

Abstract
The Cdc25A protein phosphatase drives cell cycle transitions by activating cyclin-dependent protein kinases. Failure to regulate Cdc25A leads to deregulated cell cycle progression, bypass of cell cycle checkpoints and genome instability. Ubiquitin-mediated proteolysis plays an important role in balancing Cdc25A levels. Cdc25A contains a DS82G motif whose phosphorylation is targeted by β-TrCP E3 ligase during interphase. Targeting of β-TrCP to Cdc25A requires phosphorylation of serines 79 (S79) and 82 (S82). Here, we report that casein kinase 1 alpha (CK1α) phosphorylates Cdc25A on both S79 and S82 in a hierarchical manner requiring prior phosphorylation of serine 76 by Chk1 or GSK-3β. This facilitates β-TrCP binding and ubiquitin-mediated proteolysis of Cdc25A throughout interphase and following exposure to genotoxic stress. The priming of Cdc25A by at least three kinases (Chk1, GSK-3β, CK1α), some of which also require priming, ensures diverse extra- and intra-cellular signals interface with Cdc25A to precisely control cell division.
Keywords: Cell cycle, Chk1, β-TrCP, ubiquitin
Introduction
The Cdc25A protein phosphatase is one of three Cdc25 family members in mammals. Cdc25A positively regulates both early and late cell cycle transitions by activating cyclin-dependent protein kinases (Boutros et al., 2006). The overall abundance and activity of Cdc25A is regulated throughout the cell division cycle by transcriptional and post-transcriptional mechanisms. Post-transcriptional mechanisms include reversible phosphorylation, protein-protein interactions, ubiquitin-mediated proteolysis and intracellular compartmentalization (Bernardi et al., 2000; Boutros et al., 2006; Busino et al., 2004; Chen et al., 2003). Cdc25A protein levels peak in mitosis and Cdc25A is degraded by the APC/CCdh1 E3 ligase as cells exit mitosis (Busino et al., 2004; Donzelli et al., 2002). Cdc25A transcription is activated by E2F/c-Myc in the G1 phase of the cell cycle and Cdc25A levels begin to rise in mid- to late-G1 (Galaktionov et al., 1996; Vigo et al., 1999). However, the accumulation of Cdc25A through enhanced transcription is counterbalanced by its ubiquitin-mediated proteolysis, which occurs throughout interphase and is mediated by the β-TrCP E3 ligase (Busino et al., 2004; Busino et al., 2003; Donzelli et al., 2002; Jin et al., 2003; Ray et al., 2005). Cdc25A is also rapidly targeted for ubiquitin-mediated proteolysis when cells experience genotoxic or replication stress and failure to degrade Cdc25A during a checkpoint response leads to bypass of the S- and G2-checkpoints (Falck et al., 2001; Hassepass et al., 2003; Mailand et al., 2000; Molinari et al., 2000; Zhao et al., 2002).
The importance of balancing Cdc25A accumulation with its destruction is underscored by the observation that overproduction of Cdc25A results in accelerated S phase- and mitotic-entry leading to genome instability (Bartek and Lukas, 2001; Blomberg and Hoffman, 1999; Falck et al., 2001; Mailand et al., 2000; Molinari et al., 2000; Zhao et al., 2002). In addition, overproduction of Cdc25A is observed in many human cancers and Cdc25A has been shown to be rate limiting in a mouse model of tumorigenesis (Kristjansdottir and Rudolph, 2004; Ray and Kiyokawa, 2008). Furthermore, post-transcriptional mechanisms account for Cdc25A overproduction in several breast cancer cell lines suggesting that proteins regulating Cdc25A destruction could be derailed in these cancers (Loffler et al., 2003). GSK-3β regulates Cdc25A destruction during early cell cycle phases by phosphorylating S76 and a correlation between Cdc25A overproduction and GSK-3β inactivation is observed in human tumor tissues, indicating that GSK-3β inactivation may account for Cdc25A overproduction in a subset of human tumors (Kang et al., 2008).
During interphase, β-TrCP-mediated Cdc25A destruction requires S82 within the DSG motif to be phosphorylated (Busino et al., 2003; Jin et al., 2003). Phosphorylation of DSG motif facilitates Cdc25A interactions with β-TrCP. Phosphorylation of serine and threonine residues neighboring the DSG motif including S76, S79, and T80 have also been reported to be important for Cdc25A ubiquitination by promoting phosphorylation of S82 (Donzelli et al., 2004; Goloudina et al., 2003; Hassepass et al., 2003; Jin et al., 2003; Kang et al., 2008). S76 phosphorylation is mediated by GSK-3β during early cell cycle phases and by Chk1 during S- and G2-phases (Goloudina et al., 2003; Hassepass et al., 2003; Kang et al., 2008; Zhao et al., 2002). Whereas Chk1 does not require a priming phosphorylation site, GSK-3β requires that Cdc25A first be phosphorylated on T80, which can be catalyzed by Plk3 (Kang et al., 2008). The protein kinases that phosphorylate Cdc25A on S79 and S82, the key residues that mediate β-TrCP binding, have not been identified. Here, we demonstrate that CK1α mediates interactions between β-TrCP and Cdc25A in vivo by phosphorylating Cdc25A on both S79 and S82 in a hierarchical manner that requires prior phosphorylation of S76.
Results
Validation of Cdc25A-FLuc reporter
Our original goal was to carry out high throughput screens using siRNAs against the human kinome to identify novel protein kinases that regulate Cdc25A stability and, in particular, to identify the protein kinase that phosphorylates Cdc25A on serine 82. With this goal in mind, stable cell lines that inducibly express a fusion protein between human Cdc25A and firefly luciferase (Cdc25A-FLuc) were generated to enable direct real-time monitoring of Cdc25A protein levels in cells. The promoter driving Cdc25A-FLuc expression contains a tetracycline response element regulated by rtTA (reverse tetracycline-controlled transactivator) and doxycycline (Dox), a tetracycline derivative. Expression of the Cdc25A-FLuc fusion protein was induced by addition of Dox to the culture media, and the level of Cdc25A-FLuc protein correlated with its luciferase activity (Fig. 1A, B). Furthermore, Cdc25A-FLuc protein levels were stabilized in cells incubated with either a proteosome inhibitor (MG132) or Chk1 inhibitors (UCN-01, Gö6976 and AZD7762) (Fig. 1C and data not shown). This data suggested that the Cdc25A-FLuc fusion protein was a valid substrate with which to identify novel Cdc25A regulatory kinases. As further controls, cells were incubated with siRNAs specific for protein kinases known to negatively regulate Cdc25A stability including Chk1 and GSK-3β (Fig. 2A). As expected, stabilization of the Cdc25A-FLuc reporter protein was observed under these conditions. As a negative control, cells were incubated with siRNAs specific for CK1α as CK1 inhibition was reported to have no effect on Cdc25A stability (Jin et al., 2003). Unexpectedly, enhanced bioluminescence indicative of Cdc25A-FLuc stabilization was observed in cells treated with CK1α-specific siRNAs (Fig. 2A). CK1 had been ruled out as a potential Cdc25A-regulatory kinase based on experiments performed with the casein kinase I inhibitors CKI-7 and IC261 (Jin et al., 2003).
