

Abstract
The oral microbial consortium is the most characterized polymicrobial microbial community associated with the human host. Extensive sampling of both microbial and tissue samples has demonstrated that there is a strong association between the type of microbial community found in the gingival crevice and the status of innate host mediator expression. The strong clinical association between the microbial community and the innate host response in both clinically healthy and diseased tissue suggests that the oral consortium has a direct effect on periodontal tissue expression of innate defense mediators. A preliminary study in germ-free mice has demonstrated that the oral commensal consortium has direct effect on IL-1β expression, indicating that this microbial community may contribute to the strong protective status of healthy gingival tissue. Likewise, the lipopolysaccharide composition and invasion characteristics of Porphyromonas gingivalis, an oral bacterium strongly associated with periodontitis, suggest that it may be a keystone member of the oral microbial community and facilitate a destructive change in the protective gingival innate host status.
The Microbial Oral Community Is the Most Completely Characterized Group of Bacteria That Persistently Colonize the Host
Examining the potential symbiotic relationships in the oral cavity is greatly aided by the extensive studies that have characterized the composition of dental plaque. Dental plaque is an oral microbiological consortium that forms a biofilm on the tooth and tooth root surface. The first characterization of dental plaque was performed by van Leeuwenhoek in 1683 where he described gingival bacteria as “animacules” that contributed to the beginning of the science of bacteriology (Dobell, 1958). Subsequently, descriptive studies performed throughout the twentieth century demonstrated that dental plaque was a distinct structure containing layers of different morphological types that formed on the tooth and tooth root surface in an orderly ecological succession (Socransky and Haffajee, 1994). Microbiological analyses revealed that the composition of commensal oral bacteria and the bacterial load isolated from healthy sites is significantly different from that found in diseased sites. In healthy sites the microbial load is low (102–103 isolates may be cultured from an individual healthy sulcus) (Darveau et al., 1997) consisting of mostly gram-positive streptococci (e.g., Streptococcus gordonii) and Actinomyces with about 15% gram-negative rod species, including Fusobacterium nucleatum. In contrast, characterization of the periopathogenic microbial flora has revealed that the microbial load is higher (105–108 microorganisms may be cultured from an individual pocket), and there is an increase in the number of gram-negative organisms (15–50%) (Tanner et al., 1996; Darveau et al., 1997) when compared to clinically healthy sites. Further, the relative ease of sampling from the oral cavity combined with DNA probe analysis of bacterial populations facilitated multiple cluster and community ordination statistical methods (Socransky et al., 1998) and defined the previously characterized shift from mostly gram-positive to gram-negative species (Socransky and Haffajee, 1994) that occurs in the transition from periodontal health to disease. These analyses identified periopathogenic bacteria, including Tannerella forsythia, Porphyromonas gingivalis, Treponema denticola, and Prevotella intermedia (Socransky et al., 1998), that group together in diseased sites (Ximenez-Fyvie et al., 2000). These studies also laid a foundation for studies that utilize molecular techniques to identify noncultivatable oral bacteria (Kroes et al., 1999), define oral transmission routes (Li and Caufield, 1998), and examine associations between species genotypes and disease (Griffen et al., 1999). In addition, advances in our understanding of biofilm structure have led to studies examining regulation of specific microbial adhesions (McNab et al., 2003) and how host microenvironmental conditions influence the microarchitecture of the dental plaque biofilm (Blehert et al., 2003). These studies validate that the microbial community associated with oral clinical health is not a random assortment of bacteria but rather represents a highly organized microbial consortium that has evolved to live with each other to occupy niches in the oral host environment.
Innate Host Defense Status in Clinically Healthy and Diseased Tissue
Similar to the highly organized microbial consortium associated with clinically healthy tissue, the innate defense status of this tissue is also highly organized and regulated (Fig. 1). The relative ease of tissue sampling from the oral cavity and the identification of innate host response and inflammatory molecular mediators allowed a comprehensive characterization of the tissue status in both gingival health and disease. Clinically healthy periodontal tissue contains a unique expression of select inflammatory mediators. Early histological studies (Page and Schroeder, 1976) of clinically healthy tissue demonstrated that it contains a cellular infiltrate located in juxtaposition to the colonized tooth surface (Kornman et al., 1997b). A portion of this cellular infiltrate has been described as forming a wall of neutrophils precisely located between bacteria and residing just outside the junctional epithelium, the epithelial cell surface closest to the dental plaque biofilm (Kornman et al., 1997a). Consistent with these observations molecular characterization of healthy periodontal tissue has demonstrated that IL-8, ICAM, and E-selectin are expressed in clinically healthy tissue (Moughal et al., 1992; Nylander et al., 1993; Gemmell et al., 1994; Tonetti et al., 1994; Tonetti, 1997). These inflammatory mediators are necessary for leukocyte diapedesis from the vasculature and directed movement through tissue. E-selectin expression on endothelial cells facilitates a tethering interaction between the leukocyte and the endothelial cell wall initiating the rolling stage required for leukocyte exit (Springer, 1994). IL-8 is a key neutrophil chemoattractant, and ICAM facilitates cellular adhesion. It has been demonstrated that a gradient of IL-8 and ICAM-1 expression exists in clinically healthy tissue (Tonetti et al., 1998). IL-8 expression was greatest at the most superficial junctional epithelial cell layers, and the levels of ICAM-1 increased toward areas exposed to bacterial challenges. More additional immunohistochemical and in situ studies have revealed that clinically healthy periodontal tissue also expresses human β defensin molecules 1, 2, and 3 (Lu et al., 2004, 2005) as well as soluble (Jin and Darveau, 2001) and membrane bound CD14 (Jin et al., 2004) and lipopolysaccharide binding protein (Ren et al., 2004). Lipopolysaccharide binding protein expression was greatest in the gingival epithelium (Ren et al., 2004). These innate defense proteins function in either bacterial killing or bacterial removal, consistent with the notion that healthy periodontal tissue is armed by the innate host defense system to protect against bacterial infection. Clinically healthy human gingival tissue also expresses low levels of TLR2 (Ren et al., 2005; Sugawara et al., 2006): while expression of TLR4 was reported in one study (Sugawara et al., 2006), the other (Ren et al., 2005) did not observe expression of this innate host defense receptor in healthy tissue. A more recent study (Beklen et al., 2008) describes the expression of TLR's 1–10 in both clinically healthy and diseased tissue. In addition, NOD1 and NOD2 act synergistically with select TLR's resulting in the expression of antimicrobial peptides in response to microbial challenge (Uehara and Takada, 2008). Further, gingival fibroblasts are well equipped to respond to bacterial components and may contribute to the IL-8 observed in clinically healthy tissue (Mahanonda et al., 2007). These data are all consistent with the notion that innate host defense mediator expression in clinically healthy tissue is key to the maintenance of periodontal health.
