

Abstract
Exogenous zinc can protect cardiac cells from reperfusion injury, but the exact roles of endogenous zinc in the pathogenesis of reperfusion injury and in adenosine A2 receptor activation-induced cardioprotection against reperfusion injury remain unknown. Adenosine A1/A2 receptor agonist 5′-(N-ethylcarboxamido) adenosine (NECA) given at reperfusion reduced infarct size in isolated rat hearts subjected to 30 min ischemia followed by 2 h of reperfusion. This effect of NECA was partially but significantly blocked by the zinc chelator N,N,N′,N′-tetrakis-(2-pyridylmethyl) ethylenediamine (TPEN), and ZnCl2 given at reperfusion mimicked the effect of NECA by reducing infarct size. Total tissue zinc concentrations measured with inductively coupled plasma optical emission spectroscopy (ICPOES) were decreased upon reperfusion in rat hearts and this was reversed by NECA. NECA increased intracellular free zinc during reperfusion in the heart. Confocal imaging study showed a rapid increase in intracellular free zinc in isolated rat cardiomyocytes treated with NECA. Further experiments revealed that NECA increased total zinc levels upon reperfusion in mitochondria isolated from isolated hearts. NECA attenuated mitochondrial swelling upon reperfusion in isolated hearts and this was inhibited by TPEN. Similarly, NECA prevented the loss of mitochondrial membrane potential (ΔΨm) caused by oxidant stress in cardiomyocytes. Finally, both NECA and ZnCl2 inhibited the mitochondrial metabolic activity. NECA-induced cardioprotection against reperfusion injury is mediated by intracellular zinc. NECA prevents reperfusion-induced zinc loss and relocates zinc to mitochondria. The inhibitory effects of zinc on both the mPTP opening and the mitochondrial metabolic activity may account for the cardioprotective effect of NECA.
Keywords: A2 receptor, intracellular zinc, NECA, reperfusion injury, mPTP, mitochondrial metabolism
1. Introduction
Cardioprotection against ischemia/reperfusion injury has been successfully obtained experimentally when protective interventions are applied before onset of experimental ischemia. However, since pretreatment is impossible in the clinical setting of acute myocardial infarction (AMI), the use of the pre-ischemic interventions are clinically impractical. Thus, to successfully save myocardium in patients with AMI, protective interventions must be effective when applied after ischemia or at the onset of reperfusion. Recent experimental studies have shown that activation of adenosine A2 receptors at reperfusion with adenosine analogues can protect the heart from ischemia/reperfusion injury [1-4]. Although it remains unknown if A2 receptor agonists are effective in the clinical settings of AMI, a thorough understanding of the exact mechanism by which A2 receptor stimulation leads to cardioprotection at reperfusion will help us develop successful strategies for prevention of ischemia/reperfusion injury caused by AMI.
In addition to its essential role in maintaining the structure and function of many proteins, enzymes, and transcriptional factors [5], zinc has also been demonstrated to play an important role in cellular signaling by regulating activities of several essential signaling kinases such as phosphatidylinositol 3-kinase (PI3K) [6], Akt/PKB [7], extracellular signal-regulated kinase (ERK) [7], and glycogen synthase kinase 3β (GSK-3β) [8]. Since all these kinases are involved in the mechanism underlying cardioprotection against myocardial reperfusion injury, zinc may be cardioprotective at reperfusion. Indeed, our recent studies have demonstrated that exogenous zinc protected cardiac H9c2 cells from reperfusion injury by targeting the mitochondrial permeability transition pore (mPTP) opening via Akt and GSK-3β [9, 10]. We also demonstrated that nitric oxide (NO) prevented mitochondrial oxidant damage by mobilizing intracellular zinc via the cGMP/PKG signaling pathway [11]. Recently, Karagulova et al. reported that the maintenance of myocardial zinc status is cardioprotective at reperfusion in isolated rat hearts [12]. These observations suggest that intracellular zinc levels may play an important role in myocardial reperfusion injury. Since adenosine can produce NO through A2 receptors [13] and NO mobilizes intracellular zinc [11], it is possible that intracellular zinc plays a role in A2 receptor activation induced cardioprotection at reperfusion.
The purpose of the current study was to determine if intracellular zinc plays a role in the cardioprotective effect of 5′-(N-ethylcarboxamido) adenosine (NECA), an adenosine analogue that has been demonstrated to protect the heart when applied at reperfusion by activating A2 receptors, and, if so, how NECA regulates intracellular zinc status upon reperfusion. Then we sought to define the potential mechanism by which zinc mediates the cardioprotective effect of A2 receptor activation. Our study demonstrates that NECA prevents the loss of zinc from cardiac tissue and dramatically increases mitochondrial zinc upon reperfusion. Intracellular zinc is responsible for the cardioprotective effect of NECA at reperfusion and exogenous zinc given at reperfusion mimicked the action of NECA by reducing infarct size. Zinc may mediate the cardioprotective effect of NECA by targeting the mPTP and the mitochondrial electron transport chain.
2. Methods
This study conforms to the NIH Guide for the Care and Use of Laboratory Animals (NIH publication NO. 85-23, revised 1996).
2.1 Perfusion of isolated rat heart
Male Wistar rats (250-350 g) were anesthetized with thiobutabarbital sodium (100 mg/kg i.p.). The hearts were removed rapidly and mounted on a Langendorff apparatus. The hearts were perfused with Krebs-Henseleit buffer containing (in mM) 118.5 NaCl, 4.7 KCl, 1.2 MgSO4, 1.8 CaCl2, 24.8 NaHCO3, 1.2 KH2PO4, and 10 glucose, which was heated to 37°C and gassed with 95 % O2/5 % CO2. A latex balloon connected to a pressure transducer was inserted into the left ventricle through the left atrium. The left ventricular pressure and heart rate were continuously recorded with a PowerLab system (ADInstruments, Mountain View, CA). A 5-0 silk suture was placed around the left coronary artery, and the ends of the suture were passed through a small piece of soft vinyl tubing to form a snare. All hearts were allowed to stabilize for at least 20 min. Ischemia was induced by pulling the snare and then fixing it by clamping the tubing with a small hemostat. Total coronary artery flow was measured by timed collection of the perfusate dripping from the heart into a graduated cylinder.
2.2 Measurement of infarct size
At the end of each experiment, the coronary artery was reoccluded, and fluorescent polymer microspheres (2-9 μM in diameter, Duke Scientific Corp) were infused to demarcate the risk zone as the tissue without fluorescence. The hearts were weighed, frozen and cut into 1 mm slices. The slices were incubated in 1 % triphenyltetrazolium chloride (TTC) in sodium phosphate buffer at 37°C for 20 min. The slices were immersed in 10 % formalin to enhance the contrast between stained (viable) and unstained (necrotic) tissue and then squeezed between glass plates spaced exactly 1 mm apart. The myocardium at risk was identified by illuminating the slices with U.V. light. The infarcted and risk zone regions were traced on a clear acetate sheet and quantified with ImageTool. The areas were converted into volumes by multiplying the areas by slice thickness. Infarct size is expressed as a percentage of the risk zone.