Figure 1. Validation of Cdc25A-FLuc fusion reporter.
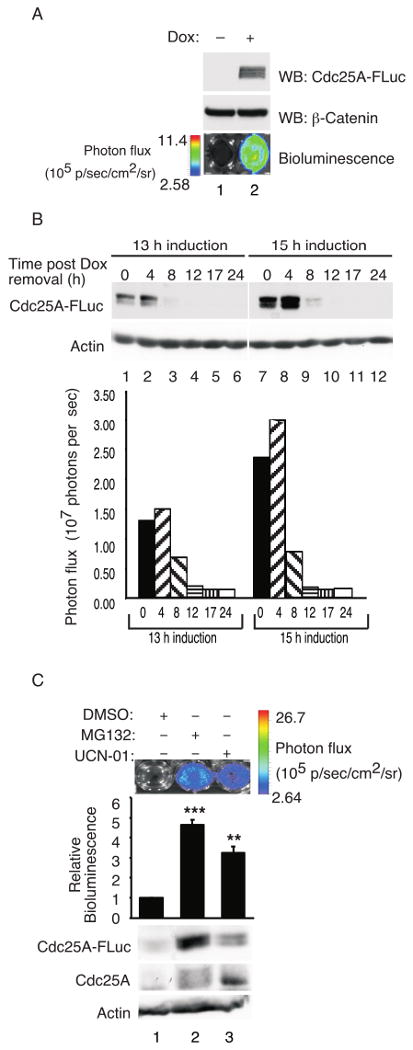
(A) HeLa Tet-on Cdc25A-FLuc cells were cultured in the presence of 2 μg/ml doxycycline (Dox) for 16 h followed by D-Luciferin for 10 min. Cells were imaged using a charge-coupled device (CCD) camera-based bioluminescence imaging system (IVIS 100; Xenogen Corp). The color overlay on the images represents the photons/sec/cm2/steradian (p/s/cm2/sr) as indicated by the color scale next to the images. Cells were harvested immediately after imaging and analyzed for the Cdc25A-FLuc reporter protein by Western blotting using a Cdc25A-specific antibody. Lysates were probed for β-Catenin as a loading control. (B) HeLa Tet-on Cdc25A-FLuc cells were incubated in the culture media containing 2 μg/ml Dox for 13 h or 15 h. Cells were then cultured in the absence of Dox for the indicated times and analyzed for Cdc25A-FLuc by Western blotting or for luciferase activity by bioluminescence imaging. Lysates were probed for actin as a loading control. (C) HeLa Tet-on Cdc25A-FLuc cells were cultured in media containing 2 μg/ml Dox. After 16 h the culture media was removed and cells were cultured in Dox-free media containing 10 μg/ml cycloheximide and either MG132 (50 μM) or UCN-01 (500 nM) for 90 min, D-Luciferin was then added and cells were imaged 10 min later. Immediately after imaging, cells were lysed for Western blotting. Quantification of bioluminescence signal plotted as mean ± SEM (n = 3). P-values from Student's t-test are shown when significantly different from control. Asterisks indicate significant p-values (**<0.01; ***<0.001).
Figure 2. CK1α negatively regulates Cdc25A stability in vivo.
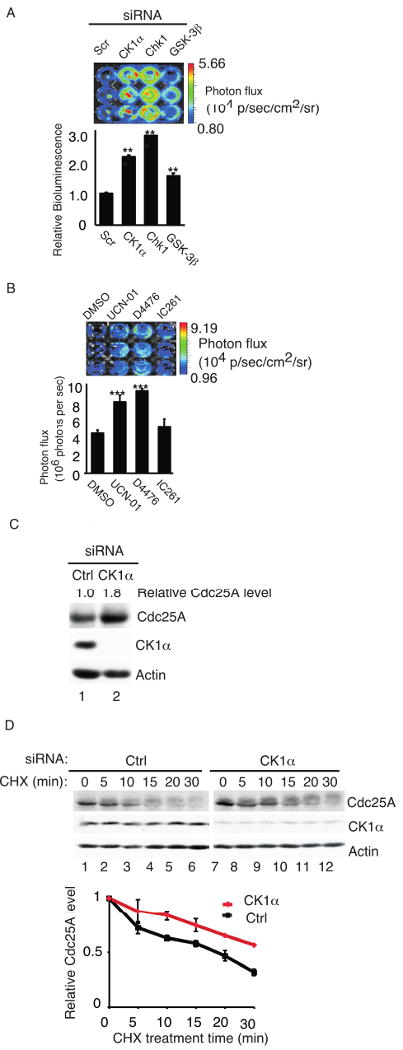
(A) HeLa Tet-on Cdc25A-FLuc cells were mock transfected or transfected with control siRNA (Scr) or siRNAs specific for Chk1, GSK-3β or CK1α for 48 h and then cultured in the presence of 2 μg/ml Dox for an additional 16 h. Cells were incubated with D-Luciferin and imaged 10 min later. Quantification of bioluminescence signal plotted as mean ± SEM (n = 3). P-values from Student's t-test are shown when significantly different from control. Asterisks indicate significant p-values (** <0.01; ***<0.001). (B) HeLa Tet-on Cdc25A-FLuc cells were cultured in media containing 2 μg/ml Dox. After 16 h the culture media was removed and cells were cultured in Dox-free media containing 10 μg/ml cycloheximide and either DMSO, UCN-01 (500 nM), D4476 (75 μM) or IC261 (50 μM) for 1 h, D-Luciferin was then added and cells were imaged 10 min later. Quantification of bioluminescence signal was plotted as mean ± SEM (n = 3). P-values from Student's t-test are shown when significantly different from control. Asterisks indicate significant p-values (***<0.001). The color overlay on the images represents the photons/sec/cm2/steradian (p/s/cm2/sr) as indicated by the color scale next to the images. (C) HeLa cells were transfected with control siRNA (Ctrl) or siRNA targeting CK1α for 48 h followed by Western blotting for the indicated proteins. (D) HeLa cells were transfected with control- (Ctrl) or CK1α-siRNA for 48 h followed by cycloheximide (CHX, 10 μg/ml) for the indicated times. Proteins were resolved by SDS-PAGE and analyzed by Western blotting. A representative Western blot is shown and quantification from 3 independent experiments is shown graphically. Standard error of the mean is shown as error bars along the y-axis.