FIG. 1.
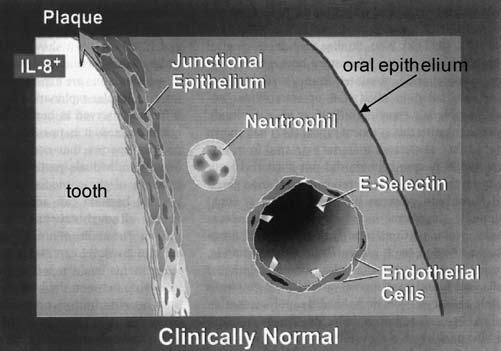
Innate host defense status in clinically normal tissue. Recently, it has been demonstrated that clinically healthy tissue displays low level expression of select inflammatory mediators. The expression of E selectin on the vascular endothelium, for example, is believed to facilitate leukocyte exit from the vasculature into surrounding tissue where they remove bacteria. A gradient of IL-8 expression (indicated by shades of gray) exists in normal tissue to guide leukocytes to the site of bacterial colonization. Recent evidence (Darveau et al., unpublished) suggests that the biofilm of gingival health may provide the stimulus for expression of these mediators, suggesting a commensal relationship between the host and these bacteria. This figure is based on the work of Tonetti et al. (1994) and Moughal et al. (1992). Reprinted with permission from Darveau et al. (1997).
The contribution of the innate defense status observed in clinically healthy tissue to periodontal health is highly significant. Loss of the protective neutrophilic barrier function either by congenital deficiency (Page et al., 1987; Waldrop et al., 1987; Carrassi et al., 1989; Hart et al., 1994) or by chemical induction with antimitotic agents such as cyclophosamide (Attström and Schroeder, 1979; Sallay et al., 1984; Hemmerle and Frank, 1991; Yoshinari et al., 1994) invariably leads to disease. Further, studies have shown that the lack of an intact innate host defense system may be responsible for the significantly increased incidence of severe periodontitis observed in diabetic patients (type I and type II) and tobacco users (Bergstrom et al., 1988; MacFarlane et al., 1992; Offenbacher et al., 1996; Zambon, 1996; Salvi et al., 1997).
Similar to the association found between the commensal oral microbial flora and the innate host defense status of clinically healthy tissue, there is a strong correlation between the periopathogenic microbial flora and a destructive inflammatory response (Ximenez-Fyvie et al., 2000). The innate defense status found in tissue obtained from periodontitis sites is significantly different from that found in healthy tissue (Fig. 2). Periodontitis is associated with the expression of more and different inflammatory mediators compared to clinically healthy tissue. This has contributed to the notion that periodontal disease is a result of both bacterial and host response factors (Page et al., 1997). Although there is increased expression of IL-8 and ICAM, the characteristic expression pattern of these mediators observed in clinically healthy tissue is absent in tissue obtained from periodontitis sites (Tonetti et al., 1994, 1998). There is no IL-8 gradient, but rather patches of intense IL-8 expression are observed in the gingival epithelium with areas of no or little expression (Tonetti et al., 1994). ICAM and E selectin are both expressed at much higher levels than that observed in healthy tissue (Tonetti et al., 1994). In addition, there is an increase in the expression levels of both TLR2 and TLR4 in periodontitis sites compared to clinically healthy tissue (Ren et al., 2005; Sugawara et al., 2006). The increase in these innate host receptors may have profound effects on the innate host response of periodontal tissue. Evidence that these mediators contribute to the characteristic loss of connective tissue and the alveolar bone that surrounds and supports the tooth root associated with periodontal disease comes from studies that demonstrate inflammatory mediator levels decrease after successful treatment (Offenbacher et al., 1986; Masada et al., 1990). Further, administration of antiinflammatory drugs that reduce levels of these mediators can suppress bone and tissue destruction (Offenbacher et al., 1987), and nonsteroidal antiinflammatory drugs that block prostaglandin synthesis can arrest tissue destruction (Offenbacher et al., 1987). Finally, removal of dental plaque remains the most effective mechanism of restoring an appropriate innate host response in periodontitis patients (Page et al., 1997), providing more evidence that the bacterial composition associated with periodontitis is directly responsible for a dysfunctional innate host response.
FIG. 2.
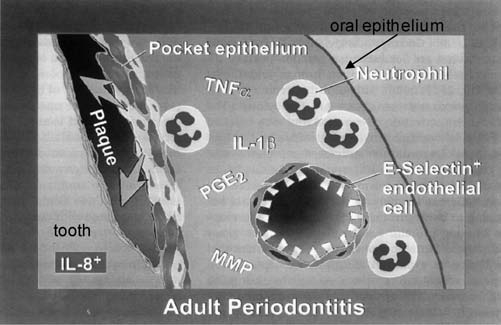
Innate host defense status in adult periodontitis. In adult periodontitis, the molecular mediators of inflammation that are expressed in clinically healthy tissue are expressed at higher levels, and new mediators are present. The gradient of IL-8 expression found in healthy tissue is disrupted (see Fig. 1 legend), and a pocket epithelium forms. This figure is based on the work of Tonetti et al. (1994) and Moughal et al. (1992). TNFα, tumor necrosis factor α; PGE2, prostaglandin E2; MMP, matrix metalloproteinase. Reprinted with permission from Darveau et al. (1997).