2.3 Isolation of adult rat cardiomyocytes
Rat cardiomyocytes were isolated enzymatically [13]. Male Wistar rats weighing 250-350 g were anesthetized with thiobutabarbital sodium (100mg/kg, i.p.). A midline thoracotomy was performed and the heart was removed and rapidly mounted on a Langendorff apparatus. The heart was perfused in a non-recirculating mode with Krebs-Henseleit buffer (37°C) containing (in mM) NaCl 118, NaHCO3 25, KCl 4.7, KH2PO4 1.2, MgSO4 1.2, CaCl2 1.25, and glucose 10 for 5 min to wash out blood. The buffer was bubbled with 95 % O2/5 % CO2. Then the heart was perfused with a calcium-free buffer that contained all of the above components except CaCl2. After 5 min of perfusion, collagenase (type II) was added to the buffer (0.1 %) and the heart was perfused in a recirculating mode for ∼15 min. The heart was removed from the apparatus and the ventricles were placed into a beaker containing the calcium-free buffer. The ventricles were agitated in a shaking bath (37°C) at a rate of 50 cycles/min until individual cells were released. The released cells were suspended in an incubation buffer containing all the components of the calcium-free buffer, 1 % bovine serum albumin, 30 mM HEPES, 60 mM taurine, 20 mM creatine, and amino acid supplements at 37°C. Calcium was gradually added to the buffer containing the cells to a final concentration of 1.2 mM. The cells were filtered through nylon mesh and centrifuged briefly. Finally the cells were suspended in culture medium M199 for 4 h before experiments.
2.4 Mitochondrial isolation
Mitochondria and cytosolic fractions were isolated by differential centrifugation as previously described [14]. Cardiac samples (or isolated cardiomyocytes) were homogenized in a buffer containing 250 mM sucrose, 10 mM Tris-HCl (pH 7.4), 1 mM EDTA, 1 mM Na3VO4, 1 mM NaF, and protease inhibitor cocktail. The homogenate was centrifuged at 1000g for 10 minutes to remove nuclei and debris. The supernatant was centrifuged at 10000g for 30 minutes. The resultant supernatant was subsequently centrifuged at 10000g for 1 hour to yield the cytosolic fraction. The 10000g pellet, corresponding to the mitochondrial fraction, was resuspended and centrifuged again at 10000g for 30 minutes.
2.5 Measurement of total zinc concentrations in cardiac tissue
Cardiac tissue (or mitochondrial) lysates were scraped into 125 μl RIPA buffer and 1.0 ml of 3N HCl/10 % trichloroacetic acid (TCA) and hydrolyzed at 70 °C for 24 h. The concentration of zinc was quantified using inductively coupled plasma optical emission spectroscopy (ICPOES, Model Optima 4300D, Perkin Elmer, Norwalk, CT) at a wavelength of 206.200. A multi-element standard (Spex Certiprep, Metuchen, NJ) was used to calibrate the instrument. The limits of detection approximated 1 ppb [15]. Zinc levels were expressed as % changes in zinc concentration s during reperfusion relative to the values measured immediately before the onset of reperfusion (0′).
2.6 Imaging of free zinc in cardiac tissue
Cardiac tissue free zinc was measured with the Zn2+-selective fluorescence probe TSQ, as previously described [12]. Briefly, cardiac tissues collected from the risk zone were frozen in OCT to preserve tissue elements. Cryosections of 30 μm were obtained at -20°C using a cryomicrotome. Thawed tissue sections were loaded with 5 μM TSQ for 20 min, washed, and mounted on the stage of a fluorescence microscope (Olympus). TSQ was excited at 360 nm and fluorescence between 420 and 480 nm was captured.
2.7 Confocal imaging of intracellular zinc in cardiomyocytes
Intracellular Zn2+ was detected with Newport Green DCF [11]. Cardiomyocytes cultured in a specific temperature-controlled culture dish were incubated with 2 μM Newport Green DCF diacetate in standard Tyrode solution containing (mM) NaCl 140, KCl 6, MgCl2 1, CaCl2 1, HEPES 5, and glucose 5.8 (pH 7.4) for 20 min. Cells were then mounted on the stage of an Olympus FV 500 laser scanning confocal microscope. The green fluorescence was excited with the 488 nm line of argon-krypton laser and imaged through a 525 nm long-path filter. Temperature was maintained at 37°C with Delta T Open Dish Systems (Bioptechs, Butler, PA). The images recorded on a computer were quantified using Image J.
2.8 Confocal Imaging of ΔΨm
ΔΨm was measured using confocal microscopy as previously documented in our laboratory [13]. Briefly, cells were incubated with tetramethylrhodamine ethyl ester (TMRE, 100 nM) in standard Tyrode solution for 20 min. The red fluorescence was excited with a 543 nm line of argon-krypton laser line and imaged through a 560 nm long-path filter.
2.9 Measurement of mitochondrial swelling
Intact mitochondria (0.3 mg/ml) isolated from cardiac samples taken 10 min after the onset of reperfusion were suspended in a buffer containing (in mM) 120 KCl, 10 Tris·HCl, 5 KH2PO4, and 20 MOPS. Mitochondrial swelling was induced by 200 μM CaCl2. Mitochondrial swelling was assessed spectrophotometrically as a decrease in absorbance at 520 nm (A520) [16].
2.10 MTT-reduction assay
The basis of this assay is the cleavage (reduction) of the yellow 3-(4,5-dimethyl-2thiazolyl)-2,5-diphenyltetrazolium bromide (MTT) to blue formazan crystals by complex I of the mitochondrial electron transport chain [17]. An increase in absorbance represents an increase in MTT reduction and thus an increase in mitochondrial metabolic activity. Isolated rat cardiomyocytes cultured in a 24-well plate were treated with NECA or ZnCl2 for 10 min. After the treatment, MTT (50 μg/ml) was added to each well. The absorbance was measured with a plate reader (VERSAmax, Molecular Devices) at 570 nm with a reference wavelength of 620 nm at 37°C.
2.11 Mitochondrial complex I activity assay
Complex I enzymatic activity microplate assay kit (Mitoscience, Eugene, Oregon) was used to determine the activity of the complex I. Mitochondrial OXPHOS Complex I was immunocaptured and the activity was determined at 450 nm by following the oxidation of NADH to NAD+.