Therefore, we tested CK1 inhibitors for their ability to stabilize Cdc25A-FLuc protein. D4476 was chosen because it is a more potent and selective CK1 inhibitor than either IC261 or CKI-7 (Bain et al., 2007; Rena et al., 2004) and IC261 was also tested. IC261 shows selectivity for CK1δ and CK1ε over CK1α (Behrend et al., 2000). As seen in Fig. 2B, enhanced bioluminescence indicative of Cdc25A-FLuc stabilization was observed in D4476-treated cells but not in IC261-treated cells. These results suggest that CK1 family members other than CK1δ and CK1ε may function to negatively regulate Cdc25A stability in vivo. To test this hypothesis more directly, levels of endogenous Cdc25A were monitored in CK1α-depleted cells. As seen in Fig. 2C, an approximate 2-fold increase in endogenous Cdc25A protein was observed in CK1α-depleted cells. In addition, the half-life of Cdc25A was increased in cells knocked down for CK1α (Fig. 2D). In contrast, endogenous Cdc25A was not stabilized in cells depleted for CK1δ, ε or γ1 (Fig. S3).
CK1 phosphorylates Cdc25A on serine 82 in vitro
A domain bordered by amino acids 76 to 88 has been shown to regulate the ubiquitin-mediated proteolysis of Cdc25A during interphase and to contain a novel β-TrCP recognition motif or phosphodegron (Fig. 3A) (Busino et al., 2003; Jin et al., 2003). The novel phosphodegron in Cdc25A utilizes pS79 and pS82, rather than pS82 and pS88, for β-TrCP binding (Jin et al., 2003). Six potential phosphorylation sites reside within this 14 amino acid domain. Cdc25A has been shown to be phosphorylated on S76 and T80 and phosphorylation of S79, S82 and S88 have been inferred based on mutagenesis studies and phosphopeptide competition experiments (Goloudina et al., 2003; Hassepass et al., 2003; Jin et al., 2003; Kang et al., 2008). Antibodies specific for known and predicted phosphorylation sites were generated (Fig. 3B) and used to demonstrate that Cdc25A is phosphorylated on S79, S82 and S88 in addition to S76 and T80 (Fig. 3C). Specificity was verified by testing each phospho-specific antibody for its ability to recognize WT Cdc25A but not the corresponding alanine-mutant protein by Western blotting (Fig. 3C). It was also determined that neither S79 phosphorylation nor substitution of alanine for serine at position 79 interfered with the ability of the phospho-S82 antibody to recognize Cdc25A when it is phosphorylated on S82 (Fig. S1) and neither S76 phosphorylation nor substitution of alanine for serine at position 76 interfered with the ability of the phospho-S79 antibody to recognize Cdc25A when it is phosphorylated on S79 (Fig. S2).
Figure 3. Phosphorylation of Cdc25A on residues surrounding and inclusive of the β-TrCP binding domain.
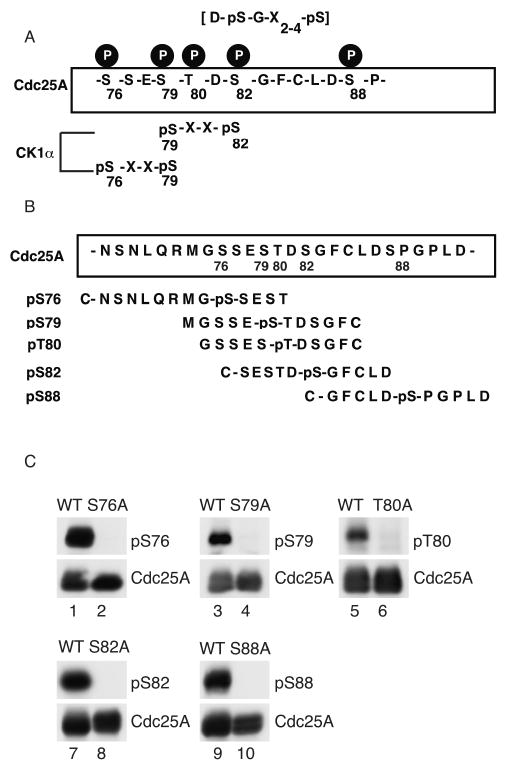
(A) Schematic representation of amino acids 76-89 of Cdc25A illustrating phosphorylated residues, CK1 consensus phosphorylation motifs and the phosphodegron (DSG) motif. (B) Peptides used to make phospho-specific antibodies. (C) Cells expressing WT and mutant forms of Cdc25A were lysed and analyzed by Western blotting with phospho-Cdc25A anti body.
Kinase assays were performed to determine if CK1 is able to directly phosphorylate Cdc25A on S82 in vitro (Fig. 4A). Wild-type Cdc25A and phosphorylation-site mutants were purified as GST fusion proteins from bacteria and incubated with purifed CK1. Phosphorylation of S82 was monitored by Western blotting with the phospho-S82 antibody. As seen in Fig. 4A, CK1 phosphorylated Cdc25A on S82 (lane 1) and substitution of alanine for serine at position 88 did not effect S82 phosphorylation (lane 3).
Figure 4. CK1α regulates S82 phosphorylation in vivo.
