

Catalytic asymmetric oxidations, most notably dihydroxylations,1 epoxidations,2-4 and aminohydroxylations,5 have proven to be versatile transforms for the installation of chiral functionality onto non-chiral alkene substrates. In all three cases, the methodology has progressed and matured to the extent that the transformations are routinely applied in organic synthesis.
Notably absent from this arsenal of transformations are examples of synthetically useful, conceptually related asymmetric electrophilic olefin halogenation reactions. With the intention of addressing this long-standing problem, we instituted a program geared towards the development of a reagent-controlled asymmetric halogenation of olefins.
Recently, a polyene cascade induced by a stoichiometric chiral iodonium source was disclosed in an elegant work by Ishihara et al.6a An efficient Co-salen catalyzed iodoetherification has also been reported by Kang et al.6b Nonetheless, an efficient catalytic asymmetric halolactonization reaction has been elusive. In contrast to the number of examples of substrate controlled stereoselective halolactonizations,7 reagent controlled processes are rare, and have only begun to emerge recently. The development of such a methodology would provide access to richly functionalized chiral halolactones in one step from achiral alkenoic acids.
The first reagent controlled enantioselective halolactonization was reported in 1992 by the Taguchi group, where an alkenoic acid was cyclized by action of iodine and a stoichiometric equivalent of a chiral titanium complex, returning an iodolactone in 65% ee.8 Subsequently, a number of examples have appeared that employ stoichiometric or super-stoichiometric amounts of chiral amine promoters. Typically, these methodologies employ a dimeric iodonium salt as the chiral halogen source (i.e. [(L*)2I+]Y-, where L* is a chiral amine).8-12 Two of the most selective examples of this strategy were presented by Wirth11,12 and Rousseau.9 Aside from the disadvantage of committing up to ~5 equiv of chiral promoter, these approaches were marred by low enantioselectivities (15 to 45% ee). Interestingly, all of these disclosures produce iodolactones. Reports on chloro and bromolactonizations are absent, except for a single example where a bromolactone was produced in 5% ee with a chiral bromonium/pyridine dimer.13
Recently, Gao and coworkers reported the only catalytic protocol for the iodolactonization of alkenoic acids, whereby trans-5-aryl-4-pentenoic acids were cyclized in the presence of iodine and 30 mol% of a cinchonidine-derived quaternary ammonium salt under PTC conditions.14 Iodolactones were returned in a nearly 1:1 ratio of δ and γ isomers with marginal enantioselectivities (δ = 16% ee, γ = 31% ee).
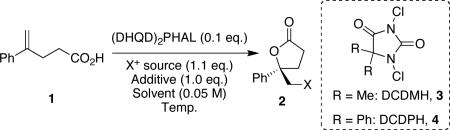
We now report the discovery of an organocatalytic enantioselective protocol for the chlorolactonization of 4-substituted 4-pentenoic acids. This methodology represents the first example of a catalytic process that returns chiral halolactones in synthetically useful enantioselectivities.
We were encouraged by the discovery that (DHQD)2PHAL15-17 provided good conversions and modest enantioselectivity in the bromolactonization of alkenoic acid 1 (Table 1, entry 1). When 1 was treated with 10 mol % of (DHQD)2PHAL in the presence of NBS in CH2Cl2, the desired bromolactone was isolated in quantitative yield with a modest 22% ee. Although lowering the reaction temperature to −20 °C produced an inferior result (entry 2), an exhaustive solvent screen subsequently revealed that CHCl3 was optimal, returning the bromolactone with an improved selectivity of 35% ee (entry 3).
Table 1.
