has direct association with a poor prognosis in pancreatic cancer patients [35]. In an earlier study using microsatellite analysis, Kimura et al., have studied chromosome deletions in 40 tissue samples and 19 PC cells lines [34]. In this study two commonly deleted regions (region A between D12S81 and D12S1719 at 12q21 at a frequency of 67.5%, and region B was located between D12S360 and D12S78 at 12q22-q23.1 at a frequency of 60%) on the long arm of the chromosome were consistently observed. This study concluded that such frequent deletions observed in chromosome 12 may provide new avenues for isolating tumor suppressor genes. However, whether this deletion is responsible for differential expression of Par-4 in PC needs to be further investigated. As mentioned earlier, Par-4 is under the influence of multiple genes among which kras is the major one [36]. It is a well known that somatic mutations in kras is high in many cancers including PC therefore, how kras regulates Par-4 and what is its relevance in PC are important questions and knowledge of the dynamics of this interaction will certainly benefit the overall understanding of this deadly disease.
Regulation of Par-4 by oncogenic KRAS
Like other members of the kras family, the kras protein is a GTPase and is an early player in many signal transduction pathways [37]. While the protein product of the un-mutated kras gene performs an essential function in normal tissue signaling, mutated kras genes are potent oncogenes that play a significant role in the development of many cancers [38]. This gene is a Kirsten kras oncogene homolog from the mammalian kras gene family. A single amino acid substitution is responsible for an activating mutation. The transforming protein that results is implicated in various malignancies, including lung adenocarcinoma, mucinous adenoma, ductal carcinoma of the pancreas and colorectal carcinoma [38–42]. Several germline kras mutations have been found to be associated with Noonan syndrome [43] and cardio-facio-cutaneous syndrome [44], while somatic kras mutations are found at high rates in colon cancer [45].
The kras gene family encodes small GTPases involved in regulation of cytoplasmic signaling pathways in response to diverse extracellular signals. KRAS gene point mutations of codons 12, 13, and 61 are found frequently in human cancers and result in persistent activation of kras due to impaired GTPase activity [46]. A consequence of constitutive kras activation is downstream changes in gene expression, which in turn act to modulate a variety of processes, including proliferation, differentiation, angiogenesis, and apoptosis [46]. Understanding how kras subverts death is critical to understanding the role of kras in cancer. kras is known to cause a shift in the growth/apoptosis balance to favor growth over death and in doing so promotes cell proliferation. kras achieves growth-favoring conditions in part by altering the expression of pro-apoptotic genes to promote tumorigenesis. Thus, oncogenic kras protects cells from apoptosis by modulation of a variety of apoptosis related proteins, including down-regulation of Par-4 expression [47]. Recently it was demonstrated that Par-4 expression was down regulated when rat epithelial cells were transfected with oncogenic Kras (V12) and in the same study it was further shown that this down-regulation of Par-4 was due to the hypermethylation of Par-4 promoter through a MEK-dependent pathway and this methylation was reversed by 5-aza-2′-deoxycytidine and restored Par-4 expression [48]. On the contrary, using mouse fibroblast NIH3T3 cell, Barradas et al. reported that the treatment with 5-aza-2′-deoxycytidine did not restore Par-4 expression by oncogenic K-ras, rather it was concluded that down regulation of Par-4 might be a result of an oncogenic Ras product that produces a potent signal through Raf/ζPKC-MEK pathway, independent of both p53 and p16/19 [49]. Based on these 2 reported studies the mechanisms underlying PAR-4 downregulation in a Ras oncogenic background might be exclusively dependent on cell type and H-ras or K-ras mutational status. Since pancreatic tumors harbor kras mutations abundantly and the tumor is epithelial in origin, it might be speculated that Par-4 down regulation in these tumors is due to hypermethylation of Par-4 promoter. The amount of information available certainly points to a very strong correlation between kras and Par-4 and is described in the forthcoming passage.
Regulation of PAR-4 by KRAS in PC
Par-4 is known to selectively inhibit the oncogenic kras-dependent NF-kB function, the activation of which makes the tumors more resistant towards commonly used chemotherapeutics [50]. In addition, Par-4 binds the atypical protein kinase C isoforms (aPKCs), which serves to inhibit their enzymatic activity [51]. Therefore, one of the mechanisms whereby Par-4 induces apoptosis is through its ability to block the aPKC-IKK axis which, in turn, inhibits the anti apoptotic protein NF-κB. Based on the functional role of Par-4 in cell death and because pancreatic tumors harbor high incidence of kras mutations, it is natural that the down regulation of Par-4 due to mutations in kras will result in the impairment or dysregulation of apoptotic mechanism and thus render selective survival advantage to pancreatic tumors. This is highlighted by the fact that survival rates of PC patients are low compared with other malignancies and recurrence and metastasis after treatment remain high. In a retrospective study by Ahmed et al, [18] Par-4 expression levels were analyzed in multiple models that include established pancreatic tumor cell lines, normal pancreatic tissues, frozen tumor tissues and paraffin-embedded pancreatic adenocarcinoma samples by Real Time RT-PCR, Western blot analysis and immunohistochemistry. In this study kras mutational status was analyzed by allele-specific oligonucleotide-hybridization. Expression levels of Par-4 were correlated with the kras mutational status and clinical characteristics. Modulation of endogenous Par-4 was tested by transiently expressing oncogenic kras in a wild-type kras pancreatic cancer cell line, BxPC-3. The study found that cell lines with kras mutations showed low levels of Par-4 when compared to a normal pancreatic tissue [18]. Only 30% of the frozen tumors, showed appreciable up regulation of Par-4 mRNA and the remaining showed significant down regulation when compared to normal pancreatic tissue. Similar results were observed in the paraffin-embedded tumors, where a significant number of samples showed down regulation of Par-4 protein and this down regulation of Par-4 correlated significantly with kras mutational status [18]. In addition, the presence of Par-4 mRNA or protein expression in pancreatic tumors correlated with prolonged survival. Transient over expression of oncogenic kras in wild-type-kras BxPC-3 cells significantly downregulated the endogenous Par-4 protein levels and conferred accelerated growth. We also investigated for the basal levels for Par-4 in a spectrum of PC cell lines available in ATCC and our observation correlated with cell line data of Ahmed et al where wild type kras containing cell line (BxPC-3) showed higher Par-4 protein expression [52;53]. Overall the above discussed findings point to a very strong correlation between kras mutational status and Par-4 and the absence of this apoptotic marker is a negative prognostic factor for PC patient undergoing standard therapy.
Regulation of Par-4 by Bcl-2 and its significance in PC
The bcl-2 gene is a key determinant of neoplastic cell expansion whose oncogenic activity has been ascribed primarily to its ability to promote cell survival [54]. It was first cloned as the t(14:18) translocation breakpoint from follicular lymphoma [55]. This translocation results in the juxtaposition of the bcl-2 gene in front of the immunoglobulin heavy-chain enhancer that leads to aberrant expression. Par-4, although not a classical transcription factor, is co-expressed along with Bcl-2 in the normal prostate basal epithelia [56]. Following differentiation of these cells to luminal/secretory epithelia, Par-4 expression is decreased and mainly restricted to the nucleus, and Bcl-2 expression is lost. As described earlier, ectopic expression of Par-4 led to the down-modulation of Bcl-2 protein expression in NIH3T3 mouse fibroblasts and in PC3 prostate cancer cell lines [57]. An inverse correlation in the expression of Bcl-2 and Par-4 also has been reported in the xenografts of human androgen-dependent and androgen-independent prostate cancer developed in mice as well as in acute lymphocytic leukemia [57;58]. Based on the relationship of Bcl-2 and Par-4 it is logical that targeting Bcl-2 would certainly benefit Par-4 mediated apoptosis.