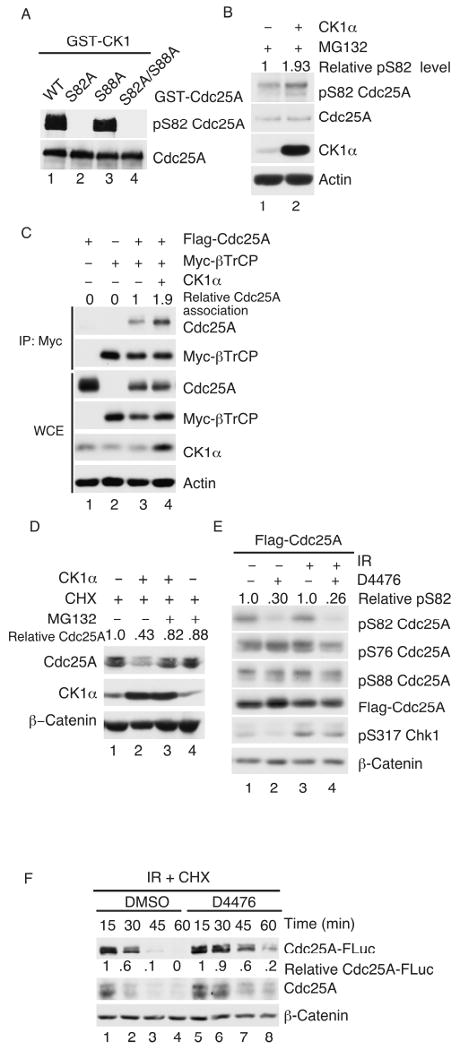
(A) WT and mutant forms of Cdc25A were purified as GST fusion proteins from bacteria and kinase assays were performed in vitro in the presence of CK1. Reaction products were resolved by SDS-PAGE and analyzed by Western blotting for the indicated proteins. (B) HeLa cells transfected with plasmids encoding either CK1α or V5-LacZ for 20 h were treated with 50 μM MG132 for 4 h, lysed and analyzed by Western blotting. (C) MDA-MB-231 cells transfected with plasmids encoding tagged forms of Cdc25A and β-TrCP and untagged CK1α for 20 h were lysed and resolved directly by SDS-PAGE (WCE) or were incubated first with Myc agarose to precipitate β-TrCP. Precipitates were then resolved by SDS-PAGE followed by Western blotting. (D) HeLa cells transfected with plasmids encoding V5-LacZ (lane 1) or CK1α (lane 2) for 20 h were treated with 10 μg/ml cycloheximide (CHX) for 15 min in the presence or absence of MG132 (50 μM). Lysates were prepared and analyzed for the indicated proteins by Western blotting. (E) HeLa cells transfected with plasmids encoding Flag-Cdc25A for 20 h were mock-irradiated or exposed to 10 Gy IR followed by D4476 (75 μM) for 30 min. Lysates were prepared and analyzed by Western blotting. (F) HeLa Tet-on Cdc25A-FLuc cells were cultured in media containing 2 μg/ml Dox for 16 h. Cells were then placed in Dox-free media and exposed to 10 Gy IR in the presence of 10 μg/ml cycloheximide together with either DMSO or D4476 (75 μM). Lysates were prepared at the indicated times after IR and were analyzed by Western blotting for the indicated proteins.
CK1 regulates phosphorylation of Cdc25A on serine 82 in vivo
If CK1α phosphorylates Cdc25A on S82 in vivo, changes in level or activity of CK1α should affect S82 phosphorylation. Indeed, enhanced S82 phosphorylation of endogenous (Fig. 4B) and ectopic (Fig. S4A) Cdc25A was observed in cells overproducing CK1α. Overproduction of CK1α did not enhance phosphorylation of Cdc25A on S76, T80, S88 or S123 (data not shown). Reduced S82 phosphorylation was detected in cells depleted for CK1α (Fig. S4B). Similarly, inhibition of CK1 kinase activity also decreased S82 phosphorylation of Cdc25A (Fig. S4C). Whereas 50 μM D4476 was sufficient to reduce S82 phosphorylation in vivo, concentrations of IC261 as high as 100 μM had minimal effects on S82 phosphorylation. Conversely, S82 phosphorylation was not affected in cells treated with either TBB (CK2 inhibitor) or KN-93 (CaMKII inhibitor) (Fig. S4D).
S82 phosphorylation has been shown to mediate β-TrCP binding, which in turn, promotes ubiquitin-mediated proteolysis of Cdc25A (Busino et al., 2003; Jin et al., 2003). Thus, CK1α overproduction was expected to enhance interactions between Cdc25A and β-TrCP. As seen in Fig. 4C, an increase in association between β-TrCP and Cdc25A was observed in CK1α-overexpressing cells comparing lanes 3 and 4. Enhanced β-TrCP binding is predicted to promote Cdc25A turnover and indeed, lower levels of Cdc25A were also observed in CK1α-overproducing cells (Fig. 4D) and CK1α knockdown increased the half-life of Cdc25A (Fig. 2D). Taken together, these results provide strong evidence that CK1α directly regulates phosphorylation of Cdc25A on S82 and promotes β-TrCP binding in vivo.
CK1 regulates S82 phosphorylation following exposure to genotoxic stress
Ionizing radiation has been shown to increase the phosphorylation of Cdc25A on S82 (Busino et al., 2003), which in turn promotes Cdc25A proteolysis. We asked if inhibition of CK1 would reduce S82 phosphorylation and extend the half-life of Cdc25A in irradiated cells. As seen in Fig. 4E, S82 phosphorylation of Cdc25A was significantly reduced after D4476 treatment in both mock-irradiated (lane 2) and irradiated (lane 4) cells. D4476 did not affect levels of S76 or S88 phosphorylation under either condition. These experiments were conducted in the absence of cycloheximide so levels of ectopic Cdc25A did not change significantly upon irradiation. In the presence of cycloheximide, D4476 was observed to extend the half-life of both endogenous and ectopic Cdc25A (Cdc25A-FLuc) in irradiated cells (Fig. 4F). These results indicate that CK1 regulates S82 phosphorylation in normal cycling cells and in cells experiencing genotoxic stress.
CK1 phosphorylates S79 to prime for subsequent phosphorylation of S82
CK1, like GSK-3β, requires a priming phosphate in its substrates, which is introduced by a priming kinase. CK1 often phosphorylates the serine residue (underlined) in the motif pS-X-X-S where pS is phosphoserine (Flotow et al., 1990). Phosphorylation of S79 could serve as a priming phosphorylation site for S82 phosphorylation and we determined that S79 is phosphorylated in vivo (Fig. 3C). Interestingly, sequences inclusive of and surrounding S79 also conform to one of the CK1 phosphorylation consensus motifs (Fig. 3A). Thus, kinase assays were performed to determine if CK1α was also capable of phosphorylating S79. As seen in Fig. 5A, CK1α phosphorylated Cdc25A on both S79 and S82 by not S88 in vitro. Importantly, CK1α was unable to phosphorylate S82 when serine 79 was changed to alanine.
Figure 5. CK1α regulates S79 phosphorylation.