The (DHQD)2PHAL mediated halolactonization.
 | |||||
|---|---|---|---|---|---|
| Entry | X+ Sourcea | Solvent | Additive | °C | % eeb |
| 1 | NBS | CH2Cl2 | - | RT | 22 |
| 2 | NBS | CH2Cl2 | - | −20 | 14 |
| 3 | NBS | CHCl3 | - | RT | 35 |
| 4 | NCS | CH2Cl2 | - | RT | 39 |
| 5 | NCS | CHCl3 | - | RT | 65 |
| 6 | NCS | CHCl3 | - | −40 | NRc |
| 7 | DCDMH | CHCl3 | - | RT | 71 |
| 8 | DCDMH | CHCl3 | - | −40 | 83 |
| 9 | DCDMH | CHCl3 | PhCO2H | −40 | 86 |
| 10 | DCDMH | CHCl3/Hex (1:1) | - | −40 | 86 |
| 11 | DCDMH | CHCl3/Hex (1:1) | PhCO2H | −40 | 89 |
| 12 | DCDPH | CHCl3/Hex (1:1) | PhCO2H | −40 | 89 |
X = Br for entries 1-3, and Cl for entries 4-12.
as judged by chiral HPLC or GC analysis.
NR = no reaction after 3 hours.
A substantial improvement in selectivity was realized when the analogous chlorolactonization was investigated, employing NCS in lieu of NBS. In this case, reaction with 10 mol % of catalyst in CHCl3 returned the desired chlorolactone with a dramatically enhanced selectivity of 65% ee (entry 5). Analogous to the bromolactonization, chlorolactonization in DCM returned the chlorolactone with an eroded 39% ee (entry 4).
Although we were initially discouraged by the sluggish chlorolactonization when the temperature of the reaction was lowered to −40 °C (no lactone was detected by TLC, entry 6), we were emboldened by the discovery that the more reactive 1,3-dichloro-5,5-dimethylhydantoin (3, DCDMH) afforded a faster and more stereoselective reaction at −40 °C (entry 8, quant. conversion, 83% ee; for absolute stereochemical assignment of product see Supporting Information). The reaction at room temperature was less stereoselective (entry 7). Extensive parallel additive and co-solvent screens revealed that both 1.0 equiv of benzoic acid additive (entry 9) and a CHCl3/hexane (1:1) solvent system (entry 10) each independently increased the selectivity of the chlorolactonization to 86% ee. When the co-solvent and additive effects were combined, the desired lactone was produced in 89% ee (entry 11). This selectivity was maintained when the analogous DCDPH (4)18 was applied as the terminal chlorine source (entry 12). We deferred to DCDPH, since it returned higher isolated yields during the course of scale-up experiments. Importantly, it appears that the N,N-dichlorohydantoins are uniquely situated between NBS and NCS in reactivity. They are reactive enough relative to NCS to allow for reaction at low temperature yet are attenuated relative to NBS, thus quelling a prevailing non-selective background reaction.
Next, a panel of p-substituted pentenoic acids was subjected to the optimal reaction conditions (Table 2). Phenyl substituted lactone 6a was generated in 86% yield and 89% ee. The quasienantiomeric (DHQ)2PHAL returned the enantiomeric lactone 6b in a reduced 75% yield and 77% ee. The reduced selectivity in the latter case is likely due to the diastereomeric relationship between the DHQD and DHQ catalysts. Lactone 6c was returned in nearly quantitative yield, but with essentially no selectivity. Evidently, the p-methoxy substituent promotes the facile ring opening of the putative chloronium intermediate, precluding a high degree of selectivity. Lactone 6d, harboring a less donating p-methyl group, was returned with a much improved 80% ee. Chloro and fluoro substituted lactones 6e and 6f were generated in good yield with 88% and 89% ee, respectively. Paratrifluoromethyl substituted 6g was isolated with 90% ee. Biphenyl substituted substrate 5h returned chlorolactone 6h in 83% ee. Larger aryl substituents resulted in reduced selectivities. Namely, 2-naphthyl lactone 6i was returned in 72% ee. Finally, replacing the 4-aryl substituent in 5 with a cyclohexyl group returned lactone 6j in 55% yield with a substantially lower 43% ee.
Table 2.