PC has been shown to over express Bcl-2 and its family members. Therefore, blocking Bcl-2 activity could become a novel therapeutic strategy for pancreatic cancer [59;60]. At the level of the mitochondria deregulated expression of members of the BCL-2 protein family causes apoptotic resistance in PC [61]. This protein family consists of two pro-apoptotic subgroups (BAX, BAK, BOK), the BH3-only subgroup (BAD, BIK, BID, BIM, BMF, HRK, NOXA, PUMA) and the anti-apoptotic subgroup (BCL-2, BCL-XL, BCL-W, MCL-1, A1) [62]. In most cases, the mitochondrial cell death pathway requires the BAX-like group as the BH3-only group. The BH3-only proteins act as sensors of cellular stress, directly antagonizing anti-apoptotic BCL-2 members and activated BAX-like proteins are permeabilizing the outer mitochondrial membrane.
Resistance towards apoptosis at the mitochondrial level in PC is conferred by at least two anti-apoptotic BCL-2 family members, BCL-XL and MCL-1. Both proteins are known to be over expressed in PC through MAPK signaling in PC cells [63]. Whether MCL-1 is also induced by the NF-κB pathway in PC cells, as it has been shown for BCL-XL, is unknown at the moment. In a murine model of PC, the BCL-XL promoter was shown to integrate the epidermal growth factor receptor (EGF-R) signal. Here, EGF-R signaling contributes to the activation of the transcription factors NF-κB and STAT3, both activating the BCL-XL gene [64]. Therefore, in order for efficient apoptosis in this model inhibition of both transcription factors is needed. It is well documented that Bcl-2 functions through heterodimerization with proapoptotic members of the Bcl-2 family to prevent mitochondrial pore formation and prevent cytochrome c release and initiation of apoptosis. However, there are more evidences showing that Bcl-2 may play an oncogenic role through survival pathways other than its function at the mitochondrial membrane. It has been reported that Bcl-2 activates NF-kB by a signaling mechanism that involves Raf-1/MEKK-1–mediated activation of IKKβ [65]. Earlier it has been shown that overexpression of Bcl-2 increased the activity of AKT and IKK as well as NF-kB transcriptional activity in pancreatic cancer [66]. These studies provide strong evidence in support of multiple effects of Bcl-2 in not only acting as an anti-apoptotic factor but also suppressing Par-4 and well as influencing NF-kB which is beyond its classical role in cell survival.
Influence of NF-kB on PAR-4
NF-kB (nuclear factor kappa-light-chain-enhancer of activated B cells) is a protein complex that controls the transcription of DNA [67]. NF-κB is found in almost all animal cell types and is involved in cellular responses to stimuli such as stress, cytokines, free radicals, ultraviolet irradiation, oxidized LDL, and bacterial or viral antigens. It plays a key role in regulating the immune response to infection. Conversely, incorrect regulation of NF-κB has been linked to cancer, inflammatory and autoimmune diseases, septic shock, viral infection, and improper immune development. NF-κB has also been implicated in processes of synaptic plasticity and memory [68].
NF-κB plays an essential role in oncogenesis and in the resistance of tumor cells including PC to ionizing radiation and chemotherapy [69]. The transcription potential of NF-κB, a heterodimer between subunits p65 (RelA) and p50, is fully activated by translocation to the nucleus and phosphorylation of RelA. Of interest is the fact that the transcriptional activity of NF-kB is an essential anti-apoptotic target of Par-4 [70]. Interestingly, Par-4 inhibits kras- and Raf-induced NF-kB transcriptional activity, and tumor necrosis factor alpha-induced NF-kB activation. Inhibition of NF-kB activity by super repressor IκBα is sufficient to induce apoptosis in NIH 3T3 cells expressing oncogenic kras [71]. As expected, over expression of Par-4 is sufficient to induce apoptosis in these cells. However, in androgen-independent prostate cancer cells, inhibition of NF-kB transcriptional activity and activation of the Fas pathway are both required for Par-4-induced apoptosis [50]. Fas (CD95) is a member of the tumor necrosis factor receptor family of death receptors that is activated by binding to Fas ligand (FasL), leading to the formation of death-inducing signaling complex (DISC) and the subsequent activation of caspase 8 and downstream caspases and apoptosis [72]. Par-4 activates the Fas pathway by promoting the Fas/FasL translocation to the cell membrane and by protecting the Fas apoptotic pathway from the inhibitory effects of ζ protein kinase C. NF-kB is found to be activated in numerous malignancies including PC and significant amount of information including the one described above points to a strong influence of NF-kB on Par-4. Therefore, it is hypothesized that downregulation of NF-kB by chemopreventive or targeted agents may benefit the overall apoptotic potential of Par-4.
Targeting PAR-4 for pancreatic cancer therapy
Our laboratory has been at the forefront in studying the effect of multiple natural agents as well as synthetic small molecule inhibitors on PAR-4. A primary screen for basal PAR-4 levels in PC cells showed differential expression of this apoptotic protein [52]. Indeed cells with wild type kras such as BxPC-3 had high PAR-4 expression compared to the ones with mutant kras status. These results laid the foundation for future experiments targeting PAR-4 for induction of apoptosis in PC. Our studies were based on the hypothesis that cells with differential PAR-4 status will respond differentially to apoptosis inducing agents. Initially we tested whether some of the well recognized chemopreventive agents such as resveratrol, quercetin, curcumin, epigallocatechin gallate (EGCG), 3,3-diindolylmethane (formulation of DIM which has greater bioavailability, B-DIM obtained from BioResponse, Inc., and from this point forward we will use B-DIM), genistein, biocahnin, delphinidin, plumbagin and thymoquinone could induce or affect localization of Par-4 in PC cells. Studies from our laboratory and others have shown 3,3′-diindolylmethane (DIM) shows potent anti-proliferative activities against a variety of cancers including PC which was linked to down regulation of NF-kB [73–79]. Among all the chemopreventive agents tested only B-DIM induced Par-4 in PC that in turn lead to growth inhibition and apoptosis in PC. In depth mechanistic studies revealed that the activation of Par-4 leads to cell death in PC via inactivation of Akt/Bcl-2/Bcl-xL signaling pathways (Figure 1)[52]. B-DIM is known to target NF-kB which is a repressor of Par-4. Therefore, it is possible that the Par-4 enhancement may be due to NF-kB downregulation by B-DIM. However, direct evidence is still not available and such studies are currently being undertaken in our laboratory. Nevertheless, this was the first and only study of its kind to prove that Par-4 could be activated by natural chemopreventive agents in PC leading to cell death.
Figure 1. Activation of Par-4 leads to apoptosis in PC.