2.12 Experimental protocols
All hearts were subjected to a 30 min regional ischemia followed by 2 h of reperfusion. Cardiac samples were collected from the risk zone upon reperfusion (0, 5, 10, and 30 min after the onset of reperfusion). Infusion of either NECA or ZnCl2 was started 5 min before the onset of reperfusion and continued for 35 min. Since ZnCl2 is cell impermeable, we treated hearts (cells) with ZnCl2 in the presence of the zinc ionophore pyrithione. Infarct size was measured 2 h after start of reperfusion. In the experiments monitoring changes in intracellular Zn2+ levels in cardiomyocytes, NECA was given immediately after baseline (0′) measurements, whereas the inhibitors were applied 10 min prior to the application of NECA. To determine the effect of ZnCl2 on ΔΨm, cardiomyocytes were exposed to 100 μM H2O2 for 20 min. ZnCl2 was given 5 min before exposure to H2O2. Inhibitors were given 10 min prior to the exposure to ZnCl2. To evaluate MTT reduction rate, cardiomyocytes were exposed to NECA or ZnCl2 for 10 min.
2.13 Statistical Analysis
Data are expressed as mean ± SEM and were obtained from 4 to 8 separate experiments. Statistical significance was determined using Student t-test or ANOVA followed by Tukey's test. A value of P < 0.05 was considered as statistically significant.
3. Results
NECA (100 nM) given at reperfusion significantly reduced infarct size (14.1 ± 1.9 % of risk zone) compared to the control (37.9 ± 3.2 % of risk zone) (Fig. 1). The anti-infarct effect of NECA was partially but significantly blocked by the Zn2+ chelator N,N,N′,N′-tetrakis-(2-pyridylmethyl) ethylenediamine (TPEN, 10 μM) (25.0 ± 0.6 % of risk zone), suggesting a role of zinc in the action of NECA. In support, treatment of hearts with ZnCl2 (5 μM) in the presence of the zinc ionophore pyrithione (2 μM) at reperfusion mimicked the effect of NECA by reducing infarct size (20.2 ± 0.4 % of risk zone). ZnCl2 (5 μM) alone failed to reduce infarct size.
Figure 1.
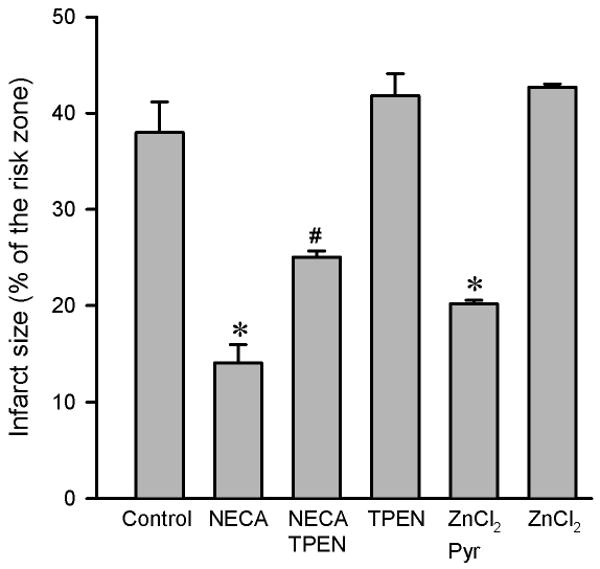
Infarct size in isolated rat hearts. All hearts were subjected to 30 min regional ischemia followed by 2 h of reperfusion. NECA (100 nM, n = 7) given at reperfusion reduced infarct size compared the control (n = 6). The anti-infarct effect of NECA was partially but significantly inhibited by the zinc chelator TPEN (10 μM, n = 6) and ZnCl2 (5 μM, n = 6) in the presence of the zinc ionophore pyrithione (Pyr, 2 μM) limited infarct size. TPEN (n = 6) and ZnCl2 (n = 6) alone did not alter infarct size. * p < 0.05 vs. control; # p < 0.05 vs. NECA
To determine how NECA regulates cardiac zinc levels upon reperfusion, we measured the time course of changes in cardiac tissue zinc levels during reperfusion. Fig. 2 shows that reperfusion dramatically decreased tissue total zinc levels (control). In contrast, hearts treated with NECA showed a much higher Zn2+ levels during reperfusion compared to the control, indicating that NECA can prevent reperfusion-induced zinc loss. Similarly, compared to the control, NECA increased TSQ fluorescence intensity in cardiac tissue upon reperfusion, indicating that more free (labile) zinc ions (Zn2+) exist in the heart treated with NECA (Fig. 3). The effect of NECA on TSQ fluorescence was reversed by the zinc chelator TPEN (Fig. 3). This effect of NECA on free zinc levels could be attributable to either an increase in zinc release from its binding sites or the decreased loss of zinc by NECA. Therefore, we then tested if NECA could mobilize zinc in isolated rat cardiomyocytes under physiological conditions. As shown in Fig. 4, cardiomyocytes exposed to NECA for 10 min showed a significant increase in Newport Green fluorescence intensity (132.5 ± 3.9 % of baseline) compared to the control (101.3 ± 2.8 % of baseline), indicating that NECA can mobilize intracellular zinc from its binding sites. This effect of NECA was abolished by both the NOS inhibitor NG-nitro-L-arginine methyl ester (L-NAME) (200 μM) and the protein kinase G (PKG) inhibitor KT5823 (1 μM), implying that NECA mobilizes zinc through the NO/PKG pathway. To determine the effect of NECA on mitochondrial zinc levels upon reperfusion, we measured zinc concentration in mitochondria isolated from the risk zone. Upon reperfusion, mitochondrial zinc levels were decreased in untreated hearts (control) (Fig. 5). In contrast, hearts treated with NECA showed a marked increase in mitochondrial zinc, indicating that NECA can relocate zinc from cytosol into mitochondria.
Figure 2.
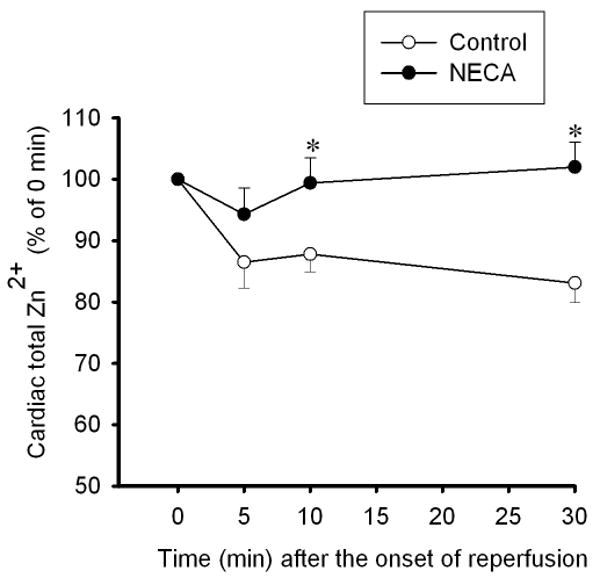
Time course of changes in total zinc concentrations at reperfusion in rat hearts subjected to 30 min regional ischemia followed by 2 h of reperfusion. Reperfusion triggered zinc loss from the hearts (control n = 6), which was reversed by NECA (100 nM, n = 6). * p < 0.05 vs. control.