(DHQD)2PHAL mediated asymmetric chlorolactonization.
 | |||||
|---|---|---|---|---|---|
| Rb | %yieldc | %eed | Rb | %yieldc | %eed |
| Ph; 6a | 86 | 89 | p-F-C6H4; 6f | 81 (78) | 89 (86) |
| Ph; 6b (ent-6a) | 75 | 77e | p-CF3-C6H4; 6g | 61 | 90 |
| p-OMe-C6H4; 6c | 99 | <5 | p-Ph-C6H4; 6h | 59 | 83 (80) |
| p-Me-C6H4; 6d | 86 (82)f | 80 (82)f | 2-Napth; 6i | 92 (83) | 72 (72) |
| p-Cl-C6H4; 6e | 80 | 88 | Cy; 6j | 55 | 43 |
Reaction times; 30 min for products 6a-6f and 6j, 90 min for products 6g-6i (as judged by TLC).
Stereochemistry was determined by chemical correlation (see Supplementary Information).
Isolated yield after column chromatography.
As judged by chiral GC or HPLC analysis.
Reaction was performed with 0.1 equiv of (DHQ)2PHAL.
Values in parentheses are yields and % ee when 0.01 eq. (DHQD)2PHAL was employed.
We have also evaluated the transformation when 0.01 equiv of (DHQD)2PHAL was employed for a selection of the substrates in Table 2. Even with a low catalyst loading of just 1 mol %, lactones 6d, 6f, 6h, and 6i were returned in 82%, 86%, 80%, and 72% ee, respectively (Table 2, values in parentheses). In each case the enantioselectivities approximately match those realized with 10 mol % catalyst.
During the study, a few nuances regarding the mechanism of the transformation have come to light. Early on, we noted that the selectivity of the process was influenced by the gross structural features of the achiral terminal halogen source. Two interesting trends emerged. First, an increase in selectivity was realized as the steric demand of the C-5 substituents on the terminal chlorine source increased. While lactone 6f was generated in 81% ee by action of unsubstituted 7, the selectivity steadily increased on employing the dimethyl (3, 84% ee), methylphenyl (8, 85% ee), and diphenyl (4, 89% ee) chlorine sources (Scheme 1).
Scheme 1.

Cyclization of 5f with various N-chlorohydantoins.
These results seem to belie any cursory mechanistic musings that conceptualized the process hinging on the transfer of an active chlorenium equivalent from the hydantoin to the alkaloid prior to delivery to the substrate. One would expect that such a process ought to return 6f with roughly the same degree of stereoinduction irrespective of the gross structure of the terminal chlorenium source. Conversely, the results in Scheme 1 may indicate a more intimate relationship between the terminal halogen source and the catalyst.
Indeed, we were able to observe an associative complex between unsubstituted hydantoin 7 and the catalyst in a stoichiometric 1H NMR experiment (Scheme 2). In the event, the equivalent C-5 protons of 7 (s, 4.35 ppm) were split into a clean AB quartet (4.30 ppm, JAB = 16.5 Hz) on incubation with an equivalent of (DHQD)2PHAL and benzoic acid (2 equiv) in CDCl3 at −40 °C. This result clearly indicates an association between the N,N-dichlorohydantoin chlorine source and the chiral catalyst. On slowly warming the sample to RT, the association collapses and the AB quartet converges to the expected singlet at 4.35 ppm.
Scheme 2.

Observation of hydantoin/catalyst complex by NMR.
Although premature, we are tempted to suggest complex 11a, invoking a hydrogen bond mediated association between the protonated catalyst and the chlorenium source. Alternatively one might imagine a tight ion-pair between the chlorinated catalyst and the monochloro anion of the hydantoin chlorine source (see structure 11b). Although the putative reactive complex awaits further investigation, we believe that the associative complex between catalyst and N,N-dichlorohydantoin disclosed herein is a crucial element in the asymmetric delivery of the chlorine atom.