Therefore, there are strong correlations between the type of microbial consortium found in the gingival pocket and the status of innate mediator expression in the adjacent periodontal tissue. Since the gingival epithelium does not contain tight junctions, bacterial components shed from the dental plaque biofilm can penetrate gingival tissue (Schwartz et al., 1972; Moore et al., 1986; Wilson et al., 1986; McCoy et al., 1987; Hamada et al., 1990), providing a mechanism by which this tissue may sample the oral bacterial plaque composition. It seems likely, therefore, that the oral microbial consortiums found in clinically healthy and diseased tissues contribute to both the highly orchestrated innate mediator expression found in healthy sites and the destructive innate mediator expression found in diseased tissues.
The Use Germ-Free Mice to Define Direct Consortium Effects on Gingival Tissue
The most definitive approach to determine the contribution of the oral microbial consortiums to the innate host defense status in the periodontium is the use of germ-free mice. For example, germ-free mice have been employed to determine the contribution of commensal bacteria to normal intestinal innate defense, and immune development has been carefully studied with the use of germ-free mice (Gordon and Pesti, 1971; Umesaki and Setoyama, 2000; Hooper et al., 2001; Xu and Gordon, 2003; Macpherson and Harris, 2004). Germ-free mice that are completely devoid of bacteria are generated by sterile Caesarean section and raised aseptically in an isolator with sterile filtered air and are housed using sterile food, water, and bedding. Germ-free mice are distinct from specific-pathogen-free mice that are only devoid of known mouse pathogens and contain intestinal bacteria (Macpherson and Harris, 2004).
It has been shown that commensal bacteria are required for the complete development of Peyer's patches, the lamina propria, and the intraepithelial spaces, all three of the main immune elements found in the intestine (Duncan and Edberg, 1995; Falk et al., 1998). Studies in germ-free mice have revealed that the commensal bacteria induce angiogenesis, contributing to the development of the complex vascular beds found just underneath the mucosal surface (Stappenbeck et al., 2002). Further, it has been found that constitutive ICAM-1 expression in these vessels is also regulated by the presence of the commensal microbiota (Komatsu et al., 2000). In fact, the state of controlled inflammation that normally exists in the intestine has been attributed to both the quality and quantity of intestinal commensal microorganisms (Chadwick and Anderson, 1992; Cebra, 1999). These studies demonstrate that commensal colonization of the intestine orchestrates selective expression of innate host defense components facilitating a mature tissue state that provides immune protection for the host.
However, little is known concerning the contribution of oral commensal bacteria to the armed protective state observed in healthy human periodontal tissue. Germ-free mice have been extensively utilized by oral researchers, however, in the context of caries and periodontitis disease models (Niederman et al., 2001) as opposed to elucidation of direct effects of bacterial colonization on innate host mediator expression in periodontal tissue. Nevertheless, these early disease models in germ-free mice combined with periodontitis mouse models of infection provide evidence that mouse commensal bacteria influence periodontal tissue innate host mediator expression. For example, the P. gingivalis gavage model developed by Baker (Hart et al., 2004) has shown that prior antibiotic treatment to reduce the commensal load is necessary to facilitate P. gingivalis colonization of the oral cavity, mRNA expression of many innate defense mediators is elevated in healthy gingival mouse tissue (Hart et al., 2004), and there are differences in the susceptibility to periodontal infection that are genetically inherited among different inbred mouse strains (Baker et al., 2000). However, these studies cannot differentiate between the passive colonization resistance effect of commensal colonization and the direct development of the protective or destructive innate defense status found in periodontal tissue by oral commensal bacteria.
We have conducted a preliminary investigation directly comparing innate host mediator expression in germ-free and conventionally reared mice (Dixon et al., 2004). It was found that IL-1β levels were significantly higher in conventionally reared mice, consistent with the notion that commensal bacteria in the periodontium, similar to the intestine, actively participate in establishing the innate mediator expression of clinically healthy or normal periodontal tissue. The reasons only IL-1β was identified as being differentially expressed in germ-free and conventionally reared mice included a limited number of samples examined in the pilot study. The presence of elevated amounts of this inflammatory mediator in healthy conventionally reared animals compared to germ-free controls may appear paradoxical since it has been associated with the development of periodontitis (Masada et al., 1990). However, we suspect that the presence of IL-1β in clinically healthy periodontal tissue may serve as a priming mechanism for several different cell types found in the periodontium. These data suggest that the host genetically programs select cytokine expression in the absence of bacterial colonization that then may be altered depending upon the number and type of bacterial species colonizing host tissue.
P. gingivalis Responds to Hemin by Generating a TLR4 Antagonistic Lipid A Structure
We have been examining the potential contribution of P. gingivalis, an oral bacterium strongly associated with periodontitis, to altering the innate defense status of periodontal tissue. A likely candidate for modulating innate mediator expression in host tissue is lipopolysaccharide (LPS). Indeed, LPS has been termed a pattern recognition receptor ligand for the innate host defense system (Medzhitov and Janeway, 2000). The concept of pattern recognition, originally put forward by Janeway (Janeway, 1992), proposes that the host has evolved receptors that recognize common conserved structures found in a variety of different microbes. LPS is present in all gram-negative bacteria, is essential for bacterial viability (one with notable exception [Steeghs et al., 1998]), and contains a highly conserved lipid A structure consisting of a phosphorylated beta-(1,6)-glucosamine disaccharide substituted with hydroxylated and nonhydroxylated fatty acids (Takada and Kotani, 1992), completely filling the criteria for innate host recognition. Consistent with its proposed role as a sentry employed by the host to monitor bacteria infection, in vitro studies have confirmed that whole bacteria and their respective isolated LPSs yield similar responses (Darveau et al., 1991; Somerville et al., 1996), and in vivo studies have validated the important role LPS serves in host recognition of bacterial infection (Khan et al., 1998; Somerville et al., 1999; Haziot et al., 2001). Clearly, innate host recognition of LPS is a key initiating event for the subsequent clearance of gram-negative bacteria from infected host tissues.