Figure 3.
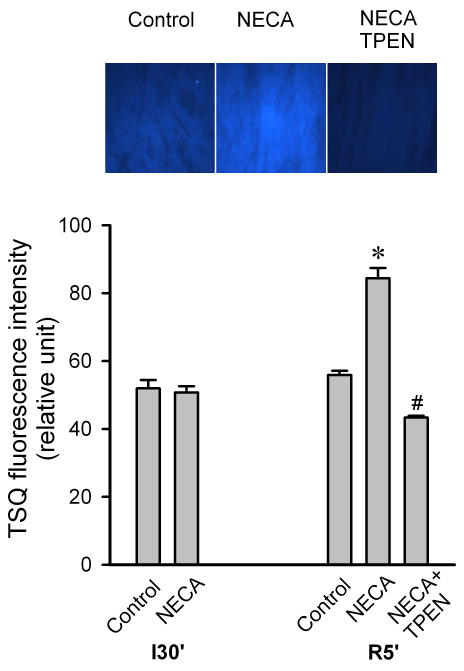
Top, Fluorescence images of TSQ 5 min after the onset of reperfusion in isolated rat hearts. Myocardial tissue Cryosections were stained with the free zinc indicator TSQ. Bottom, Summarized data for TSQ fluorescence intensity 5 min after the onset of reperfusion. Compared to the control (n = 3), the heart treated with NECA (100 nM, n = 3) showed a marked increase in TSQ fluorescence intensity at reperfusion, indicating that NECA increases intracellular free zinc at reperfusion. The effect of NECA on TSQ fluorescence was reversed by the zinc chelator TPEN (10 μM). * p < 0.05 vs. control; # p < 0.05 vs. NECA.
Figure 4.
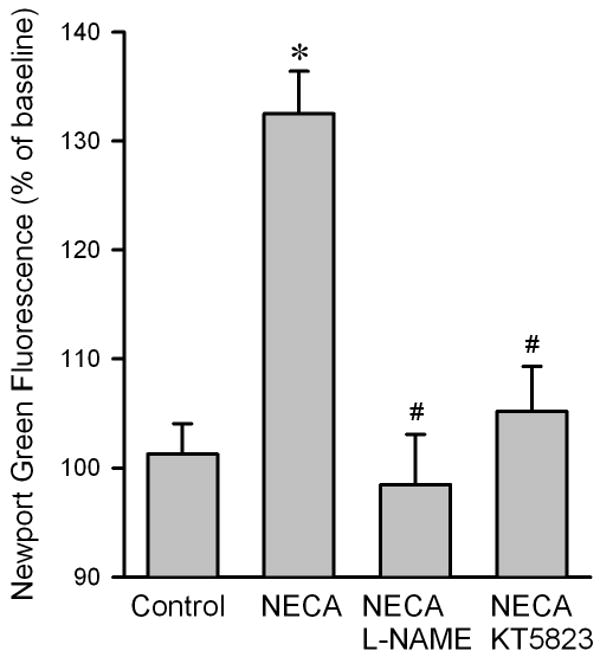
Summarized data for Newport Green DCF fluorescence intensity 10 min after exposure to NECA in isolated rat cardiomyocytes. NECA (100 nM, n = 6) markedly increased the fluorescence intensity compared to control (n = 6), an effect that was reversed by both the NOS inhibitor L-NAME (200 μM, n = 6) and the PKG inhibitor KT5823 (1 μM, n = 6), indicating that NECA mobilizes intracellular zinc through the NO/PKG signaling pathway. At least 5 cells were analyzed in each experiment. * p < 0.05 vs. control; # p < 0.05 vs. NECA.
Figure 5.
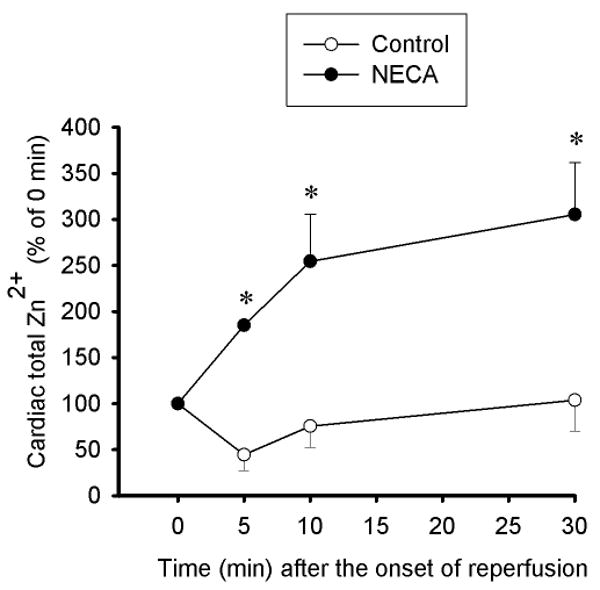
Time course of changes in total zinc concentrations at reperfusion in mitochondria (2.25 μg protein per μl) isolated from rat hearts subjected to 30 min regional ischemia followed by 2 h of reperfusion. Compared to the control (n = 5) in which mitochondrial zinc was decreased, NECA (100 nM, n = 5) increased mitochondrial zinc upon reperfusion. * p < 0.05 vs. control.
To test if NECA modulates the mPTP opening via zinc, we measured the mitochondrial membrane potential (ΔΨm) by loading rat cardiomyocytes with TMRE (Fig. 6). Exposure of cardiomyocytes to 100 μM H2O2 resulted in a marked decrease in TMRE fluorescence, indicating that oxidant stress induces the mPTP opening. In contrast, cardiomyocytes treated with NECA showed a much smaller decrease in the red fluorescence, implying that NECA can prevent the pore opening. This effect of NECA was partially but significantly blocked by the zinc chelator TPEN, suggesting that zinc may mediate the action of NECA by modulating the pore opening. To corroborate the role of zinc in the inhibitory effect of NECA on the mPTP opening, we evaluated the effect of NECA on mitochondrial swelling upon reperfusion in isolated rat hearts by measuring light scattering at A520. Compared to the control (0.266 ± 0.020), mitochondria isolated from the heart treated with NECA had a higher value of A520 (0.530 ± 0.036), indicating that NECA can modulate the mPTP opening at reperfusion (Fig. 2). This effect of NECA was inhibited by TPEN (0.385 ± 0.018) (Fig. 7), indicating that the preventive effect of NECA on the mPTP opening is mediated by zinc. Treatment of hearts with ZnCl2 in the presence of pyrithione at reperfusion mimicked the action of NECA by increasing the value of A520 (0.448 ± 0.025). To test if the treatment of the heart with ZnCl2 could increase mitochondrial zinc levels, we measured mitochondrial zinc levels with ICPOES upon reperfusion. ZnCl2 in the presence of pyrithione markedly in creased mitochondrial zinc levels 10 min after the onset of reperfusion compared to the control (543.2 ± 41.3 % vs. 74.7 ± 26.7 % of baseline), suggesting that increasing cytosolic zinc with ZnCl2 leads to an increase in mitochondrial zinc levels.