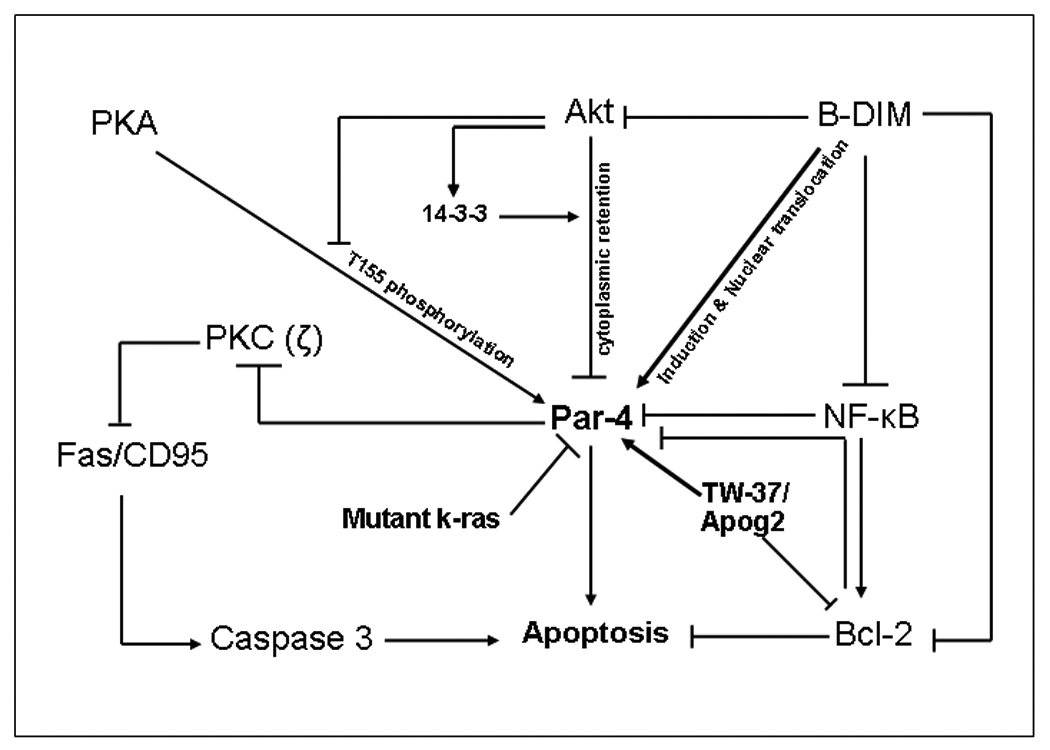
Par-4 is under multiple controls the major ones being mutations in kras along with Akt, Bcl-2, and NF-kB over expression. Akt suppresses Par-4 through phosphorylation and promoting 14-3-3 binding that restricts Par-4 in the cytoplasm. Akt also blocks T155 phosphorylation by PKA in cancer cells. Elevated NF-kB in cancer cells can induce Bcl2 that is known to down-regulate PAR-4. Chemopreventive agent B-DIM can activate Par-4 and also induce Par-4 nuclear translocation by down regulating NF-kB (consequently suppressing Bcl-2) and inhibition of Akt. Apog2 and TW-37 (small molecule inhibitors of Bcl-2) induce Par-4 that ultimately leads to apoptosis. Par-4 can also transduce classical apoptosis pathways like Fas/CD95-mediated cell death by interaction with atypical PKC (ζ). (PC Pancreatic Cancer, B-DIM 3,3’diindolylmethane, Apog2 Apogossypolone, PKA protein kinase A).
In recent years, the predictive anti-tumor therapeutic effect with combinations of different classes of chemotherapeutic agents has been attempted in the clinical setting, but owing to high toxicity and acquired chemo-resistance, the expected therapeutic benefit could not be accomplished. In our opinion both de novo and acquired resistance to therapy could be overcome by employing rational combination therapy, where toxic agents could be used in lower doses but the efficacy of treatment could be increased by novel non-toxic agent that may have different mechanism of action. To this end we have tested whether gemcitabine or cisplatin could induce Par-4 either alone or in combination with B-DIM. Our published results clearly demonstrate that therapeutic drugs such as gemcitabine and cisplatin, either alone or in combination with B-DIM, were able to induce Par-4 in Colo-357, L3.6pl as well as BxPc-3 cell lines [52]. We also hypothesized that the activation of Par-4 by a novel non-toxic agent could lead to sensitization of pancreatic cancer cells to conventional chemotherapeutic agents such as gemcitabine or cisplatin. Based on this rationale, we sought to assess the efficacy of B-DIM, in combination with gemcitabine or cisplatin against three pancreatic cancer cell lines [52]. Our findings provided additional novel mechanistic insights underlying sensitization of pancreatic cancer cells to gemcitabine or cisplatin. Western blot analysis was done which revealed that combination of B-DIM with gemcitabine or cisplatin resulted in the appearance of cleaved PARP. Our results clearly show a marked decrease in Bcl-2 upon treatment with B-DIM or drug alone as well as their combination. Similar decreased expression was observed for another anti-apoptotic factor Bcl-xl. On the other hand a significantly induced expression of pro-apoptotic factor Bax was observed which again confirms that the combination treatment results in apoptotic cell death. These observations strongly suggest that B-DIM sensitizes the pancreatic Colo-357, L3.6pl as well as BxPC-3 cells to gemcitabine as well as cisplatin-induced apoptosis, which is due to the activation of Par-4. Several lines of evidence suggest that the Akt pathway is intricately linked to regulation of apoptosis as well as prognosis [80]. The conventional notion is that Akt enhances survival of tumor cells through transduction of anti-apoptotic and proliferative signals. The mechanism of Akt activation in pancreatic cancer remains unknown, although a majority of pancreatic cancer cell lines that have been examined harbor constitutively activated Akt. Akt was found to be activated (i.e., phosphorylated at Ser 473) in both the Colo-357 as well as L3.6pl cell lines tested, which was abrogated by B-DIM treatment. It is therefore logical to speculate that the observed suppression of cell viability and augmentation of apoptotic cell death by B-DIM could be attributed to its dual effect on targeted disruption of Akt phosphorylation of its substrate, which could be another mechanism by which B-DIM sensitizes pancreatic cancer cells killing to conventional therapeutics. Although apoptosis by B-DIM, cisplatin/gemcitabine combination occurs with simultaneous induction of Par-4, it is still quite premature to conclude whether Par-4 plays any major or integral role in this event; although our results are encouraging and warrants further in-depth mechanistic studies in the future. In conclusion, our findings suggest that B-DIM at non-toxic doses induces Par-4 in pancreatic cancer cells and when combined with a chemotherapeutic drug leads to cell death through apoptosis. This phenomenon can be exploited for future development of therapeutic strategies against pancreatic cancers.
Targeting PAR-4 by small molecule inhibitors of Bcl-2 for pancreatic cancer therapy
As mentioned earlier Bcl-2 expression inversely regulates Par-4. Therefore, it is imperative that downregulation of Bcl-2 would result in over expression of Par-4 ultimately leading to apoptosis and cell death. Bcl-2 is found to be over expressed in many solid tumors, it certainly is an attractive target from a therapeutic point of view. Indeed resistance to chemotherapeutics is certainly attributed to over expression of key anti-apoptotic family members. Many groups have been working to develop anticancer drugs that block the function of Bcl-2 members [81–83]. Apog2 and TW-37 are two recently developed small-molecule inhibitor of Bcl-2, targets multiple members of the Bcl-2 family and attenuates activation of Bcl-2. TW-37 was designed to target the elongated groove of antiapoptotic proteins that normally bind the BH3 domain of proapoptotic effectors such as Bid, Bax, Bim, and others. Over the last several years our laboratory has also been investigating the potential of small molecule inhibitors of Bcl-2 (such a gossypol, apogossypolone, TW-37 and AT-101) against pancreatic cancer. To test whether Bcl-2 downregulation through small molecule inhibitors will enhance the sensitivity of pancreatic cancer cells we investigated various such agents (specifically TW-37 and Apog2). In our study, we found that the treatment of different pancreatic cancer cell lines with sub-micro molar doses of ApoG2 resulted in the induction of Par-4 [53]. The induction of Par-4 was directly correlated with inhibition of cell growth induction of apoptosis as confirmed by histone/DNA ELISA and DAPI cell scoring. Interestingly, sensitivity to apoptosis was directly correlated with Par-4 expression in the four cell lines tested (R = 0.92 and R2 = 0.95). Moreover, siRNA against Par-4 abrogated apoptosis by SMI in L3.6pl and Colo-357 cells underscoring the critical role of Par-4 in inducing apoptosis in pancreatic cancer cells [53]. Further studies confirmed that Apog2 could also induce nuclear localization of Par-4 which is considered a prerequisite for Par-4-mediated apoptosis.