We have found that P. gingivalis changes its lipid A structural composition in response to the hemin concentration in the medium. Hemin binds host iron and represents the major iron acquisition system for P. gingivalis (Olczak et al., 2005). Hemin is a relevant microenvironmental factor for P. gingivalis since its concentration is low in healthy tissue and high in diseased sites where vascular ulceration leads the leakage of blood into the underlying gingival epithelium. The lipid A structural content of P. gingivalis after growth in medium containing 1 μg/mL hemin consists of a single major monophosphoryl penta-acylated lipid A cluster (centered at m/z 1690), while the lipid A content of bacteria incubated with 10 μg/mL hemin showed a significantly reduced amount of this lipid A structure and a significant increase in both monophosphoryl tetra-acylated lipid A structures (m/z 1435 and 1449) and a diphosphoryl penta-acylated lipid A cluster (centered at m/z 1770). The monophosphoryl penta- and tetra-acylated lipid A structures have been purified and shown to display distinctly different effects on endothelial cell E selectin activation (Reife et al., 2006) due to the fact that PgLPS1435/1449 is a TLR4 antagonist (Darveau et al., 1995; Yoshimura et al., 2002; Coats et al., 2003; Reife et al., 2006), whereas PgLPS1690 is a TLR4 agonist (Reife et al., 2006). Consistent with PgLPS1435/1449 and PgLPS1690 displaying different effects on E-selectin expression, it was shown that these two LPS preparations interact with the TLR4 complex differently (Reife et al., 2006). The expression of functionally distinct lipid A structures that have opposing effects on TLR4 activation indicates that P. gingivalis may utilize its lipid A structural content to modulate the innate host response in different microenvironmental conditions.
P. gingivalis May Represent a Keystone Species in the Oral Microbial Consortium
Two lines of evidence indicate that P. gingivalis may be a keystone species in the oral microbial community. Keystone species in this context means that this one bacterium serves an essential function for the entire community, similar to a differentiated cell serving a function for an entire tissue. The first is that the presence of the P. gingivalis TLR4 lipid A antagonist that can block TLR4 activation in response to several different oral microbial bacteria (Darveau et al., 1995) through competitive binding to MD-2 (Coats et al., 2005, 2007) combined with the observation that P. gingivalis releases LPS that can penetrate gingival tissue (Schwartz et al., 1972) supports the notion that the TLR4 lipid A antagonist will dampen TLR4 responses for the entire oral microbial community. This is especially relevant considering the proposal by Munford (Munford and Varley, 2006) that TLR4 sensing prevents invasion into submucosal tissue by mucosal gram-negative bacteria. Therefore, hemin may act as an environmental sensor that P. gingivalis responds to by making the lipid A TLR4 antagonist, and then this facilitates invasion of tissue and modulation of innate host defense mediator expression in response to numerous members of the oral microbial consortium. Hemin, provided to the bacteria in the form of hemoglobin, has been shown to significantly increase in concentration in diseased sites (Hanioka et al., 1990, 1991). Secondly, we have shown that when P. gingivalis invades gingival epithelial cells it blocks the epithelial cell IL-8 response to other oral bacteria (Darveau et al., 1998). We have termed this process local chemokine paralysis, since the ability to detect and locate bacterial colonization by IL-8 would be effectively paralyzed and unable to function at sites of P. gingivalis invasion. Inhibition of IL-8 accumulation by P. gingivalis at sites of bacterial epithelial cell invasion could have a devastating effect on innate host defense in the periodontium, where bacterial exposure is constant. The host may no longer be able to detect the presence of bacteria and direct leukocytes for their removal. This phenomenon represents another mechanism by which the action of a single bacterial member of the oral consortium can affect the host responses to a wide variety of different bacteria.
Disclosure Statement
No competing financial interests exist.
References
- Attström R. Schroeder H.E. Effect of experimental neutropenia on initial gingivitis in dogs. Scand J Dent Res. 1979;87:7–23. doi: 10.1111/j.1600-0722.1979.tb01935.x. [DOI] [PubMed] [Google Scholar]
- Baker P.J. Dixon M. Roopenian D.C. Genetic control of susceptibility to Porphyromonas gingivalis-induced alveolar bone loss in mice. Infect Immun. 2000;68:5864–5868. doi: 10.1128/iai.68.10.5864-5868.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beklen A. Hukkanen M. Richardson R. Konttinen Y.T. Immunohistochemical localization of Toll-like receptors 1–10 in periodontitis. Oral Microbiol Immunol. 2008;23:425–431. doi: 10.1111/j.1399-302X.2008.00448.x. [DOI] [PubMed] [Google Scholar]
- Bergstrom J. Persson L. Preber H. Influence of cigarette smoking on vascular reaction during experimental gingivitis. Scand J Dent Res. 1988;96:34–39. doi: 10.1111/j.1600-0722.1988.tb01405.x. [DOI] [PubMed] [Google Scholar]