Figure 6.
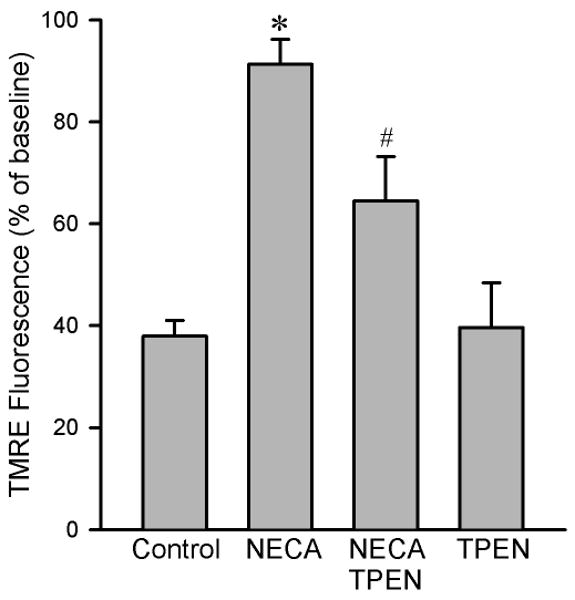
Summarized data for TMRE fluorescence intensity 20 min after exposure to 100 μM H2O2 in isolated rat cardiomyocytes. NECA (100 nM, n = 7) prevented oxidant-induced TMRE fluorescence reduction compared to the control (n = 9), an effect that was attenuated by the zinc chelator TPEN (10 μM, n = 6). TPEN alone (n = 6) did not alter the fluorescence intensity. Three to 6 cells were analyzed in each experiment. * p < 0.05 vs. control; # p < 0.05 vs. NECA.
Figure 7.
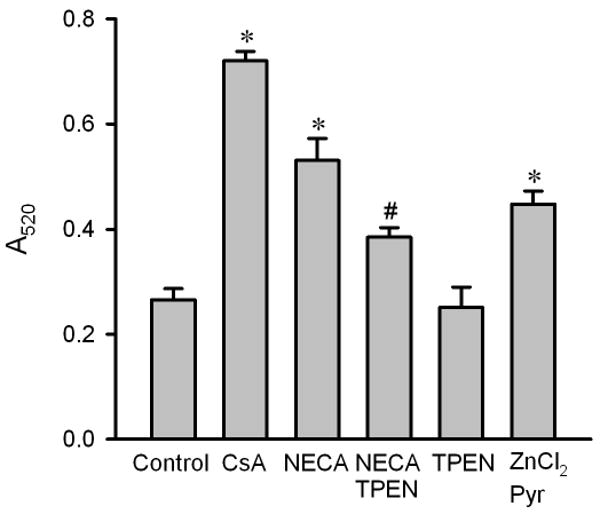
Mitochondrial swelling in isolated rat hearts. Mitochondria were isolated from myocardial samples collected at 10 min after the onset of reperfusion. Mitochondrial swelling was induced by 200 μM CaCl2 and was measured as a decrease in absorbance at 520 nm (A520) over 20 min. Cyclosporin A (CsA, 2 μM, n = 5) and NECA (100 nM, n = 6) prevented mitochondrial swelling at reperfusion compared to the control (n = 6). The action of NECA was inhibited by the zinc chelator TPEN (10 μM, n = 6). TPEN alone (n = 5) did not inhibit swelling. ZnCl2 (5 μM, n = 6) in the presence of the zinc ionophore pyrithione (Pyr, 2.5 μM) also prevented mitochondrial swelling. * p < 0.05 vs. control; # p < 0.05 vs. NECA.
To examine if zinc regulates the mitochondrial metabolic activity, we determined the effect of zinc on the redox state of complex I in isolated rat cardiomyocytes using the MTT reduction assay. As shown in Fig. 8A, Both NECA and ZnCl2 decreased MTT reduction, indicating that NECA may inhibit complex I via zinc. To confirm this result, we next measured mitochondrial complex I activity 10 min after the onset of reperfusion in isolated rat hearts. compared to the control, NECA (100 nM) significantly reduced the activity of complex I, an effect that was reversed by the zinc chelator TPEN (10 μM), indicating that NECA indeed inhibits complex I via zinc (Fig. 8B).
Figure 8.
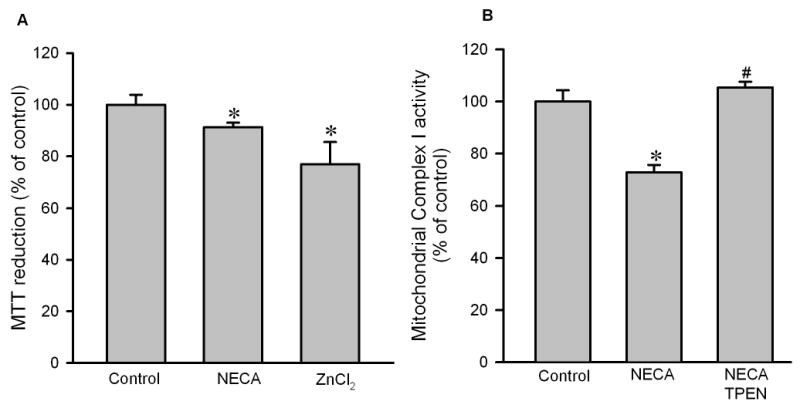
A, MTT reduction assay in isolated rat cardiomyocytes. Cells were treated with NECA (100 nM) or ZnCl2 (5 μM) in the presence of pyrithione for 10 min. Both NECA (n = 6) and ZnCl2 (n = 6) reduced MTT reduction, indicating that zinc may mediate the cardioprotective effect of NECA by inhibiting mitochondrial metabolic activity. * p < 0.05 vs. control. B, Mitochondrial complex I activity assay in isolated rat hearts subjected to 30 min regional ischemia followed by reperfusion. NECA (100 nM, n = 4) and TPEN (10 μM, n = 4)) given 5 min prior to the onset of reperfusion for 35 min. Myocardial samples were collected 10 min the onset of reperfusion. NECA reduced the activity of the complex I and this was reversed by TPEN. * p < 0.05 vs. control; # p < 0.05 vs. NECA.