One of the most promising aspects of SMIs in treating cancer is that their targets and mechanisms of action are different from those of cytotoxic drugs and radiation. This makes it feasible to combine SMIs with gemcitabine, creating a synergistic therapy, for pancreatic cancer without developing any cross-resistance or increased toxicity. As mentioned earlier the nucleoside analogue gemcitabine remains the cornerstone of neoadjuvant and adjuvant chemotherapy in pancreatic cancer, although only a partial response is achieved in a minority of patients , thus resulting in a dismal progression-free survival interval ranging from 0.9 to 4.2 months [84]. However, many forms of pancreatic cancer show initial sensitivity to gemcitabine therapy followed by the rapid development of resistance, a feature that essentially characterizes this fatal disease. Overcoming the acquired resistance in pancreatic tumors through sensitization by novel agents such as SMI may be a promising new area of research. Interestingly, the combination of TW-37 with gemcitabine resulted in enhanced cell killing [53]. Isobologram analysis of the data confirmed a synergistic mode of action between gemcitabine and TW-37, suggesting that further studies for this combination using multiple animal models of pancreatic cancer must be done in the future.
To identify the clinical relevance of our in vitro results, an initial pilot experiment was done using a xenograft animal model of pancreatic cancer. Immunohistochemical analysis of Colo-357 xenograft animal tissue stained with Par-4 antibody revealed some interesting results [53]. In the untreated control tumor tissues, we did not find any significant presence of Par-4 and correspondingly negligible apoptosis or necrosis. In contrast, in the TW-37-treated tumors, we found extensive Par-4 staining as well as high amount of necrotic cells. These observations provide evidence in support of the “proof-of-principle” for targeting Par-4 by SMIs, which could be an important and new area in the treatment of pancreatic cancer. Nevertheless, based on a recent study using tissue array on multiple human normal as well as tumor samples, it has been reported that the presence of Par-4 is correlated with longer survival of patients with pancreatic cancer [18], suggesting that the presence of Par-4 leads to enhanced killing of pancreatic cancer cells in patients during therapy. In summary, we found that the SMIs ApoG2 and TW-37 induced cell growth inhibition and apoptosis in pancreatic cancer cells by modulating a novel gene product Par-4. That the two SMIs could also sensitize pancreatic cancer cells to the cytotoxic action of gemcitabine underscores the importance of the SMIs for further development toward the treatment of pancreatic cancer by SMIs alone or in combination with conventional therapeutic agents (Figure 1).
The era of targeted therapies has generated a lot of interest in discovering better approaches for patients with pancreatic cancer. Unfortunately such an approach in PC has met with more failure than success. In the late 1990s, studies were undertaken with agents such as the farnesyltransferase inhibitors (targeting kras pathway) and matrix metalloproteinase inhibitors (targeting matrix metalloproteinase which is known to be overexpressed in PC [85]) . While these drugs did not show any single-agent activity [86], they were studied in the phase III setting combined with gemcitabine or placebo [87;88]. Results were negative with no obvious scientific explanation and no studies have gone back to look at why the farnesyltransferase inhibitors, which target -ras, were ineffective when tested clinically. Based on the well recognized observation that kras mutations suppresses Par-4, in our opinion a possible explanation for failure of these inhibitors could be attribute to the absence of Par-4 in PC patient and certainly warrants further investigation. There was also excitement about testing anti-VEGF strategies in pancreatic cancer. Bevacizumab, an anti-VEGF monoclonal antibody, had demonstrated efficacy in most tumor types in which it had been studied and had resulted in encouraging data in the phase II setting in patients with pancreatic cancer [89]. Unfortunately, two randomized phase III trials of monoclonal antibodies directed against EGFR and VEGF, were negative. Similarly, EGFR-directed therapies for pancreatic cancer show that using anti-EGFR monoclonal antibodies are unlikely to add benefit, and EGFR tyrosine kinase inhibitors add a marginal benefit [90]. While there had been preclinical rationale for studying these agents in PC patients, we now know that increased EGFR protein expression is not synonymous with EGFR pathway activation. Further, the prognostic significance of EGFR ligand expression has not been confirmed in large prospective trials. Although anti-EGFR strategies demonstrated preclinical activity in certain tumor types, the tumor models that were used may not have provided accurate guidance for the study of these agents clinically. Another important protein src (protein tyrosine kinase) that is found to be frequently activated, with greater increases during progressive stages in different cancers [91]. Src is considered an important target for different tumors including PC and numerous src inhibitors have been tested in the clinic [92]. It is interesting to note that src has been shown to downregulate Par-4 [93]. However, as it was the case with farnesyltransferase, VEGF and EGFR inhibitors, experts agree that single agent treatment of human malignancy using src inhibitor may not be successful and is destined for resistance and failure [94].
In conclusion, the major reason for the failure of targeted therapies is related to incomplete understanding and validation of molecular targets before their clinical testing. The complexities of the genetic and epigenetic changes in PC coupled by the redundancies and cross-talk in signaling pathways may explain the failure of single-pathway targeted strategies in this disease. Studies in PC will need to show modulation of the intended target molecule in vivo in patients receiving those drugs. For example, the failure of farnesyltransferase inhibition may have been a result of ineffective inhibition of the RAS activity by either compensatory pathway that activated RAS protein or by the limited benefit of targeting kras in advanced PDA. The fact that Par-4 has been shown to be a negative prognostic factor in PC patients, certainly points towards a requirement for greater in depth studies that would ultimately provide explanation of its involvement in failure of these targeted therapies. Such analysis is certainly hindered by lack of readily accessible tumor tissue from PC patients that is a significant challenge in performing in vivo target validation studies.
Expert opinion
Pancreatic cancer is a dreadful disease which so far is intractable to any currently available treatment regimens or surgery. Gemcitabine and 5 FU continue to be the two commonly used agents for this disease with modest response. The basic phenomena behind such a poor outcome for this disease are a combination of multiple factors that includes oncogenic signaling and acquired resistance. Therefore newer molecular targets and agents are the urgent need of the hour for a disease as dreadful as PC. Par-4 is a known apoptosis inducer, which unfortunately has till now been ignored as a potential therapeutic target for PC. Since PC is an oncogenic kras driven disease and kras is well known to down regulate Par-4 certainly points to a direct correlation between the two. However, more information is certainly needed on this subject. Basic mechanistic studies highlight to a crosstalk between the master transcription regulator NF-kB, the anti-apoptotic Bcl-2 family members and Par-4 the former two being main culprits for failure of chemotherapy in PC. Our preclinical results have established that Par-4 can be induced by NF-kB targeting chemopreventive agents as well as synthetic small molecule inhibitors of Bcl-2 leading to cell death and apoptosis in PC (Figure 1). More significantly, activation of Par-4 by these agents also reverts the resistance towards gemcitabine and cisplatin which is inherently observed in PC. Although B-DIM/SMI-5FU combination has not been studied earlier, it is hypothesized that a similar chemosensitization effect will be observed toward 5FU that involves induction of Par-4 and warrants further investigation. Therefore based on our results in our expert opinion, Par-4 should be further studied as a possible target candidate that would benefit PC therapy.