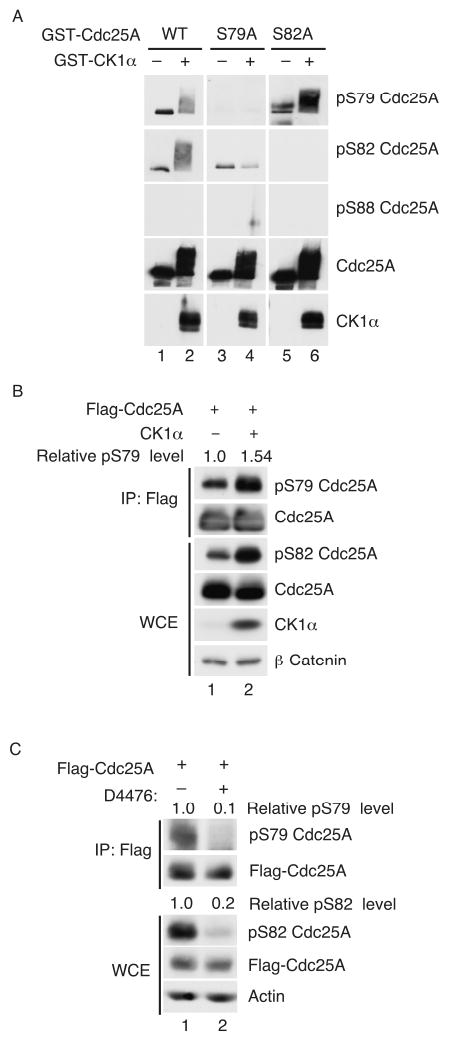
(A) WT and mutant forms of Cdc25A were purified as GST fusion proteins from bacteria and kinase assays were performed in vitro in the presence of CK1. Reaction products were resolved by SDS-PAGE and analyzed by Western blotting with the indicated antibodies. (B) Cells transfected with plasmids encoding Flag-Cdc25A and either V5-LacZ (lane 1) or CK1α (lane 2) for 20 h were lysed directly or were first incubated with Flag agarose to precipitate Flag-Cdc25A. Lysates and precipitates were resolved by SDS-PAGE and subjected to Western blotting. (C) Cells transfected with plasmids encoding Flag-Cdc25A for 20 h were treated in the absence or presence of D4476 (75 μM) for 1 h. Lysates were prepared and subjected to Western blotting.
To determine if CK1α also regulates S79 phosphorylation in vivo, CK1α was overproduced with Flag-Cdc25A and as seen in Fig. 5B, enhanced phospho-S79 levels was observed in the presence of ectopic CK1α. In addition, D4476-treatment resulted in reduced S79 phosphorylation in vivo (Fig. 5C). These results indicate that CK1α phosphorylates Cdc25A on both S79 and S82 in vivo. Taken together, our data is supportive of a model whereby S76 phosphorylation by either Chk1 or GSK-3β primes Cdc25A for phosphorylation on S79 by CK1α and this, in turn, primes Cdc25A for S82 phosphorylation also by CK1α.
Phosphorylation-dependencies within phosphodegron of Cdc25A
Phospho-specific antibodies and Cdc25A mutant proteins were used to determine phosphorylation-dependencies within the phosphodegron of Cdc25A. Phosphorylation of S76 did not require prior phosphorylation of S79, T80, S82 or S88 (Fig. 6A) and phosphorylation of S88 was independent of all other phosphorylation sites, including S76 (Fig. 6B). Phosphorylation of S82 was dependent on S76 (Fig. 6C, lane 2) and S79 (lane 3) but did not require S88 (lane 6). As seen in Fig. 6D, phosphorylation of S79 was dependent on S76 (lane 2) but did not require S82 (lane 5) or S88 (lane 6).
Figure 6. Phosphorylation dependencies that target Cdc25A to β-TrCP.
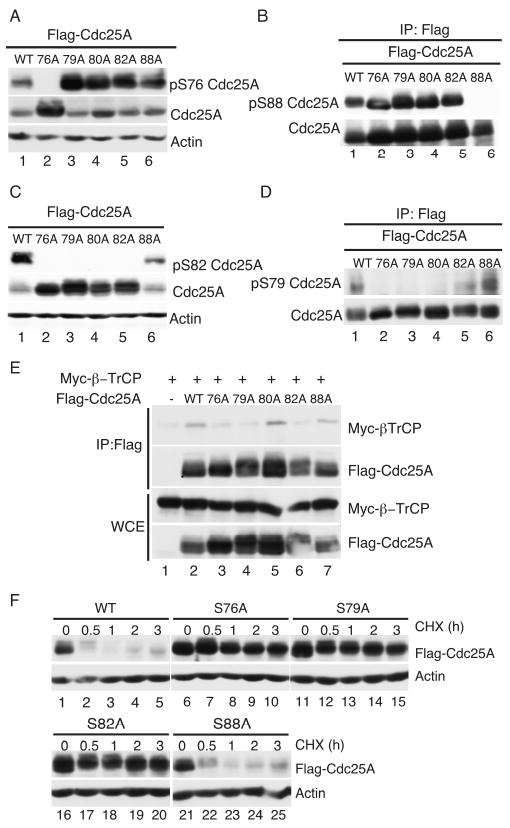
Cells transfected with plasmids expressing WT and mutant forms of Cdc25A for 20 h were lysed and lysates were resolved by SDS-PAGE. Western blotting was performed to monitor S76- (A), S88- (B), S82- (C) and S79 (D)-phosphorylation. (E) HeLa cells transfected with plasmids encoding tagged forms of Cdc25A and β-TrCP for 20 h were lysed and resolved directly by SDS-PAGE (WCE) or were incubated first with Flag agarose to precipitate Cdc25A. Precipitates were then resolved by SDS-PAGE followed by Western blotting. (F) Cells expressing WT and mutant forms of Cdc25A were either lysed immediately (time = 0) or were incubated in the presence of 10 μg/ml cycloheximide (CHX) for the indicated times prior to processing. Lysates were resolved by SDS-PAGE and subjected to Western blotting.
The Cdc25A phosphorylation-site mutants were also used to determine which residues are required for β-TrCP binding. As seen in Fig. 6E, point mutants of S76A (lane 3), S79A (lane 4) or S82A (lane 6) but not T80A (lane 5) or S88A (lane 7) were severely impaired in their ability to bind to β-TrCP. Like the S76A mutant (lanes 6-10), the S79A (lanes 11-15) and S82A (lanes 16-20) mutants, but not the S88A (lanes 21-25) mutant protein, exhibited an extended half-life in vivo (Fig. 6F). These results demonstrate that serines 76, 79 and 82 are the key regulatory residues that mediate recognition of Cdc25A by β-TrCP.