4. Discussion
This study demonstrates that NECA prevents cardiac zinc loss upon reperfusion and limits infarct size via a zinc-dependent mechanism. NECA also rapidly increases mitochondrial zinc levels upon reperfusion, which may lead to mitochondrial metabolic inhibition and suppression of the mPTP opening.
Although there are controversies on the cardioprotective effects of adenosine A2 receptor activation at reperfusion, NECA has been consistently shown to protect the heart when given at reperfusion [4, 18-20]. NECA confers cardioprotection at reperfusion mainly through activation of A2 receptors [3, 4, 21]. Our recent study has demonstrated that both A2A and A2B receptors are involved in the cardioprotective effect of NECA at reperfusion [22]. Since both A2A [23] and A2B [3] receptors contribute to the mechanism underlying postconditioning, and either A2A [2, 24] or A2B [4] receptor agonists have been proposed to protect the heart at reperfusion, a thorough investigation of NECA's cardioprotective mechanism may help us find novel strategies for prevention of reperfusion injury.
PI3K and ERK have been proposed to contribute to the cardioprotective effect of NECA at reperfusion [18] and are the essential signaling elements in the RISK pathway [25]. Recently, we have demonstrated that exogenous zinc protects cardiac H9c2 cells from reperfusion injury through PI3K/Akt and ERK [9-11]. In addition, we have reported that adenosine produces NO via A2 receptors [13] and that NO prevents oxidant-induced mitochondrial damage by mobilizing intracellular zinc in cardiomyocytes [11]. These observations led us to make the hypothesis that intracellular zinc may play a role in NECA-induced cardioprotection at reperfusion. In addition to its fundamental role in maintaining the structure and function of many proteins, enzymes, and transcription factors [5], free or labile zinc plays a crucial role in signal transduction [26]. Under physiological conditions, intracellular zinc homeostasis is tightly controlled by zinc transporters [27] and zinc deficiency is linked to a number of diseases [28]. In the present study, we found that zinc levels were decreased upon reperfusion in isolated rat hearts. Since we measured the concentration of the total cellular zinc using ICPOES instead of free or labile zinc, our data suggest a loss of zinc from the heart at reperfusion. Interestingly, NECA reversed the loss of zinc and reduced infarct size via a zinc-dependent mechanism. Moreover, exogenous zinc applied at reperfusion mimicked the cardioprotective effect of NECA by reducing infarct size. These data clearly suggest that the zinc loss is a potential cause of reperfusion injury and preservation of cardiac zinc levels at reperfusion is an important mechanism by which NECA or other adenosine A2 receptor agonists protect the heart from reperfusion injury. Moreover, our data also suggest that the maintenance of cardiac zinc levels at reperfusion by treating patients with zinc is a potential strategy for prevention of reperfusion injury in the clinical settings of AMI. A recent study showing that intracellular liable or free zinc levels were decreased in the heart subjected to ischemia/reperfusion [12] further supports our interpretation. In an early study, Powell et al. demonstrated that pre- and postischemic treatment with Zn2+ reduced cardiac dysfunction and LDH release during reperfusion in perfused rat hearts and proposed that this protective effect of zinc was attributed to prevention of ·OH formation [29]. They further found that zinc prevented mitochondrial swelling and damage during reperfusion, supporting our finding that zinc may mediate the cardioprotective effect of NECA by suppression of the mPTP opening. However, it should be noted that zinc treatment starting at the onset of reperfusion was not able to protect the heart in their preparation. The exact reason for the failure of protection remains unknown.
Zinc transporters are encoded by two gene families: ZnT and Zip. There are at least 9 ZnT and 15 Zip transporters in human cells. ZnT and Zip transporters have opposite roles in zinc homeostasis. ZnT transporters lower cytoplasmic zinc by promoting zinc efflux from cells or influx into intracellular vesicles, whereas Zip transporters increase cytoplasmic zinc by promoting zinc transport from the extracellular fluid or from intracellular vesicles into cytoplasm. Since ZnT-1 is the only mammalian zinc transporter serving as a cellular zinc exporter [27], the loss of zinc may suggest an “over-functioning” of ZnT-1 transporter upon reperfusion by promoting a rapid zinc efflux from cells. If this is true, NECA might have served as a negative regulator of ZnT-1 activity to prevent zinc loss at reperfusion. However, functional and structural disorders of cell membrane upon reperfusion may also result in “leakage” of zinc from cardiac cells. In this case, the functional and structural integrity of cardiac cells owing to NECA's protective effect may account for the preservation of cellular zinc at reperfusion. Further studies are needed to clarify the mechanism by which NECA prevents the loss of zinc from the heart at reperfusion.
In this study, NECA not only prevented the loss of zinc but also significantly increased mitochondrial zinc upon reperfusion compared the control in which mitochondrial zinc levels were decreased. This result points to that NECA can relocate zinc to mitochondria, which may be associated to the protective effect of NECA. Our data also showed that NECA increases cytosolic free zinc presumably by mobilizing zinc from its biding sites via the NO/PKG signaling. However, the preventive effect of NECA on the zinc loss may also account for the elevation of cytosolic free zinc at reperfusion. A recent study by Sensi et al. has demonstrated that both mitochondrial and cytosolic zinc pools can be mobilized largely independently of each other, with zinc release from one resulting in net zinc transfer to the other [30]. Thus, it is likely that the increased cytosolic free zinc could serve as the source of zinc relocation to mitochondria upon reperfusion in the heart treated with NECA.
Zinc has been proposed to affect mitochondrial function [31]. In the present study, we found that NECA prevents the mPTP opening upon reperfusion in a zinc-dependent manner and exogenous zinc applied at reperfusion could inhibit the pore opening. Our previous studies also demonstrated that exogenous zinc modulates the mPTP opening induced by oxidant stress [9-11]. The potential mechanism underlying the inhibitory effect of zinc on the mPTP might be phosphorylation of mitochondrial GSK-3β, since increased mitochondrial phospho-GSK-3β was associated with inhibition of the mPTP opening [32] and exogenous zinc was shown to increase mitochondrial GSK-3β phosphorylation in cardiac cells [9]. The critical role of GSK-3β in the action of zinc on the mPTP was further supported by our recent study in which NECA could increase mitochondrial GSK-3β phosphorylation in isolated rat hearts [22]. Further studies are needed to test if relocated zinc modulates the mPTP opening through GSK-3β in the heart treated with NECA.