Article highlight Box
Pancreatic cancer is a deadly disease that so far is intractable to currently available treatment regimens.
Par-4 is an apoptosis inducer that has been well described in different tumors types yet its significance has so far been ignored in pancreatic cancer.
Par-4 is negatively regulated by oncogenic kras, NF-kB and Bcl-2 all of which are found up-regulated in pancreatic cancer and contribute to acquired chemoresistance observed in this disease.
Chemopreventive agents targeting NF-kB and small molecule inhibitors of Bcl-2 can induce Par-4 leading to apoptosis in PC.
Induction of Par-4 sensitizes PC cells to chemotherapeutic induced apoptosis.
Based on the currently available evidence, Par-4 holds promise as a potential target candidate for this deadly disease.
Acknowledgment
The authors acknowledge Dr. Vivek M Rangnekar for his contribution on studies on Par-4 in pancreatic cancer.
Declaration of interest
NIH grants 5R01CA109389-03 (to RM Mohammad) and 5R01CA131151-02 and 5R01CA132794-02 to FH Sarkar are acknowledged.
Reference List
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59(4):225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Buchholz M, Gress TM. Molecular changes in pancreatic cancer. Expert Rev Anticancer Ther. 2009;9(10):1487–1497. doi: 10.1586/era.09.107. [DOI] [PubMed] [Google Scholar]
- 3.DeNicola GM, Tuveson DA. RAS in cellular transformation and senescence. Eur J Cancer. 2009;45 Suppl 1:211–216. doi: 10.1016/S0959-8049(09)70036-X. [DOI] [PubMed] [Google Scholar]
- 4.Perez-Mancera PA, Tuveson DA. Physiological analysis of oncogenic K-ras. Methods Enzymol. 2006;407:676–690. doi: 10.1016/S0076-6879(05)07053-9. [DOI] [PubMed] [Google Scholar]
- 5.Hingorani SR, Tuveson DA. In search of an early warning system for pancreatic cancer. Cancer Biol Ther. 2003;2(1):84–86. doi: 10.4161/cbt.246. [DOI] [PubMed] [Google Scholar]
- 6.Furukawa T, Horii A. [Mechanistic insights for pancreatic carcinogenesis] Nippon Rinsho. 2006;64 Suppl 1:27–31. [PubMed] [Google Scholar]
- 7.Sato Y, Nio Y, Song MM, Sumi S, Hirahara N, Minari Y, et al. p53 protein expression as prognostic factor in human pancreatic cancer. Anticancer Res. 1997;17(4A):2779–2788. [PubMed] [Google Scholar]
- 8.Bartsch DK, Sina-Frey M, Lang S, Wild A, Gerdes B, Barth P, et al. CDKN2A germline mutations in familial pancreatic cancer. Ann Surg. 2002;236(6):730–737. doi: 10.1097/00000658-200212000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moskaluk CA, Hruban RH, Kern SE. p16 and K-ras gene mutations in the intraductal precursors of human pancreatic adenocarcinoma. Cancer Res. 1997;57(11):2140–2143. [PubMed] [Google Scholar]
- 10.Popovic HM, Korolija M, Jakic RJ, Pavkovic P, Hadzija M, Kapitanovic S. K-ras and Dpc4 mutations in chronic pancreatitis: case series. Croat Med J. 2007;48(2):218–224. [PMC free article] [PubMed] [Google Scholar]
- 11.Perren A, Saremaslani P, Schmid S, Bonvin C, Locher T, Roth J, et al. DPC4/Smad4: no mutations, rare allelic imbalances, and retained protein expression in pancreatic endocrine tumors. Diagn Mol Pathol. 2003;12(4):181–186. doi: 10.1097/00019606-200312000-00001. [DOI] [PubMed] [Google Scholar]
- 12.Gerdes B, Wild A, Wittenberg J, Barth P, Ramaswamy A, Kersting M, et al. Tumor-suppressing pathways in cystic pancreatic tumors. Pancreas. 2003;26(1):42–48. doi: 10.1097/00006676-200301000-00008. [DOI] [PubMed] [Google Scholar]
- 13.Olsen CC, Schefter TE, Chen H, Kane M, Leong S, McCarter MD, et al. Results of a phase I trial of 12 patients with locally advanced pancreatic carcinoma combining gefitinib, paclitaxel, and 3-dimensional conformal radiation: report of toxicity and evaluation of circulating K-ras as a potential biomarker of response to therapy. Am J Clin Oncol. 2009;32(2):115–121. doi: 10.1097/COC.0b013e318180baa3. [DOI] [PubMed] [Google Scholar]
- 14.Jonson T, Albrechtsson E, Axelson J, Heidenblad M, Gorunova L, Johansson B, et al. Altered expression of TGFB receptors and mitogenic effects of TGFB in pancreatic carcinomas. Int J Oncol. 2001;19(1):71–81. [PubMed] [Google Scholar]
- 15.Jonson T, Gorunova L, Dawiskiba S, ndren-Sandberg A, Stenman G, ten DP, et al. Molecular analyses of the 15q and 18q SMAD genes in pancreatic cancer. Genes Chromosomes Cancer. 1999;24(1):62–71. doi: 10.1002/(sici)1098-2264(199901)24:1<62::aid-gcc9>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 16.Truty MJ, Urrutia R. Basics of TGF-beta and pancreatic cancer. Pancreatology. 2007;7(5–6):423–435. doi: 10.1159/000108959. [DOI] [PubMed] [Google Scholar]
- 17.Truty MJ, Urrutia R. Transforming growth factor-beta: what every pancreatic surgeon should know. Surgery. 2007;141(1):1–6. doi: 10.1016/j.surg.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 18.Ahmed MM, Sheldon D, Fruitwala MA, Venkatasubbarao K, Lee EY, Gupta S, et al. Downregulation of PAR-4, a pro-apoptotic gene, in pancreatic tumors harboring K-ras mutation. Int J Cancer. 2008;122(1):63–70. doi: 10.1002/ijc.23019. [DOI] [PubMed] [Google Scholar]
- 19.Sells SF, Han SS, Muthukkumar S, Maddiwar N, Johnstone R, Boghaert E, et al. Expression and function of the leucine zipper protein Par-4 in apoptosis. Mol Cell Biol. 1997;17(7):3823–3832. doi: 10.1128/mcb.17.7.3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnstone RW, See RH, Sells SF, Wang J, Muthukkumar S, Englert C, et al. A novel repressor, par-4, modulates transcription and growth suppression functions of the Wilms' tumor suppressor WT1. Mol Cell Biol. 1996;16(12):6945–6956. doi: 10.1128/mcb.16.12.6945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rangnekar VM. Apoptosis mediated by a novel leucine zipper protein Par-4. Apoptosis. 1998;3(2):61–66. doi: 10.1023/a:1009666705875. [DOI] [PubMed] [Google Scholar]
- 22.Lee JW, Hsiao WT, Lee KF, Sheu LF, Hsu HY, Hsu LP, et al. Widespread expression of prostate apoptosis response-4 in nasopharyngeal carcinoma. Head Neck. 2009 doi: 10.1002/hed.21282. [DOI] [PubMed] [Google Scholar]