- Blehert D.S. Palmer R.J., Jr. Xavier J.B. Almeida J.S. Kolenbrander P.E. Autoinducer 2 production by Streptococcus gordonii DL1 and the biofilm phenotype of a luxS mutant are influenced by nutritional conditions. J Bacteriol. 2003;185:4851–4860. doi: 10.1128/JB.185.16.4851-4860.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrassi A. Abati S. Santarelli G. Vogel G. Periodontitis in a patient with chronic neutropenia. J Periodontol. 1989;60:352–357. doi: 10.1902/jop.1989.60.6.352. [DOI] [PubMed] [Google Scholar]
- Cebra J.J. Influences of microbiota on intestinal immune system development. Am J Clin Nutr. 1999;69:1046S–1051S. doi: 10.1093/ajcn/69.5.1046s. [DOI] [PubMed] [Google Scholar]
- Chadwick V.S. Anderson R.P. Microorganisms and their products in inflammatory bowel disease. In: R.P. MacDermott., editor; W.F. Stenson., editor. Inflammatory Bowel Disease. Elsevier Science; Amsterdam: 1992. pp. 241–258. [Google Scholar]
- Coats S.R. Do C.T. Karimi-Naser L.M. Braham P.H. Darveau R.P. Antagonistic lipopolysaccharides block E. coli lipopolysaccharide function at human TLR4 via interaction with the human MD-2 lipopolysaccharide binding site. Cell Microbiol. 2007;9:1191–1202. doi: 10.1111/j.1462-5822.2006.00859.x. [DOI] [PubMed] [Google Scholar]
- Coats S.R. Pham T.T. Bainbridge B.W. Reife R.A. Darveau R.P. MD-2 mediates the ability of tetra-acylated and penta-acylated lipopolysaccharides to antagonize Escherichia coli lipopolysaccharide at the TLR4 signaling complex. J Immunol. 2005;175:4490–4498. doi: 10.4049/jimmunol.175.7.4490. [DOI] [PubMed] [Google Scholar]
- Coats S.R. Reife R.A. Bainbridge B.W. Pham T.T. Darveau R.P. Porphyromonas gingivalis lipopolysaccharide antagonizes Escherichia coli lipopolysaccharide at toll-like receptor 4 in human endothelial cells. Infect Immun. 2003;71:6799–6807. doi: 10.1128/IAI.71.12.6799-6807.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darveau R.P. Belton C.M. Reife R.A. Lamont R.J. Local chemokine paralysis, a novel pathogenic mechanism for Porphyromonas gingivalis. Infect Immun. 1998;66:1660–1665. doi: 10.1128/iai.66.4.1660-1665.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darveau R.P. Cunningham M.D. Bailey T. Seachord C. Ratcliffe K. Bainbridge B. Ability of bacteria associated with chronic inflammatory disease to stimulate E-selectin expression and promote neutrophil adhesion. Infect Immun. 1995;63:1311–1317. doi: 10.1128/iai.63.4.1311-1317.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darveau R.P. Cunningham M.D. Seachord C.L. Cassiano-Clough L. Cosand W.L. Blake J. ß-lactam antibiotics potentiate magainin 2 antimicrobial activity in vitro and in vivo. Antimicrob Agents Chemother. 1991;35:1153–1159. doi: 10.1128/aac.35.6.1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darveau R.P. Tanner A. Page R.C. The microbial challenge in periodontitis. Periodontol 2000. 1997;14:12–32. doi: 10.1111/j.1600-0757.1997.tb00190.x. [DOI] [PubMed] [Google Scholar]
- Dixon D.R. Reife R.A. Cebra J.J. Darveau R.P. Commensal bacteria influence innate status within gingival tissues: a pilot study. J Periodontol. 2004;75:1486–1492. doi: 10.1902/jop.2004.75.11.1486. [DOI] [PubMed] [Google Scholar]
- Dobell C. Antony Van Leeuwenhoek and His “Little Animals”. Russell and Russell Inc.; New York: 1958. The first observations on entozoic protozoa and bacteria; pp. 236–256. [Google Scholar]
- Duncan H.E. Edberg S.C. Host-microbe interaction in the gastrointestinal tract. Crit Rev Microbiol. 1995;21:85–100. doi: 10.3109/10408419509113535. [DOI] [PubMed] [Google Scholar]
- Falk P.G. Hooper L.V. Midtvedt T. Gordon J.I. Creating and maintaining the gastrointestinal ecosystem: what we know and need to know from gnotobiology. Microbiol Mol Biol Rev. 1998;62:1157–1170. doi: 10.1128/mmbr.62.4.1157-1170.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemmell E. Walsh L.J. Savage N.W. Seymore G.J. Adhesion molecule expression in chronic inflammatory periodontal disease tissue. J Periodontal Res. 1994;29:46–53. doi: 10.1111/j.1600-0765.1994.tb01090.x. [DOI] [PubMed] [Google Scholar]
- Gordon H.A. Pesti L. The gnotobiotic animal as a tool in the study of host microbial relationships. Bacteriol Rev. 1971;35:390–429. doi: 10.1128/br.35.4.390-429.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffen A.L. Lyons S.R. Becker M.R. Moeschberger M.L. Leys E.J. Porphyromonas gingivalis strain variability and periodontitis. J Clin Microbiol. 1999;37:4028–4033. doi: 10.1128/jcm.37.12.4028-4033.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada S. Takada H. Ogawa T. Fujiwara T. Mihara J. Lipopolysaccharides of oral anaerobes associated with chronic inflammation: chemical and immunomodulating properties. Int Rev Immunol. 1990;6:247–261. doi: 10.3109/08830189009056635. [DOI] [PubMed] [Google Scholar]
- Hanioka T. Shizukuishi S. Tsunemitsu A. Hemoglobin concentration and oxygen saturation of clinically healthy and inflamed gingiva in human subjects. J Periodontal Res. 1990;25:93–98. doi: 10.1111/j.1600-0765.1990.tb00898.x. [DOI] [PubMed] [Google Scholar]