Discussion
Orderly progression through the mammalian cell division cycle requires that the abundance of the Cdc25A protein phosphatase be tightly controlled. This is accomplished by two distinct E3 ligase complexes that target Cdc25A for ubiquitin-mediated proteolysis at distinct phases of the cell division cycle. SCFβ-TrCP operates throughout interphase to regulate Cdc25A abundance whereas APCCdh1 operates during mitotic exit to facilitate Cdc25A degradation (Busino et al., 2004; Donzelli et al., 2002; Jin et al., 2003). Binding of β-TrCP to its target substrates typically requires phosphorylation to create a phosphodegron recognition motif (Nash et al., 2001). Although S76 phosphorylation is required for Cdc25A proteolysis, it does not participate directly in β-TrCP binding (Busino et al., 2003; Jin et al., 2003). In contrast, phosphorylation of S82 has been shown to be a key mediator of β-TrCP binding (Busino et al., 2003; Jin et al., 2003). Here, CK1 is shown to catalyze the phosphorylation of S79 and S82 and to facilitate the docking of β-TrCP to Cdc25A in vivo. This conclusion is based on the following observations: pharmacological inhibition of the CK1 family or siRNA knockdown of CK1α in particular resulted in reduced S82 phosphorylation and enhanced Cdc25A stabilization in vivo; overproduction of CK1α resulted in enhanced Cdc25A phosphorylation on S79 and S82 and enhanced β-TrCP binding in vivo, and CK1 directly phosphorylated Cdc25A on both S79 and S82 in vitro. Human Cdc25A contains two motifs that regulate its interactions with β-TrCP (pS79-T-D-pS82-G and D215DGFVD220). These motifs are conserved in several higher eukaryotes (Fig. S5), with the notable exception of Xenopus Cdc25A, which exclusively utilizes the DDGΦXD motif (Kanemori et al., 2005).
Paradoxically, CK1 was previously ruled out as a Cdc25A regulatory kinase (Jin et al., 2003). The CK1 inhibitors used in that study (IC261 and CK1-7) are not as potent and selective as D4476 (used in our study). In addition, IC261 shows selectivity for the δ and ε isoforms rather than the α isoform and CK1-7 poorly penetrates cell membranes (Knippschild et al., 2005). These properties likely account for CK1 being ruled out as a Cdc25A regulator. The CK1 family contains seven family members (α, β, γ1, γ2, γ3, δ, ε) that share similar substrate specificity in vitro. Substrate selection in vivo is regulated, in part, through subcellular localization and the presence of CK1 docking sites within specific substrates (Gross and Anderson, 1998). Our data indicates that CK1α contributes significantly to S82 phosphorylation in vivo. Given that IC261 did not effect either S82 phosphorylation or Cdc25A stabilization at concentrations as high as 100 μM indicates that CK1δ and CK1ε do not significantly contribute to S82 phosphorylation in vivo. Furthermore, knockdown of CK1δ, CK1ε or CK1γ1 did not stabilize endogenous Cdc25A (Fig. S3).
Cdc25A is also phosphorylated on S88 (Fig. 3C and 6B) and there has been conflicting reports regarding the contribution made by S88 phosphorylation to interactions between β-TrCP and Cdc25A (Busino et al., 2003; Jin et al., 2003). We demonstrate that S88 phosphorylation does not rely on previous phosphorylation of S76, T80, S79 or S82 and that phosphorylation of these residues does not require S88 phosphorylation (Fig. 6A-D). Furthermore, in contrast to mutation of S76, S79 or S82, mutation of S88 does not impair β-TrCP binding to Cdc25A (Fig. 6E) nor alter the half-life of Cdc25A in vivo (Fig. 6F). The kinase(s) that phosphorylates Cdc25A on S88 has not been identified. We observed that treatment of cells with SB212190 (p38 inhibitor) or U0126 (MEK1/2) inhibitor but not LiCl (GSK-3β inhibitor) or roscovitine (Cdk inhibitor) reduced phosphorylation of Cdc25A on S88 (data not shown). This suggests that the mitogen-activated protein kinase (MAPK) signaling pathways may regulate S88 phosphorylation in vivo.
Our data is supportive of a model whereby S76 phosphorylation by either Chk1 or GSK-3β primes Cdc25A for subsequent phosphorylation on S79 by CK1 and this, in turn, primes Cdc25A for S82 phosphorylation also by CK1. This conclusion is based on the phosphorylation-dependencies observed with various Cdc25A mutant proteins in vivo (Fig. 6) and phosphorylation assays performed in vitro with purified proteins (Fig. 4A and 5A). In the case of GSK-3β phosphorylation of S76, prior phosphorylation of T80 is required and this can be catalyzed by Plk3 (Kang et al., 2008). Hierarchical phosphorylation of S76 (+/-T80), S79 and S82 facilitates the binding of β-TrCP to Cdc25A to facilitate the ubiquitin-mediated destruction of Cdc25A (Fig. 7).
Figure 7. Hierarchical phosphorylation regulates destruction of Cdc25A during interphase and in response to genotoxic stress.
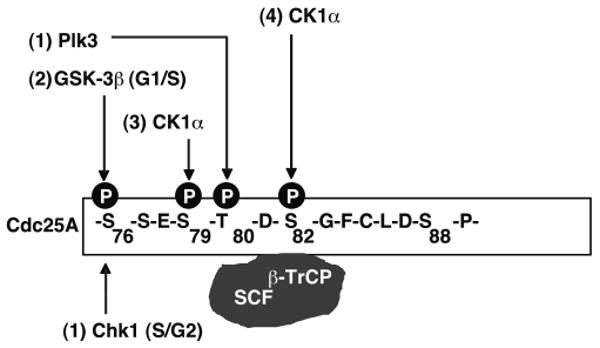
S76 phosphorylation by either Chk1 in the S/G2-phases of the cell cycle or by GSK-3β in the G1/S-phases primes Cdc25A for subsequent phosphorylation on S79 by CK1 and this, in turn, primes Cdc25A for S82 phosphorylation also by CK1. In the case of GSK-3β phosphorylation of S76, prior phosphorylation of S80 is required and this can be catalyzed by Plk3. Hierarchical phosphorylation of S76 (+/-T80), S79 and S82 enables binding of β-TrCP to Cdc25A to facilitate its ubiquitin-mediated proteolysis during interphase and in response to genotoxic stress. Inactivation of GSK-3β in various cancer cells through constitutive activation of the PI3K- or MAPK-pathways accounts for Cdc25A overproduction in a subset of tumors (Cohen and Frame, 2001; Doble and Woodgett, 2003; Zhou et al., 2004; Kang et al., 2008). One advantage of using multiple phosphorylation events, multiple kinases and multiple E3 ligases to regulate Cdc25A may be to guarantee a flexible regulation of its levels in order to impose different thresholds of Cdk activity in response to both the external and internal environment.