- 23.Gonzalez IH, Santana P, Gonzalez-Robayna I, Ferrer M, Morales V, Blanco FL, et al. Regulation of the expression of prostate apoptosis response protein 4 (Par-4) in rat granulosa cells. Apoptosis. 2007;12(4):769–779. doi: 10.1007/s10495-006-0019-7. [DOI] [PubMed] [Google Scholar]
- 24.Xie J, Guo Q. Par-4 is a novel mediator of renal tubule cell death in models of ischemia-reperfusion injury. Am J Physiol Renal Physiol. 2007;292(1):F107–F115. doi: 10.1152/ajprenal.00083.2006. [DOI] [PubMed] [Google Scholar]
- 25.Gurumurthy S, Rangnekar VM. Par-4 inducible apoptosis in prostate cancer cells. J Cell Biochem. 2004;91(3):504–512. doi: 10.1002/jcb.20000. [DOI] [PubMed] [Google Scholar]
- 26.Boghaert ER, Sells SF, Walid AJ, Malone P, Williams NM, Weinstein MH, et al. Immunohistochemical analysis of the proapoptotic protein Par-4 in normal rat tissues. Cell Growth Differ. 1997;8(8):881–890. [PubMed] [Google Scholar]
- 27.Gurumurthy S, Goswami A, Vasudevan KM, Rangnekar VM. Phosphorylation of Par-4 by protein kinase A is critical for apoptosis. Mol Cell Biol. 2005;25(3):1146–1161. doi: 10.1128/MCB.25.3.1146-1161.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garcia-Cao I, Duran A, Collado M, Carrascosa MJ, Martin-Caballero J, Flores JM, et al. Tumour-suppression activity of the proapoptotic regulator Par4. EMBO Rep. 2005;6(6):577–583. doi: 10.1038/sj.embor.7400421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burikhanov R, Zhao Y, Goswami A, Qiu S, Schwarze SR, Rangnekar VM. The tumor suppressor Par-4 activates an extrinsic pathway for apoptosis. Cell. 2009;138(2):377–388. doi: 10.1016/j.cell.2009.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cho YS, Park YG, Lee YN, Kim MK, Bates S, Tan L, et al. Extracellular protein kinase A as a cancer biomarker: its expression by tumor cells and reversal by a myristate-lacking Calpha and RIIbeta subunit overexpression. Proc Natl Acad Sci U S A. 2000;97(2):835–840. doi: 10.1073/pnas.97.2.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chakraborty M, Qiu SG, Vasudevan KM, Rangnekar VM. Par-4 drives trafficking and activation of Fas and Fasl to induce prostate cancer cell apoptosis and tumor regression. Cancer Res. 2001;61(19):7255–7263. [PubMed] [Google Scholar]
- 32.El-Guendy N, Rangnekar VM. Apoptosis by Par-4 in cancer and neurodegenerative diseases. Exp Cell Res. 2003;283(1):51–66. doi: 10.1016/s0014-4827(02)00016-2. [DOI] [PubMed] [Google Scholar]
- 33.Goswami A, Burikhanov R, de TA, Fujita N, Goswami M, Zhao Y, et al. Binding and phosphorylation of par-4 by akt is essential for cancer cell survival. Mol Cell. 2005;20(1):33–44. doi: 10.1016/j.molcel.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 34.Kimura M, Furukawa T, Abe T, Yatsuoka T, Youssef EM, Yokoyama T, et al. Identification of two common regions of allelic loss in chromosome arm 12q in human pancreatic cancer. Cancer Res. 1998;58(11):2456–2460. [PubMed] [Google Scholar]
- 35.Youssef EM, Kaneko K, Yatsuoka T, Hayashi Y, Hoshi M, Horii A, et al. Human BAC contig covering the deleted region in pancreatic cancer at 12q21. DNA Seq. 2001;11(6):541–546. doi: 10.3109/10425170109041339. [DOI] [PubMed] [Google Scholar]
- 36.Moreno-Bueno G, Fernandez-Marcos PJ, Collado M, Tendero MJ, Rodriguez-Pinilla SM, Garcia-Cao I, et al. Inactivation of the candidate tumor suppressor par-4 in endometrial cancer. Cancer Res. 2007;67(5):1927–1934. doi: 10.1158/0008-5472.CAN-06-2687. [DOI] [PubMed] [Google Scholar]
- 37.Kranenburg O. The KRAS oncogene: past, present, and future. Biochim Biophys Acta. 2005;1756(2):81–82. doi: 10.1016/j.bbcan.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 38.Riely GJ, Ladanyi M. KRAS mutations: an old oncogene becomes a new predictive biomarker. J Mol Diagn. 2008;10(6):493–495. doi: 10.2353/jmoldx.2008.080105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mao C, Qiu LX, Liao RY, Du FB, Ding H, Yang WC, et al. KRAS mutations and resistance to EGFR-TKIs treatment in patients with non-small cell lung cancer: A meta-analysis of 22 studies. Lung Cancer. 2009 doi: 10.1016/j.lungcan.2009.11.020. [DOI] [PubMed] [Google Scholar]
- 40.Cejas P, Lopez-Gomez M, Aguayo C, Madero R, de Castro CJ, Belda-Iniesta C, et al. KRAS mutations in primary colorectal cancer tumors and related metastases: a potential role in prediction of lung metastasis. PLoS One. 2009;4(12):e8199. doi: 10.1371/journal.pone.0008199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dobrzycka B, Terlikowski SJ, Kowalczuk O, Niklinska W, Chyczewski L, Kulikowski M. Mutations in the KRAS gene in ovarian tumors. Folia Histochem Cytobiol. 2009;47(2):221–224. doi: 10.2478/v10042-009-0039-6. [DOI] [PubMed] [Google Scholar]
- 42.Jang TW, Oak CH, Chang HK, Suo SJ, Jung MH. EGFR and KRAS mutations in patients with adenocarcinoma of the lung. Korean J Intern Med. 2009;24(1):48–54. doi: 10.3904/kjim.2009.24.1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schubbert S, Zenker M, Rowe SL, Boll S, Klein C, Bollag G, et al. Germline KRAS mutations cause Noonan syndrome. Nat Genet. 2006;38(3):331–336. doi: 10.1038/ng1748. [DOI] [PubMed] [Google Scholar]
- 44.Neumann TE, Allanson J, Kavamura I, Kerr B, Neri G, Noonan J, et al. Multiple giant cell lesions in patients with Noonan syndrome and cardio-facio-cutaneous syndrome. Eur J Hum Genet. 2009;17(4):420–425. doi: 10.1038/ejhg.2008.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Castagnola P, Giaretti W. Mutant KRAS, chromosomal instability and prognosis in colorectal cancer. Biochim Biophys Acta. 2005;1756(2):115–125. doi: 10.1016/j.bbcan.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 46.Grunicke HH, Maly K. Role of GTPases and GTPase regulatory proteins in oncogenesis. Crit Rev Oncog. 1993;4(4):389–402. [PubMed] [Google Scholar]
- 47.Vasudevan KM, Ranganathan P, Rangnekar VM. Regulation of Par-4 by oncogenic Ras. Methods Enzymol. 2006;407:422–442. doi: 10.1016/S0076-6879(05)07035-7. [DOI] [PubMed] [Google Scholar]