Zinc has also been suggested to be involved in inhibition of complexes of the mitochondrial electron transport chain or inhibition of matrix enzymes [33, 34]. Our data show that both NECA and zinc could decrease MTT reduction in cardiomyocytes. Because MTT is reduced by complex I of the electron transport chain within mitochondria and reduction of MTT occurs predominantly at mitochondria in cardiomyocytes [17], our finding suggests that NECA may inhibit the mitochondrial complex I activity through zinc. “Metabolic shut down and gradual wake-up” achieved by mitochondrial inhibition has been proposed to be a critical mechanism for cardioprotection [17]. Therefore, it is possible that the inhibition of mitochondria metabolic activity by zinc may also in part contribute to the action of NECA. Mitochondrial zinc may inhibit complex I by inhibiting either quinine reduction or proton translocation [35]. Further efforts are needed to test the effects of zinc on other components of the electron transport chain.
In summary (Fig. 9), NECA prevents zinc loss upon reperfusion and protects the heart from reperfusion injury via a zinc-dependent manner. Zinc translocation to mitochondria and its inhibitory actions on the mPTP opening and the mitochondrial metabolic activity may be responsible for the mechanism by which zinc mediates the cardioprotective effect of NECA. We propose that prevention of zinc loss from the heart and elevation of cardiac mitochondrial zinc levels are critical factors for cardioprotection against reperfusion injury and that an appropriate zinc supplementation at reperfusion might be a potential strategy for prevention of reperfusion injury in the clinical settings of AMI.
Figure 9.

The signaling mechanism by which intracellular zinc mediates A2 receptor activation induced cardioprotection at reperfusion.
Acknowledgments
This work was supported by National Institute of Health grant HL08336 (to Dr. Xu).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zhao ZQ, Sato H, Williams MW, Fernandez AZ, Vinten-Johansen J. Adenosine A2-receptor activation inhibits neutrophil-mediated injury to coronary endothelium. Am J Physiol. 1996;271:H1456–H1464. doi: 10.1152/ajpheart.1996.271.4.H1456. [DOI] [PubMed] [Google Scholar]
- 2.Jordan JE, Zhao ZQ, Sato H, Taft S, Vinten-Johansen J. Adenosine A2 receptor activation attenuates reperfusion injury by inhibiting neutrophil accumulation, superoxide generation and coronary endothelial adherence. J Pharmacol Exp Ther. 1997;280:301–309. [PubMed] [Google Scholar]
- 3.Philipp S, Yang XM, Cui L, Davis AM, Downey JM, Cohen MV. Postconditioning protects rabbit hearts through a protein kinase C-adenosine A2b receptor cascade. Cardiovasc Res. 2006;70:308–314. doi: 10.1016/j.cardiores.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 4.Kuno A, Critz SD, Cui L, Solodushko V, Yang XM, Krahn T, Albrecht B, Philipp S, Cohen MV, Downey JM. Protein kinase C protects preconditioned rabbit hearts by increasing sensitivity of adenosine A2b-dependent signaling during early reperfusion. J Mol Cell Cardiol. 2007;43:262–271. doi: 10.1016/j.yjmcc.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berg JM, Shi Y. The galvanization of biology: a growing appreciation for the roles of zinc. Science. 1996;271:1081–5. doi: 10.1126/science.271.5252.1081. [DOI] [PubMed] [Google Scholar]
- 6.Barthel A, Ostrakhovitch EA, Walter PL, Kampkötter A, Klotz LO. Stimulation of phosphoinositide 3-kinase/Akt signaling by copper and zinc ions: Mechanisms and consequences. Arch Biochem Biophys. 2007;463:175–182. doi: 10.1016/j.abb.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 7.An WL, Pei JJ, Nishimura T, Winblad B, Cowburn RF. Zinc-induced anti-apoptotic effects in SH-SY5Y neuroblastoma cells via the extracellular signal-regulated kinase 1/2. Brain Res Mol Brain Res. 2005;135:40–47. doi: 10.1016/j.molbrainres.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 8.Ilouz R, Kaidanovich O, Gurwitz D, Eldar-Finkelman H. Inhibition of glycogen synthase kinase-3β by bivalent zinc ions: insight into the insulin-mimetic action of zinc. Biochem Biophys Res Commun. 2002;295:102–106. doi: 10.1016/s0006-291x(02)00636-8. [DOI] [PubMed] [Google Scholar]
- 9.Chanoit G, Lee S, Xi J, Zhu M, McIntosh RA, Mueller RA, Norfleet EA, Xu Z. Exogenous zinc protects cardiac cells from reperfusion injury by targeting mitochondrial permeability transition pore through inactivation of glycogen synthase kinase-3β. Am J Physiol. 2008;295:H1227–1233. doi: 10.1152/ajpheart.00610.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee S, Chanoit G, McIntosh RA, Zvara DA, Xu Z. The molecular mechanism underlying Akt activation in zinc-induced cardioprotection. Am J Physiol. 2009;297:H569–575. doi: 10.1152/ajpheart.00293.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jang Y, Wang H, Xi J, Mueller RA, Norfleet EA, Xu Z. NO mobilizes intracellular Zn2+ via cGMP/PKG signaling pathway and prevents mitochondrial oxidant damage in cardiomyocytes. Cardiovasc Res. 2007;75:426–433. doi: 10.1016/j.cardiores.2007.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karagulova G, Yue Y, Moreyra A, Boutjdir M, Korichneva I. Protective Role of Intracellular Zinc in Myocardial Ischemia/Reperfusion Is Associated with Preservation of Protein Kinase C Isoforms. J Pharmacol Exp Ther. 2007;321:517–525. doi: 10.1124/jpet.107.119644. [DOI] [PubMed] [Google Scholar]
- 13.Xu Z, Park SS, Mueller RA, Bagnell RC, Patterson C, Boysen PG. Adenosine produces nitric oxide and prevents mitochondrial oxidant damage in rat cardiomyocytes. Cardiovasc Res. 2005;65:803–12. doi: 10.1016/j.cardiores.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 14.Baines CP, Zhang J, Wang GW, Zheng YT, Xiu JX, Cardwell EM, Bolli R, Ping P. Mitochondrial PKCepsilon and MAPK form signaling modules in the murine heart: enhanced mitochondrial PKCepsilon-MAPK interactions and differential MAPK activation in PKCepsilon-induced cardioprotection. Circ Res. 2002;90:390–397. doi: 10.1161/01.res.0000012702.90501.8d. [DOI] [PubMed] [Google Scholar]