This study identifies CK1 (CK1α) as the long sought-after protein kinase that creates a Cdc25A phosphodegron to enable β-TrCP to bind to and facilitate Cdc25A destruction during interphase and in response to genotoxic stress. In general, the CK1 kinases are constitutively active. Thus, the requirement for a priming phosphorylation by another kinase imposes restrictions on the ability of CK1 to facilitate the destruction of Cdc25A and thereby impact cell cycle progression. One advantage of using multiple phosphorylation events, multiple kinases and multiple E3 ligases to regulate Cdc25A may be to guarantee a flexible regulation of its levels in order to impose different thresholds of Cdk activity in response to both the external and internal environment. The fact that Cdc25A is primed by at least two kinases (one of which also requires substrate priming) and that the priming kinases are regulated by distinct signaling pathways, allows diverse cues to feed into the Cdc25A regulatory pathway to control cell division. For example, priming of Cdc25A by GSK-3β enables regulation of the cell division cycle by extrinsic signals that impact the PI3K- and MAPK-pathways whereas, priming of Cdc25A by Chk1 facilitates regulation of the cell division cycle in the S- and G2-phases by intrinsic checkpoint pathways that respond to replication stress and DNA damage. Whether CK1 activity is also regulated in this pathway remains to be determined. A recent report indicated that NEK11 phosphorylates Cdc25A on S79, S82 and S88 and regulates Cdc25A degradation during S and G2 phases (Melixetian et al., 2009). Paradoxically, S82 does not reside within a consensus motif for NEK11 phosphorylation and we have been unable to detect S82 phosphorylation when full length NEK11 is incubated with full length Cdc25A in vitro under conditions where MBP is robustly phosphorylated by NEK11 (data not shown). Contrary to our findings and those reported previously (Donzelli et al., 2004), Melixetian et al. (2009) did not observe an absolute dependency of S82 phosphorylation by NEK11 in vitro on prior S76 phosphorylation. Future experiments are required to resolve these discrepancies.
Materials and Methods
Cell lines and reagents
HeLa cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Invitrogen) supplemented with 10% bovine growth serum (BGS, Hyclone), 1 mM glutamine, and 100 U/ml each of penicillin and streptomycin. HeLa Tet-on cells (BD Biosciences-Clontech) were cultured in DMEM supplemented with 10% Tet system approved FBS (Clontech), 1 mM glutamine, 100 U/ml each of penicillin and streptomycin, and Geneticin (Clontech, 100 μg/ml). HeLa Tet-on Cdc25A-FLuc stable cells were cultured in the above media plus hygromycin B (200 μg/ml, Invitrogen). MDA-MB-231 cells were cultured in DMEM supplemented with 10% BGS, 1mM glutamine, 1mM sodium pyruvate, and 100 U/ml penicillin and streptomycin. MG132 and cycloheximide (Sigma Chemical Co.) were dissolved in DMSO and 1× PBS respectively. Kinase inhibitors used in this study include D4476, IC261, Gö6976, TBB and KN-93 (Calbiochem), UCN-01 (Sigma Chemical Co.) and AZD7762 (Zabludoff et al., 2008) (Axon Medchem BV). D4476 was prepared as described (Rena et al., 2004).
Antibodies
Monoclonal anti-Cdc25A antibodies (Ab-3, Neomarkers, and Ab2357, Abcam) were used to detect human Cdc25A. Other antibodies used in this study include: anti-actin (Sigma Chemical Co), anti-Myc (A-14 and 9E-10, Santa Cruz Biotechnology), anti-β-Catenin (BD Transduction), anti-CK1α (C-19, Santa Cruz Biotechnology), anti-phospho-S45 β-Catenin (Biosource), anti-Chk1 (G4, Santa Cruz Biotechnology), and anti-pS317 Chk1 (Cell Signaling Technology). Antibodies specific for Cdc25A phosphorylated on T80 and S123 were generated by immunizing rabbits with the coupled phosphopeptides C-GSSES-pT-DSGFC and C-LKRSH-pS-DSLD, respectively (Kang et al., 2008). Antibodies specific for Cdc25A phosphorylated on S76 (C-NSNLQRMG-pS-SEST), S79 (MGSSE-pS-TDSGFC), S82 (C-SESTD-pS-GFCLD) and S88 (C-GFCLD-pS-PGPLD) were generated by immunizing rabbits with the indicated phosphopeptides followed by affinity purification (Abgent). Bound primary antibodies were reacted with horseradish peroxidase (HRP)-conjugated goat anti-mouse antibody (Jackson), HRP-goat-anti-rabbit antibody (Zymed), or HRP-rabbit anti-goat antibody (Zymed) and detected by ECL (GE Healthcare).
Peptide competition
Cell lysates prepared from control cells and cells overproducing Flag-Cdc25A(WT) were resolved by SDS-PAGE and transferred to nitrocellulose following by incubation in Blocking buffer (5% milk/1× TBST). HPLC purified peptides (Tufts University Core Facility, Boston, MA) were incubated with 2 μg/ml of phospho-antibodies at concentrations between 4 -40 μg/ml in 1× TBST overnight at 4°C. Western blotting was then performed.
Expression plasmids
Generation of pcDNA3-Myc-Cdc25A-FLuc: the stop codon of Cdc25A was eliminated and an Xba I site was inserted at the C-terminus of Cdc25A. The resulting plasmid pcDNA3-Myc-Cdc25A (Xba I) was digested with Xba I and Xho I and ligated to sequences encoding codon-optimized firefly luciferase (FLuc) that had been isolated as an Xba I-Xho I fragment from the pGL-3 control plasmid (Promega). Sequences encoding Cdc25A-FLuc but lacking the Myc-tag were isolated from pcDNA3-Myc-Cdc25A-FLuc and subcloned into pTRE-Tight, a tetracycline-regulated expression plasmid (Clontech) to generate pTRE-Tight-Cdc25A-FLuc. Cdc25A serine-to-alanine mutants were generated using the QuikChange XL site-directed mutagenesis kit (Stratagene) and all the mutations were verified by DNA sequencing. Plasmids encoding CK1α and Myc-β-TrCP1 were provided by Dr. Jiandong Chen (Chen et al., 2005) and Dr. Binhua P. Zhou (Zhou et al, 2004), respectively.