- 48.Pruitt K, Ulku AS, Frantz K, Rojas RJ, Muniz-Medina VM, Rangnekar VM, et al. Ras-mediated loss of the pro-apoptotic response protein Par-4 is mediated by DNA hypermethylation through Raf-independent and Raf-dependent signaling cascades in epithelial cells. J Biol Chem. 2005;280(24):23363–23370. doi: 10.1074/jbc.M503083200. [DOI] [PubMed] [Google Scholar]
- 49.Barradas M, Monjas A, az-Meco MT, Serrano M, Moscat J. The downregulation of the pro-apoptotic protein Par-4 is critical for Ras-induced survival and tumor progression. EMBO J. 1999;18(22):6362–6369. doi: 10.1093/emboj/18.22.6362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ranganathan P, Rangnekar VM. Regulation of cancer cell survival by Par-4. Ann N Y Acad Sci. 2005;1059:76–85. doi: 10.1196/annals.1339.046. [DOI] [PubMed] [Google Scholar]
- 51.Moscat J, az-Meco MT. Par-4 keeps the atypical PKCs at bay. Cell Cycle. 2003;2(2):71–72. [PubMed] [Google Scholar]
- 52.Azmi AS, Ahmad A, Banerjee S, Rangnekar VM, Mohammad RM, Sarkar FH. Chemoprevention of pancreatic cancer: characterization of Par-4 and its modulation by 3,3' diindolylmethane (DIM) Pharm Res. 2008;25(9):2117–2124. doi: 10.1007/s11095-008-9581-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Azmi AS, Wang Z, Burikhanov R, Rangnekar VM, Wang G, Chen J, et al. Critical role of prostate apoptosis response-4 in determining the sensitivity of pancreatic cancer cells to small-molecule inhibitor-induced apoptosis. Mol Cancer Ther. 2008;7(9):2884–2893. doi: 10.1158/1535-7163.MCT-08-0438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lessene G, Czabotar PE, Colman PM. BCL-2 family antagonists for cancer therapy. Nat Rev Drug Discov. 2008;7(12):989–1000. doi: 10.1038/nrd2658. [DOI] [PubMed] [Google Scholar]
- 55.Hua C, Zorn S, Jensen JP, Coupland RW, Ko HS, Wright JJ, et al. Consequences of the t(14;18) chromosomal translocation in follicular lymphoma: deregulated expression of a chimeric and mutated BCL-2 gene. Oncogene Res. 1988;2(3):263–275. [PubMed] [Google Scholar]
- 56.Cheema SK, Mishra SK, Rangnekar VM, Tari AM, Kumar R, Lopez-Berestein G. Par-4 transcriptionally regulates Bcl-2 through a WT1-binding site on the bcl-2 promoter. J Biol Chem. 2003;278(22):19995–20005. doi: 10.1074/jbc.M205865200. [DOI] [PubMed] [Google Scholar]
- 57.Qiu G, Ahmed M, Sells SF, Mohiuddin M, Weinstein MH, Rangnekar VM. Mutually exclusive expression patterns of Bcl-2 and Par-4 in human prostate tumors consistent with down-regulation of Bcl-2 by Par-4. Oncogene. 1999;18(3):623–631. doi: 10.1038/sj.onc.1202344. [DOI] [PubMed] [Google Scholar]
- 58.Boghaert ER, Sells SF, Walid AJ, Malone P, Williams NM, Weinstein MH, et al. Immunohistochemical analysis of the proapoptotic protein Par-4 in normal rat tissues. Cell Growth Differ. 1997;8(8):881–890. [PubMed] [Google Scholar]
- 59.Hamacher R, Schmid RM, Saur D, Schneider G. Apoptotic pathways in pancreatic ductal adenocarcinoma. Mol Cancer. 2008;7:64. doi: 10.1186/1476-4598-7-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schneider G, Hamacher R, Eser S, Friess H, Schmid RM, Saur D. Molecular biology of pancreatic cancer--new aspects and targets. Anticancer Res. 2008;28(3A):1541–1550. [PubMed] [Google Scholar]
- 61.Evans JD, Cornford PA, Dodson A, Greenhalf W, Foster CS, Neoptolemos JP. Detailed tissue expression of bcl-2, bax, bak and bcl-x in the normal human pancreas and in chronic pancreatitis, ampullary and pancreatic ductal adenocarcinomas. Pancreatology. 2001;1(3):254–262. doi: 10.1159/000055820. [DOI] [PubMed] [Google Scholar]
- 62.Azmi AS, Mohammad RM. Non-peptidic small molecule inhibitors against Bcl-2 for cancer therapy. J Cell Physiol. 2009;218(1):13–21. doi: 10.1002/jcp.21567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Boucher MJ, Morisset J, Vachon PH, Reed JC, Laine J, Rivard N. MEK/ERK signaling pathway regulates the expression of Bcl-2, Bcl-X(L), and Mcl-1 and promotes survival of human pancreatic cancer cells. J Cell Biochem. 2000;79(3):355–369. [PubMed] [Google Scholar]
- 64.Greten FR, Weber CK, Greten TF, Schneider G, Wagner M, Adler G, et al. Stat3 and NF-kappaB activation prevents apoptosis in pancreatic carcinogenesis. Gastroenterology. 2002;123(6):2052–2063. doi: 10.1053/gast.2002.37075. [DOI] [PubMed] [Google Scholar]
- 65.Regula KM, Baetz D, Kirshenbaum LA. Nuclear factor-kappaB represses hypoxia-induced mitochondrial defects and cell death of ventricular myocytes. Circulation. 2004;110(25):3795–3802. doi: 10.1161/01.CIR.0000150537.59754.55. [DOI] [PubMed] [Google Scholar]
- 66.Wang Z, Song W, Aboukameel A, Mohammad M, Wang G, Banerjee S, et al. TW-37, a small-molecule inhibitor of Bcl-2, inhibits cell growth and invasion in pancreatic cancer. Int J Cancer. 2008;123(4):958–966. doi: 10.1002/ijc.23610. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 67.Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene. 2006;25(51):6680–6684. doi: 10.1038/sj.onc.1209954. [DOI] [PubMed] [Google Scholar]
- 68.Tian B, Brasier AR. Identification of a nuclear factor kappa B-dependent gene network. Recent Prog Horm Res. 2003;58:95–130. doi: 10.1210/rp.58.1.95. [DOI] [PubMed] [Google Scholar]
- 69.Baud V, Karin M. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov. 2009;8(1):33–40. doi: 10.1038/nrd2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chendil D, Das A, Dey S, Mohiuddin M, Ahmed MM. Par-4, a pro-apoptotic gene, inhibits radiation-induced NF kappa B activity and Bcl-2 expression leading to induction of radiosensitivity in human prostate cancer cells PC-3. Cancer Biol Ther. 2002;1(2):152–160. doi: 10.4161/cbt.61. [DOI] [PubMed] [Google Scholar]