This associative complex serves not only to impart the enantioselectivity observed in the transformation, but also to significantly accelerate the transformation. Indeed, the uncatalyzed background chlorolactonization is exceedingly slow under identical reaction conditions (effectively no conversion after 24 h). This stands in stark contrast to the catalyzed reactions, which are complete in a 30-90 min timeframe. In fact, the rate of the reaction seems to be chiefly governed by the solubility of the substrate. Remarkably, we have observed that p-fluoro substituted substrate 5f is converted to lactone 6f (89% ee) in less than two minutes!
Secondly, comparison of the results with DCDMH (3) and its N-1 and N-3 monomethylated derivatives (9 and 10 respectively, Scheme 1) and NMR studies indicate an intriguing synergistic role of the two chlorine atoms in the dichlorohydantoin chlorine sources that prove most selective in the transformation. Monitoring the reaction with NMR, revealed that the chlorine atom from the more activated N-3 position, flanked by two carbonyls, was transferred leading to the chlorolactone, apparent from the immediate production of the N1-Cl-hydantoin side product (see supporting information). Comparison of the data for DCDMH 3 and its mono-methylated derivatives 9 and 10 highlight the importance of the less active N-1 chlorine atom. It seems that the N-1 chlorine atom in the dichlorohydantoins serves to inductively activate the N-3 chlorine towards electrophilic delivery. Replacing the N-1 chlorine with a methyl group (9) resulted in an inferior transformation returning 6f in a lower yield (50%) and enantioselectivity of 78% ee (Scheme 1). Further evidence suggesting that the N-1 chlorine is not readily transferred to the product lactone is the exceedingly poor yield of 6f realized when monochlorohydantoin 10 is employed (7% yield, 72% ee). Taken together, these data suggest that the N-3 chlorine is delivered to the substrate exclusively, while the static N-1 chlorine serves to enhance the reactivity of the chlorine source through the inductive withdrawal of electron density.
Currently, the role played by the benzoic acid additive is less clear. Considering the hydrogen bond-mediated complex 11a (Scheme 2), the additive might serve to ensure adequate protonation of the alkaloid, thus facilitating the formation of the hydantoin/catalyst complex. This role ought to be particularly important late in the reaction as the reduced hydantoin product consumes the acidic proton from the starting alkenoic acid. Alternatively, the benzoic acid may serve to protonate the hydantoin carbonyl, thus facilitating chlorenium transfer to the substrate (in 11a) or to the catalyst (in 11b).
We have evaluated the enantioselectivity of the transformation as a function of time. In the chlorolactonization of 5f (leading to p-fluoro substituted 6f) we noted that the enantioselectivity of the transformation does not change as a function of time or conversion, monitoring the reaction as early as 30 s after initiation. Additionally, a catalyst aging period of 0 min to 1 h prior to the addition of the substrate does not cause a change in the observed enantioselectivity. Finally, doping the reaction mixture with enantiopure 6f failed to increase the observed enantioselectivity, thus ruling out autocatalysis phenomena. Taken together, these data suggest that non-linear catalysis behavior is not operative in this system.
We have also developed a convenient one-pot protocol for the transformation of these chiral chlorolactones into enantioenriched 1,1-disubstituted epoxy alcohols (Scheme 3). Lithium borohydride reduction of lactone 6f (86% ee) followed by sodium hydroxide mediated cyclization of the resulting chlorohydrin intermediate returned 1,1-disubstituted epoxy alcohol 12 in good yield and importantly without any appreciable loss of enantiopurity. Significantly, 4-arylpenten-1-ols have proven to be difficult substrates for conventional asymmetric epoxidation protocols.19
Scheme 3.

One-pot conversion of 6f to chiral epoxyalcohol 12.
In summary, we have discovered a novel organocatalytic asymmetric chlorolactonization that returns chiral chlorolactones by action of (DHQD)2PHAL and DCDPH. This methodology represents the first example of a catalytic, enantioselective halolactonization that proceeds with synthetically useful enantioselectivities. Current efforts are aimed at understanding the mechanism of the transformation and the details of enantioselection in order to expand the substrate scope and to improve the enantioselectivity. We believe that a thorough understanding of the associative catalyst/chlorohydantoin complexation will likely allow for the development of related transformations.