- Hanioka T. Shizukuishi S. Tsunemitsu A. Changes in hemoglobin concentration and oxygen saturation in human gingiva with decreasing inflammation. J Periodontol. 1991;62:366–369. doi: 10.1902/jop.1991.62.6.366. [DOI] [PubMed] [Google Scholar]
- Hart G.T. Shaffer D.J. Akilesh S. Brown A.C. Moran L. Roopenian D.C. Quantitative gene expression profiling implicates genes for susceptibility and resistance to alveolar bone loss. Infect Immun. 2004;72:4471–4479. doi: 10.1128/IAI.72.8.4471-4479.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart T.C. Shapira L. Van Dyke T.E. Neutrophil defects as risk factors for periodontal diseases. J Periodontol. 1994;65:521–529. doi: 10.1902/jop.1994.65.5s.521. [DOI] [PubMed] [Google Scholar]
- Haziot A. Hijiya N. Gangloff S.C. Silver J. Goyert S.M. Induction of a novel mechanism of accelerated bacterial clearance by lipopolysaccharide in CD14-deficient and Toll-like receptor 4-deficient mice. J Immunol. 2001;166:1075–1078. doi: 10.4049/jimmunol.166.2.1075. [DOI] [PubMed] [Google Scholar]
- Hemmerle J. Frank R.M. Bacterial invasion of periodontal tissues after experimental immunosuppression in rats. J Biol Buccale. 1991;19:271–282. [PubMed] [Google Scholar]
- Hooper L.V. Wong M.H. Thelin A. Hansson L. Falk P.G. Gordon J.I. Molecular analysis of commensal host-microbial relationships in the intestine. Science. 2001;291:881–884. doi: 10.1126/science.291.5505.881. [DOI] [PubMed] [Google Scholar]
- Janeway C.J. The immune system evolved to discriminate infectious nonself from noninfectious self. Immunol Today. 1992;13:11–16. doi: 10.1016/0167-5699(92)90198-G. [DOI] [PubMed] [Google Scholar]
- Jin L. Darveau R.P. Soluble CD14 levels in gingival crevicular fluid of subjects with untreated adult periodontitis. J Periodontol. 2001;72:634–640. doi: 10.1902/jop.2001.72.5.634. [DOI] [PubMed] [Google Scholar]
- Jin L. Ren L. Leung W.K. Darveau R.P. The in vivo expression of membrane-bound CD14 in periodontal health and disease. J Periodontol. 2004;75:578–585. doi: 10.1902/jop.2004.75.4.578. [DOI] [PubMed] [Google Scholar]
- Khan S.A. Everest P. Servos S. Foxwell N. Zahringer U. Brade H. A lethal role for lipid A in Salmonella infections. Mol Microbiol. 1998;29:571–579. doi: 10.1046/j.1365-2958.1998.00952.x. [DOI] [PubMed] [Google Scholar]
- Komatsu S. Berg R.D. Russell J.M. Nimura Y. Granger D.N. Enteric microflora contribute to constitutive ICAM-1 expression on vascular endothelial cells. Am J Physiol Gastrointest Liver Physiol. 2000;279:G186–G191. doi: 10.1152/ajpgi.2000.279.1.G186. [DOI] [PubMed] [Google Scholar]
- Kornman K.S. Crane A. Wang H.Y. di Giovine F.S. Newman M.G. Pirk F.W. The interleukin-1 genotype as a severity factor in adult periodontal disease. J Clin Periodontol. 1997a;24:72–77. doi: 10.1111/j.1600-051x.1997.tb01187.x. [DOI] [PubMed] [Google Scholar]
- Kornman K.S. Page R.C. Tonetti M.S. The host response to the microbial challenge in periodontitis: assembling the players. Periodontol 2000. 1997b;14:112–143. doi: 10.1111/j.1600-0757.1997.tb00191.x. [DOI] [PubMed] [Google Scholar]
- Kroes I. Lepp P.W. Relman D.A. Bacterial diversity within the human subgingival crevice. Proc Natl Acad Sci USA. 1999;96:14547–14552. doi: 10.1073/pnas.96.25.14547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. Caufield P.W. Arbitrarily primed polymerase chain reaction fingerprinting for the genotypic identification of mutans streptococci from humans. Oral Microbiol Immunol. 1998;13:17–22. doi: 10.1111/j.1399-302x.1998.tb00745.x. [DOI] [PubMed] [Google Scholar]
- Lu Q. Jin L. Darveau R.P. Samaranayake L.P. Expression of human beta-defensins-1 and -2 peptides in unresolved chronic periodontitis. J Periodontal Res. 2004;39:221–227. doi: 10.1111/j.1600-0765.2004.00727.x. [DOI] [PubMed] [Google Scholar]
- Lu Q. Samaranayake L.P. Darveau R.P. Jin L. Expression of human beta-defensin-3 in gingival epithelia. J Periodontal Res. 2005;40:474–481. doi: 10.1111/j.1600-0765.2005.00827.x. [DOI] [PubMed] [Google Scholar]
- MacFarlane G.D. Herzberg M.C. Wolff L.F. Hardie N.A. Refractory periodontitis associated with abnormal polymorphonuclear leukocyte phagocytosis and cigarette smoking. J Periodontol. 1992;63:908–913. doi: 10.1902/jop.1992.63.11.908. [DOI] [PubMed] [Google Scholar]
- Macpherson A.J. Harris N.L. Interactions between commensal intestinal bacteria and the immune system. Nat Rev Immunol. 2004;4:478–485. doi: 10.1038/nri1373. [DOI] [PubMed] [Google Scholar]
- Mahanonda R. Sa-Ard-Iam N. Montreekachon P. Pimkhaokham A. Yongvanichit K. Fukuda M.M. IL-8 and IDO expression by human gingival fibroblasts via TLRs. J Immunol. 2007;178:1151–1157. doi: 10.4049/jimmunol.178.2.1151. [DOI] [PubMed] [Google Scholar]
- Masada M.P. Persson R. Kenney J.S. Lee S.W. Page R.C. Allison A.C. Measurement of interleukin-1 alpha and -1 beta in gingival crevicular fluid: implications for the pathogenesis of periodontal disease. J Periodontal Res. 1990;25:156–163. doi: 10.1111/j.1600-0765.1990.tb01038.x. [DOI] [PubMed] [Google Scholar]