- 71.El-Guendy N, Zhao Y, Gurumurthy S, Burikhanov R, Rangnekar VM. Identification of a unique core domain of par-4 sufficient for selective apoptosis induction in cancer cells. Mol Cell Biol. 2003;23(16):5516–5525. doi: 10.1128/MCB.23.16.5516-5525.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mollinedo F, Gajate C. Fas/CD95 death receptor and lipid rafts: new targets for apoptosis-directed cancer therapy. Drug Resist Updat. 2006;9(1–2):51–73. doi: 10.1016/j.drup.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 73.Ahmad A, Kong D, Sarkar SH, Wang Z, Banerjee S, Sarkar FH. Inactivation of uPA and its receptor uPAR by 3,3'-diindolylmethane (DIM) leads to the inhibition of prostate cancer cell growth and migration. J Cell Biochem. 2009;107(3):516–527. doi: 10.1002/jcb.22152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ali S, Varghese L, Pereira L, Tulunay-Ugur OE, Kucuk O, Carey TE, et al. Sensitization of squamous cell carcinoma to cisplatin induced killing by natural agents. Cancer Lett. 2009;278(2):201–209. doi: 10.1016/j.canlet.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 75.Ali S, Banerjee S, Ahmad A, El-Rayes BF, Philip PA, Sarkar FH. Apoptosis-inducing effect of erlotinib is potentiated by 3,3'-diindolylmethane in vitro and in vivo using an orthotopic model of pancreatic cancer. Mol Cancer Ther. 2008;7(6):1708–1719. doi: 10.1158/1535-7163.MCT-08-0354. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 76.Rahman KM, Ali S, Aboukameel A, Sarkar SH, Wang Z, Philip PA, et al. Inactivation of NF-kappaB by 3,3'-diindolylmethane contributes to increased apoptosis induced by chemotherapeutic agent in breast cancer cells. Mol Cancer Ther. 2007;6(10):2757–2765. doi: 10.1158/1535-7163.MCT-07-0336. [DOI] [PubMed] [Google Scholar]
- 77.Li Y, Wang Z, Kong D, Murthy S, Dou QP, Sheng S, et al. Regulation of FOXO3a/beta-catenin/GSK-3beta signaling by 3,3'-diindolylmethane contributes to inhibition of cell proliferation and induction of apoptosis in prostate cancer cells. J Biol Chem. 2007;282(29):21542–21550. doi: 10.1074/jbc.M701978200. [DOI] [PubMed] [Google Scholar]
- 78.Kong D, Li Y, Wang Z, Banerjee S, Sarkar FH. Inhibition of angiogenesis and invasion by 3,3'-diindolylmethane is mediated by the nuclear factor-kappaB downstream target genes MMP-9 and uPA that regulated bioavailability of vascular endothelial growth factor in prostate cancer. Cancer Res. 2007;67(7):3310–3319. doi: 10.1158/0008-5472.CAN-06-4277. [DOI] [PubMed] [Google Scholar]
- 79.Bhuiyan MM, Li Y, Banerjee S, Ahmed F, Wang Z, Ali S, et al. Down-regulation of androgen receptor by 3,3'-diindolylmethane contributes to inhibition of cell proliferation and induction of apoptosis in both hormone-sensitive LNCaP and insensitive C4-2B prostate cancer cells. Cancer Res. 2006;66(20):10064–10072. doi: 10.1158/0008-5472.CAN-06-2011. [DOI] [PubMed] [Google Scholar]
- 80.Tokunaga E, Oki E, Egashira A, Sadanaga N, Morita M, Kakeji Y, et al. Deregulation of the Akt pathway in human cancer. Curr Cancer Drug Targets. 2008;8(1):27–36. doi: 10.2174/156800908783497140. [DOI] [PubMed] [Google Scholar]
- 81.Chonghaile TN, Letai A. Mimicking the BH3 domain to kill cancer cells. Oncogene. 2008;27 Suppl 1:S149–S157. doi: 10.1038/onc.2009.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang G, Nikolovska-Coleska Z, Yang CY, Wang R, Tang G, Guo J, et al. Structure-based design of potent small-molecule inhibitors of anti-apoptotic Bcl-2 proteins. J Med Chem. 2006;49(21):6139–6142. doi: 10.1021/jm060460o. [DOI] [PubMed] [Google Scholar]
- 83.Zeitlin BD, Joo E, Dong Z, Warner K, Wang G, Nikolovska-Coleska Z, et al. Antiangiogenic effect of TW37, a small-molecule inhibitor of Bcl-2. Cancer Res. 2006;66(17):8698–8706. doi: 10.1158/0008-5472.CAN-05-3691. [DOI] [PubMed] [Google Scholar]
- 84.Prost P, Ychou M, Azria D. [Gemcitabine and pancreatic cancer] Bull Cancer. 2002;89:S91–S95. Spec No. [PubMed] [Google Scholar]
- 85.Keleg S, Buchler P, Ludwig R, Buchler MW, Friess H. Invasion and metastasis in pancreatic cancer. Mol Cancer. 2003;2:14. doi: 10.1186/1476-4598-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Macdonald JS, McCoy S, Whitehead RP, Iqbal S, Wade JL, III, Giguere JK, et al. A phase II study of farnesyl transferase inhibitor R115777 in pancreatic cancer: a Southwest oncology group (SWOG 9924) study. Invest New Drugs. 2005;23(5):485–487. doi: 10.1007/s10637-005-2908-y. [DOI] [PubMed] [Google Scholar]
- 87.Van CE, Van d V, Karasek P, Oettle H, Vervenne WL, Szawlowski A, et al. Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J Clin Oncol. 2004;22(8):1430–1438. doi: 10.1200/JCO.2004.10.112. [DOI] [PubMed] [Google Scholar]
- 88.Bramhall SR, Schulz J, Nemunaitis J, Brown PD, Baillet M, Buckels JA. A double-blind placebo-controlled, randomised study comparing gemcitabine and marimastat with gemcitabine and placebo as first line therapy in patients with advanced pancreatic cancer. Br J Cancer. 2002;87(2):161–167. doi: 10.1038/sj.bjc.6600446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kindler HL, Friberg G, Singh DA, Locker G, Nattam S, Kozloff M, et al. Phase II trial of bevacizumab plus gemcitabine in patients with advanced pancreatic cancer. J Clin Oncol. 2005;23(31):8033–8040. doi: 10.1200/JCO.2005.01.9661. [DOI] [PubMed] [Google Scholar]
- 90.Xiong HQ, Rosenberg A, LoBuglio A, Schmidt W, Wolff RA, Deutsch J, et al. Cetuximab, a monoclonal antibody targeting the epidermal growth factor receptor, in combination with gemcitabine for advanced pancreatic cancer: a multicenter phase II Trial. J Clin Oncol. 2004;22(13):2610–2616. doi: 10.1200/JCO.2004.12.040. [DOI] [PubMed] [Google Scholar]
- 91.Stehelin D, Fujita DJ, Padgett T, Varmus HE, Bishop JM. Detection and enumeration of transformation-defective strains of avian sarcoma virus with molecular hybridization. Virology. 1977;76(2):675–684. doi: 10.1016/0042-6822(77)90250-1. [DOI] [PubMed] [Google Scholar]
- 92.Summy JM, Gallick GE. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 2003;22(4):337–358. doi: 10.1023/a:1023772912750. [DOI] [PubMed] [Google Scholar]
- 93.Nalca A, Qiu SG, El-Guendy N, Krishnan S, Rangnekar VM. Oncogenic Ras sensitizes cells to apoptosis by Par-4. J Biol Chem. 1999;274(42):29976–29983. doi: 10.1074/jbc.274.42.29976. [DOI] [PubMed] [Google Scholar]
- 94.Gallick GE. SRC as a potential therapeutic target in solid tumor oncology. Clin Adv Hematol Oncol. 2004;2(7):435–437. [PubMed] [Google Scholar]