Supplementary Material
ACKNOWLEDGMENT
We acknowledge the ACS PRF (No. 47272-AC) and the NIH (No. R01-GM082961) for generous funding. The authors thank Ms. Aman Kulshrestha for collecting the HRMS data for this manuscript. The authors wish to dedicate this manuscript to the memory of Professor Peter Wagner.
Footnotes
Supporting Information Available:. General experimental procedure for the chlorolactonization of alkenoic acids, and full spectroscopic data for each product. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Kolb HC, Vannieuwenhze MS, Sharpless KB. Chem. Rev. 1994;94:2483. [Google Scholar]
- 2.Katsuki T. Adv. Synth. Catal. 2002;344:131. [Google Scholar]
- 3.Katsuki T, Martin VS. Org. React. 1996;48:1. [Google Scholar]
- 4.Wong OA, Shi Y. Chem. Rev. 2008;108:3958. doi: 10.1021/cr068367v. [DOI] [PubMed] [Google Scholar]
- 5.O'Brien P. Angew. Chem., Int. Ed. 1999;38:326. [Google Scholar]
- 6.a Sakakura A, Ukai' A, Ishihara' K. Nature. 2007;445:900. doi: 10.1038/nature05553. [DOI] [PubMed] [Google Scholar]; b Kang SH, Lee SB, Park CM. J. Am. Chem. Soc. 2003;125:15748. doi: 10.1021/ja0369921. [DOI] [PubMed] [Google Scholar]
- 7.a Dowle MD, Davies DI. Chem. Soc. Rev. 1979;8:171. [Google Scholar]; b Cardillo G, Orena M. Tetrahedron. 1990;46:3321. [Google Scholar]
- 8.Kitagawa O, Hanano T, Tanabe K, Shiro M, Taguchi T. J. Chem. Soc.-Chem. Commun. 1992:1005. [Google Scholar]
- 9.Garnier JM, Robin S, Rousseau G. Eur. J. Org. Chem. 2007:3281. [Google Scholar]
- 10.Grossman RB, Trupp RJ. Can. J. Chem. 1998;76:1233. [Google Scholar]
- 11.Haas J, Bissmire S, Wirth T. Chem.-Eur. J. 2005;11:5777. doi: 10.1002/chem.200500507. [DOI] [PubMed] [Google Scholar]
- 12.Haas J, Piguel S, Wirth T. Org. Lett. 2002;4:297. doi: 10.1021/ol0171113. [DOI] [PubMed] [Google Scholar]
- 13.Cui XL, Brown RS. J. Org. Chem. 2000;65:5653. doi: 10.1021/jo000449a. [DOI] [PubMed] [Google Scholar]
- 14.Wang M, Gao LX, Mai WP, Xia AX, Wang F, Zhang SB. J. Org. Chem. 2004;69:2874. doi: 10.1021/jo035719e. [DOI] [PubMed] [Google Scholar]
- 15.Becker H, Sharpless KB. Angew. Chem., Int. Ed. 1996;35:448. [Google Scholar]
- 16.a Li G, Chang HT, Sharpless KB. Angew. Chem., Int. Ed. 1996;35:451. [Google Scholar]; b Li G, Hubert HH, Sharpless KB. Angew. Chem., Int. Ed. 1996;35:2813. [Google Scholar]
- 17.Jacobsen EN, Marko I, Mungall WS, Schroder G, Sharpless KB. J. Am. Chem. Soc. 1988;110:1968. [Google Scholar]
- 18.Whitehead DC, Staples RJ, Borhan B. Tetrahedron Lett. 2009;50:656. doi: 10.1016/j.tetlet.2008.11.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang B, Wong OA, Zhao MX, Shi Y. J. Org. Chem. 2008;73:9539. doi: 10.1021/jo801576k. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.