- McCoy S.A. Creamer H.R. Kawanami M. Adams D.F. The concentration of lipopolysaccharide on individual root surfaces at varying times following in vivo root planing. J Periodontol. 1987;58:393–399. doi: 10.1902/jop.1987.58.6.393. [DOI] [PubMed] [Google Scholar]
- McNab R. Ford S.K. El-Sabaeny A. Barbieri B. Cook G.S. Lamont R.J. LuxS-based signaling in Streptococcus gordonii: autoinducer 2 controls carbohydrate metabolism and biofilm formation with Porphyromonas gingivalis. J Bacteriol. 2003;185:274–284. doi: 10.1128/JB.185.1.274-284.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzhitov R. Janeway C., Jr. Innate immune recognition: mechanisms and pathways. Immunol Rev. 2000;173:89–97. doi: 10.1034/j.1600-065x.2000.917309.x. [DOI] [PubMed] [Google Scholar]
- Moore J. Wilson M. Kieser J.B. The distribution of bacterial lipopolysaccharide (endotoxin) in relation to periodontally involved root surfaces. J Clin Periodontol. 1986;13:748–751. doi: 10.1111/j.1600-051x.1986.tb00877.x. [DOI] [PubMed] [Google Scholar]
- Moughal N.A. Adonogianaki E. Thornhill M.H. Kinane D.F. Endothelial cell leukocyte adhesion molecule-1 (ELAM-1) and intercellular adhesion molecule-1 (ICAM-1) expression in gingival tissue during health and experimentally-induced gingivitis. J Periodontal Res. 1992;27:623–630. doi: 10.1111/j.1600-0765.1992.tb01746.x. [DOI] [PubMed] [Google Scholar]
- Munford R.S. Varley A.W. Shield as signal: lipopolysaccharides and the evolution of immunity to gram-negative bacteria. PLoS Pathog. 2006;2:e67. doi: 10.1371/journal.ppat.0020067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederman R. Westernoff T. Lee C. Mark L.L. Kawashima N. Ullman-Culler M. Infection-mediated early-onset periodontal disease in P/E-selectin-deficient mice. J Clin Periodontol. 2001;28:569–575. doi: 10.1034/j.1600-051x.2001.028006569.x. [DOI] [PubMed] [Google Scholar]
- Nylander K. Danielsen B. Fejerskov O. Dabelsteen E. Expression of the endothelial leukocyte adhesion molecule-1 (ELAM-1) on endothelial cells in experimental gingivitis in humans. J Periodontol. 1993;64:355–357. doi: 10.1902/jop.1993.64.5.355. [DOI] [PubMed] [Google Scholar]
- Offenbacher S. Braswell L.D. Loos A.S. Johnson H.G. Hall C.M. McClure H. Effects of flurbiprofen on the progression of periodontitis in Macaca mulatta. J Periodontal Res. 1987;22:473–481. doi: 10.1111/j.1600-0765.1987.tb02058.x. [DOI] [PubMed] [Google Scholar]
- Offenbacher S. Katz V. Fertik G. Collins J. Boyd D. Maynor G. Periodontal infection as a possible risk factor for preterm low birth weight. J Periodontol. 1996;67:1103–1113. doi: 10.1902/jop.1996.67.10s.1103. [DOI] [PubMed] [Google Scholar]
- Offenbacher S. Odle B.M. Van Dyke T.E. The use of crevicular fluid prostaglandin E2 levels as a predictor of periodontal attachment loss. J Periodontal Res. 1986;21:101–112. doi: 10.1111/j.1600-0765.1986.tb01443.x. [DOI] [PubMed] [Google Scholar]
- Olczak T. Simpson W. Liu X. Genco C.A. Iron and heme utilization in Porphyromonas gingivalis. FEMS Microbiol Rev. 2005;29:119–144. doi: 10.1016/j.femsre.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Page R.C. Beatty P. Waldrop T.C. Molecular basis for the functional abnormality in neutrophils from patients with generalized prepubertal periodontitis. J Periodontal Res. 1987;22:182–183. doi: 10.1111/j.1600-0765.1987.tb01562.x. [DOI] [PubMed] [Google Scholar]
- Page R.C. Offenbacher S. Schroeder H.E. Seymour G.J. Kornman K.S. Advances in the pathogenesis of periodontitis: summary of developments, clinical implications and future directions. Periodontol 2000. 1997;14:216–248. doi: 10.1111/j.1600-0757.1997.tb00199.x. [DOI] [PubMed] [Google Scholar]
- Page R.C. Schroeder H.E. Pathogenesis of inflammatory periodontal disease. A summary of current work [Review] Lab Investig. 1976;33:235–249. [PubMed] [Google Scholar]
- Reife R.A. Coats S.R. Al-Qutub M. Dixon D.M. Braham P.A. Billharz R.J. Porphyromonas gingivalis lipopolysaccharide lipid A heterogeneity: differential activities of tetra- and penta-acylated lipid A structures on E-selectin expression and TLR4 recognition. Cell Microbiol. 2006;8:857–868. doi: 10.1111/j.1462-5822.2005.00672.x. [DOI] [PubMed] [Google Scholar]
- Ren L. Jin L. Leung W.K. Local expression of lipopolysaccharide-binding protein in human gingival tissues. J Periodontal Res. 2004;39:242–248. doi: 10.1111/j.1600-0765.2004.00732.x. [DOI] [PubMed] [Google Scholar]
- Ren L. Leung W.K. Darveau R.P. Jin L. The expression profile of lipopolysaccharide-binding protein, membrane-bound CD14, and toll-like receptors 2 and 4 in chronic periodontitis. J Periodontol. 2005;76:1950–1959. doi: 10.1902/jop.2005.76.11.1950. [DOI] [PubMed] [Google Scholar]
- Sallay K. Listgarten M. Sanavi F. Ring I. Nowotny A. Bacterial invasion of oral tissues of immunosuppressed rats. Infect Immun. 1984;43:1091–1093. doi: 10.1128/iai.43.3.1091-1093.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvi G.E. Lawrence H.P. Offenbacher S. Beck J.D. Influence of risk factors on the pathogenesis of periodontitis. Periodontol 2000. 1997;14:173–201. doi: 10.1111/j.1600-0757.1997.tb00197.x. [DOI] [PubMed] [Google Scholar]