- 15.Deng Z, Dailey L, Soukup J, Stonehuerner J, Richards J, Callaghan K, Yang F, Ghio A. Zinc transport by respiratory epithelial cells and interaction with iron homeostasis. BioMetals. 2009 doi: 10.1007/s10534-009-9227-2. [DOI] [PubMed] [Google Scholar]
- 16.Wang G, Liem DA, Vondriska TM, Honda HM, Korge P, Pantaleon DM, Qiao X, Wang Y, Weiss JN, Ping P. Nitric Oxide Donors Protect the Murine Myocardium Against Infarction via Modulation of Mitochondrial Permeability Transition. Am J Physiol. 2005;288:H1290–5. doi: 10.1152/ajpheart.00796.2004. [DOI] [PubMed] [Google Scholar]
- 17.Viola HM, Arthur PG, Hool LC. Evidence for regulation of mitochondrial function by the L-type Ca2+ channel in ventricular myocytes. J Mol Cell Cardiol. 2009;46:1016–1026. doi: 10.1016/j.yjmcc.2008.12.015. [DOI] [PubMed] [Google Scholar]
- 18.Yang XM, Krieg T, Cui L, Downey JM, Cohen MV. NECA and bradykinin at reperfusion reduce infarction in rabbit hearts by signaling through PI3K, ERK, and NO. J Mol Cell Cardiol. 2004;36:411–21. doi: 10.1016/j.yjmcc.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 19.Förster K, Paul I, Solenkova N, Staudt A, Cohen M, Downey J, Felix S, Krieg T. NECA at reperfusion limits infarction and inhibits formation of the mitochondrial permeability transition pore by activating p70S6 kinase. Basic Res Cardiol. 2006;101:319–326. doi: 10.1007/s00395-006-0593-4. [DOI] [PubMed] [Google Scholar]
- 20.Xu Z, Downey JM, Cohen MV. AMP 579 reduces contracture and limits infarction in rabbit heart by activating adenosine A2 receptors. J Cardiovasc Pharmacol. 2001;38:474–81. doi: 10.1097/00005344-200109000-00016. [DOI] [PubMed] [Google Scholar]
- 21.Kuno A, Solenkova NV, Solodushko V, Dost T, Liu Y, Yang XM, Cohen MV, Downey JM. Infarct limitation by a protein kinase G activator at reperfusion in rabbit hearts is dependent on sensitizing the heart to A2b agonists by protein kinase C. Am J Physiol. 2008;295:H1288–1295. doi: 10.1152/ajpheart.00209.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xi J, McIntosh RA, Shen X, Lee S, Chanoit G, Criswell H, Zvara DA, Xu Z. Adenosine A2A and A2B receptors work in concert to induce a strong protection against reperfusion injury in rat hearts. J Mol Cell Cardiol. 2009 doi: 10.1016/j.yjmcc.2009.08.009. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morrison RR, Tan XL, Ledent C, Mustafa SJ, Hofmann PA. Targeted deletion of A2A adenosine receptors attenuates the protective effects of myocardial postconditioning. Am J Physiol. 2007;293:H2523–2529. doi: 10.1152/ajpheart.00612.2007. [DOI] [PubMed] [Google Scholar]
- 24.Rork TH, Wallace KL, Kennedy DP, Marshall MA, Lankford AR, Linden J. Adenosine A2A receptor activation reduces infarct size in the isolated, perfused mouse heart by inhibiting resident cardiac mast cell degranulation. Am J Physiol. 2008;295:H1825–1833. doi: 10.1152/ajpheart.495.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hausenloy DJ, Yellon DM. New directions for protecting the heart against ischaemia-reperfusion injury: targeting the Reperfusion Injury Salvage Kinase (RISK)-pathway. Cardiovasc Res. 2004;61:448–60. doi: 10.1016/j.cardiores.2003.09.024. [DOI] [PubMed] [Google Scholar]
- 26.Beyersmann D, Haase H. Functions of zinc in signaling, proliferation and differentiation of mammalian cells. Biometals. 2001;14:331–41. doi: 10.1023/a:1012905406548. [DOI] [PubMed] [Google Scholar]
- 27.Liuzzi JP, Cousins RJ. Mammalian zinc transporters. Annu Rev Nutr. 2004;24:151–172. doi: 10.1146/annurev.nutr.24.012003.132402. [DOI] [PubMed] [Google Scholar]
- 28.Masaaki M, Toshio H. Intracellular zinc homeostasis and zinc signaling. Cancer Sci. 2008;99:1515–1522. doi: 10.1111/j.1349-7006.2008.00854.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Powell SR, Hall D, Aiuto L, Wapnir RA, Teichberg S, Tortolani AJ. Zinc improves postischemic recovery of isolated rat hearts through inhibition of oxidative stress. Am J Physiol. 1994;266:H2497–507. doi: 10.1152/ajpheart.1994.266.6.H2497. [DOI] [PubMed] [Google Scholar]
- 30.Sensi SL, Ton-That D, Sullivan PG, Jonas EA, Gee KR, Kaczmarek LK, Weiss JH. Modulation of mitochondrial function by endogenous Zn2+ pools. Proc Natl Acad Sci U S A. 2003;100:6157–62. doi: 10.1073/pnas.1031598100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Korichneva I. Zinc Dynamics in the Myocardial Redox Signaling Network. Antioxid Redox Signal. 2006;8:1707–1721. doi: 10.1089/ars.2006.8.1707. [DOI] [PubMed] [Google Scholar]
- 32.Nishihara M, Miura T, Miki T, Tanno M, Yano T, Naitoh K, Ohori K, Hotta H, Terashima Y, Shimamoto K. Modulation of the mitochondrial permeability transition pore complex in GSK-3β-mediated myocardial protection. J Mol Cell Cardiol. 2007;43:564–570. doi: 10.1016/j.yjmcc.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 33.Brown AM, Kristal BS, Effron MS, Shestopalov AI, Ullucci PA, Sheu KFR, Blass JP, Cooper AJL. Zn2+ Inhibits alpha -Ketoglutarate-stimulated Mitochondrial Respiration and the Isolated alpha -Ketoglutarate Dehydrogenase Complex. J Biol Chem. 2000;275:13441–13447. doi: 10.1074/jbc.275.18.13441. [DOI] [PubMed] [Google Scholar]
- 34.Michele L, Tiziana C, Anna Maria S, Michele M, Francesco B, Sergio P. Interaction of Zn2+ with the bovine-heart mitochondrial bc1 complex. Eu J Biochem. 1991;197:555–561. doi: 10.1111/j.1432-1033.1991.tb15944.x. [DOI] [PubMed] [Google Scholar]
- 35.Sharpley MS, Hirst J. The Inhibition of Mitochondrial Complex I (NADH:Ubiquinone Oxidoreductase) by Zn2+ J Biol Chem. 2006;281:34803–34809. doi: 10.1074/jbc.M607389200. [DOI] [PubMed] [Google Scholar]