- Schwartz J. Stinson F.L. Parker R.B. The passage of tritiated bacterial endotoxin across intact gingival crevicular epithelium. J Periodontol. 1972;43:270–276. doi: 10.1902/jop.1972.43.5.270. [DOI] [PubMed] [Google Scholar]
- Socransky S.S. Haffajee A.D. Evidence of bacterial etiology: a historical perspective. Periodontol 2000. 1994;5:7–25. doi: 10.1111/j.1600-0757.1994.tb00016.x. [DOI] [PubMed] [Google Scholar]
- Socransky S.S. Haffajee A.D. Cugini M.A. Smith C. Kent R.L., Jr. Microbial complexes in subgingival plaque. J Clin Periodontol. 1998;25:134–144. doi: 10.1111/j.1600-051x.1998.tb02419.x. [DOI] [PubMed] [Google Scholar]
- Somerville J.E., Jr. Cassiano L. Bainbridge B. Cunningham M.D. Darveau R.P. A novel Escherichia coli lipid A mutant that produces an antiinflammatory lipopolysaccharide. J Clin Investig. 1996;97:359–365. doi: 10.1172/JCI118423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somerville J.E., Jr. Cassiano L. Darveau R.P. Escherichia coli msbB gene as a virulence factor and a therapeutic target. Infect Immun. 1999;67:6583–6590. doi: 10.1128/iai.67.12.6583-6590.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer T.A. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- Stappenbeck T.S. Hooper L.V. Gordon J.I. Developmental regulation of intestinal angiogenesis by indigenous microbes via Paneth cells. Proc Natl Acad Sci USA. 2002;99:15451–15455. doi: 10.1073/pnas.202604299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steeghs L. den Hartog R. Dden Boer A. Zomer B. Roholl P. van der Ley P. Meningitis bacterium is viable without endotoxin. Nature. 1998;392:449–450. doi: 10.1038/33046. [DOI] [PubMed] [Google Scholar]
- Sugawara Y. Uehara A. Fujimoto Y. Kusumoto S. Fukase K. Shibata K. Toll-like receptors, NOD1, and NOD2 in oral epithelial cells. J Dent Res. 2006;85:524–529. doi: 10.1177/154405910608500609. [DOI] [PubMed] [Google Scholar]
- Takada H. Kotani S. Bacterial Endotoxic Lipopolysaccharides. CRC Press; Boca Raton: 1992. [Google Scholar]
- Tanner A. Kent R. Maiden M.F. Taubman M.A. Clinical, microbiological and immunological profile of healthy, gingivitis and putative active periodontal subjects. J Periodontal Res. 1996;31:195–204. doi: 10.1111/j.1600-0765.1996.tb00484.x. [DOI] [PubMed] [Google Scholar]
- Tonetti M.S. Molecular factors associated with compartmentalization of gingival immune responses and transepithelial neutrophil migration. J Periodontal Res. 1997;32:104–109. doi: 10.1111/j.1600-0765.1997.tb01389.x. [DOI] [PubMed] [Google Scholar]
- Tonetti M.S. Imboden M.A. Gerber L. Lang N.P. Laisue J. Mueller C. Localized expression of mRNA for phagocyte-specific hemotactic cytokines in human periodontal infections. Infect Immun. 1994;62:4005–4014. doi: 10.1128/iai.62.9.4005-4014.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonetti M.S. Imboden M.A. Lang N.P. Neutrophil migration into the gingival sulcus is associated with transepithelial gradients of interleukin-8 and ICAM-1. J Periodontol. 1998;69:1139–1147. doi: 10.1902/jop.1998.69.10.1139. [DOI] [PubMed] [Google Scholar]
- Uehara A. Takada H. Synergism between TLRs and NOD1/2 in oral epithelial cells. J Dent Res. 2008;87:682–686. doi: 10.1177/154405910808700709. [DOI] [PubMed] [Google Scholar]
- Umesaki Y. Setoyama H. Structure of the intestinal flora responsible for development of the gut immune system in a rodent model. Microbes Infect. 2000;2:1343–1351. doi: 10.1016/s1286-4579(00)01288-0. [DOI] [PubMed] [Google Scholar]
- Waldrop T.C. Anderson D.C. Hallmon W.W. Schmalstieg F.C. Jacobs R.L. Periodontal manifestations of the heritable Mac-1, LFA-1, deficiency syndrome. Clinical, histopathologic and molecular characteristics. J Periodontol. 1987;58:400–416. doi: 10.1902/jop.1987.58.6.400. [DOI] [PubMed] [Google Scholar]
- Wilson M. Moore J. Kieser J.B. Identity of limulus amoebocyte lysate-active root surface materials from periodontally involved teeth. J Clin Periodontol. 1986;13:743–747. doi: 10.1111/j.1600-051x.1986.tb00876.x. [DOI] [PubMed] [Google Scholar]
- Ximenez-Fyvie L.A. Haffajee A.D. Socransky S.S. Comparison of the microbiota of supra- and subgingival plaque in health and periodontitis. J Clin Periodontol. 2000;27:648–657. doi: 10.1034/j.1600-051x.2000.027009648.x. [DOI] [PubMed] [Google Scholar]
- Xu J. Gordon J.I. Inaugural article: honor thy symbionts. Proc Natl Acad Sci USA. 2003;100:10452–10459. doi: 10.1073/pnas.1734063100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura A. Kaneko T. Kato Y. Golenbock D.T. Hara Y. Lipopolysaccharides from periodontopathic bacteria Porphyromonas gingivalis and Capnocytophaga ochracea are antagonists for human toll- like receptor 4. Infect Immun. 2002;70:218–225. doi: 10.1128/IAI.70.1.218-225.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshinari N. Kameyama Y. Aoyama Y. Nishiyama H. Noguchi T. Effect of long-term methotrexate-induced neutropenia on experimental periodontal lesion in rats. J Periodontal Res. 1994;29:393–400. doi: 10.1111/j.1600-0765.1994.tb01240.x. [DOI] [PubMed] [Google Scholar]
- Zambon J.J. Periodontal diseases: microbial factors. Ann Periodontol. 1996;1:879–925. doi: 10.1902/annals.1996.1.1.879. [DOI] [PubMed] [Google Scholar]