Generation of HeLa Tet-on Cdc25A-FLuc stable cell lines and bioluminescence imaging
pTRE-Tight-Cdc25A-FLuc was co-transfected with a selection plasmid pTK-Hyg (Clontech) into HeLa Tet-on cells using Lipofectamine 2000 (Invitrogen). Twenty-four hours later, cells were trypsinized and seeded into 10-cm tissue culture dishes. Selection media containing hygromycin B was added 24 h later. Media was changed every 3 days until the drug-resistant colonies became visible. Cells were incubated with D-Luciferin (150 μg/ml in PBS, BIOSYNTH International) at 37°C for 10 min followed by imaging using an IVIS 100 imaging system (Xenogen). Luciferase activity (photons/sec/cm2/steradian) was quantitated using Living Image Software (Xenogen). Those colonies that scored positive for bioluminescence were eliminated. Cells were then rinsed in PBS and incubated in culture media containing 2 μg/ml of doxycycline (Dox, Clontech). Twenty-four hours later, D-Luciferin was added to the media and colonies were re-imaged. Colonies that displayed Dox-inducible bioluminescence were isolated and expanded for further analysis. The stable cell line was maintained in selection medium containing hygromycin B and geneticin.
Kinase assays
Wild-type and mutant forms of Cdc25A were produced in bacteria as GST-fusion proteins and purified as described previously (Chen et al., 2003). The elution of GST-Cdc25A proteins was performed as described (Kang et al., 2008). 100 ng CK1 (GST-CK1α or GST-CK1δ from Sigma Chemical Co.) was diluted in kinase reaction buffer (50 mM Tris-HCl, pH 7.4, 2 mM DTT, 10 mM MgCl2 and 120 μM ATP) followed by the addition of 1 μg soluble GST-Cdc25A. Reactions were incubated at 30°C for 30 min, boiled in 1× loading buffer at 95°C for 10 min and then resolved by SDS-PAGE followed by Western blotting.
Transfection
Approximately 1 × 106 HeLa cells were seeded in p60 plates 16-20 h before transfection. Cells at 95% confluence were transfected with 2 to 4 μg plasmids using Lipofectamine 2000. 24 h after transfection, cells were harvested or subjected to the indicated treatments prior to harvest and analysis. For RNAi transfection, HeLa cells (3× 105) were seeded in p60 dishes 20 h before transfection. Cells were transfected with 50 nM CK1α, GSK-3β (Dharmacon) or Chk1 siRNA (Zhao et al., 2002) when they reached 20-30% confluency using DharmaFECT 1 or 3 (Dharmacon). Forty-eight hours after transfection, cells were treated as indicated and then harvested for Western blotting. Scrambled siRNA or luciferase siRNA (GL3, Dharmacon) was used as transfection controls. For half-life experiments, siRNA-transfected cells were incubated with 10 μg/ml cycloheximide 48 h after siRNA-transfection. Cells were harvested at various times after cycloheximide treatment and analyzed by Western blotting. MG132 additions were made 48 h after siRNA transfection and cells were harvested 2 h later.
Cell lysis and analysis
Cells were lysed in mammalian cell lysis buffer (MCLB) [50 mM Tris-HCl pH 8.0, 2 mM DTT, 5 mM EDTA, 0.5% NP-40, 100 mM NaCl, 1 mM microcystin (Sigma Chemical Co), 1 mM sodium orthovanadate, 2 mM PMSF] supplemented with protease inhibitor cocktail (Sigma Chemical Co.) and phosphatase inhibitor cocktail (Calbiochem). Clarified lysates were resolved directly by SDS-PAGE and transferred to nitrocellulose membranes for Western blotting. Alternatively, cell lysates containing 0.5 to 1 mg of total cell protein were pre-cleared with protein A beads (Santa Cruz Biotechnology) for 1 h at 4°C. Pre-cleared lysates were incubated with anti-Flag agarose (Sigma Chemical Co.) or anti-Myc agarose (9E-10, Santa Cruz Biotechnology) for 1 h at 4°C. Immunoprecipitates were then washed 6 times with MCLB. Proteins were visualized using the ECL reagent (GE Healthcare Life Sciences), and densitometry analysis was performed using ImageJ software (Abramoff et al., 2004).
Supplementary Material
Acknowledgments
Drs. Van Leung-Pineda and Tiebang Kang are thanked for helpful suggestions. Chris Ryan is thanked for editorial assistance. We acknowledge Chris Ryan, Yonghao Hou, Mei-Shya Chen, Dr. Jiandong Chen, and Dr. Binhua P. Zhou for providing expression plasmids. This work was supported by grants from the National Institutes of Health (GM047017 and P50 CA94056). We thank the p50 Molecular Imaging Center and the Alvin J. Siteman Cancer Center at Washington University School of Medicine for the use of the High Throughput Core. The Siteman Cancer Center is supported in part by an NCI Cancer Center Support Grant #P30 CA91842. H.P.-W. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Conflict of interest: The authors declare no conflict of interest.
References
- Abramoff MD, Magelhaes PJ, Ram SJ. Image processing with Image J. Biophotonics International. 2004;11:36–42. [Google Scholar]
- Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, et al. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartek J, Lukas J. Pathways governing G1/S transition and their response to DNA damage. FEBS Lett. 2001;490:117–122. doi: 10.1016/s0014-5793(01)02114-7. [DOI] [PubMed] [Google Scholar]
- Behrend L, Milne DM, Stoter M, Deppert W, Campbell LE, Meek DW, et al. IC261, a specific inhibitor of the protein kinases casein kinase 1-delta and -epsilon, triggers the mitotic checkpoint and induces p53-dependent postmitotic effects. Oncogene. 2000;19:5303–13. doi: 10.1038/sj.onc.1203939. [DOI] [PubMed] [Google Scholar]
- Bernardi R, Lieberman DA, Hoffman B. Cdc25A stability is controlled by the ubiquitin-proteasome pathway during cell cycle progression and terminal differentiation. Oncogene. 2000;19:2447–2454. doi: 10.1038/sj.onc.1203564. [DOI] [PubMed] [Google Scholar]
- Blomberg I, Hoffman I. Ectopic expression of Cdc25A accelerates the G1/S transition and leads to premature activation of cyclin E-and cyclin A-dependent kinases. Mol Cell Biol. 1999;19:6183–6194. doi: 10.1128/mcb.19.9.6183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutros R, Dozier C, Ducommun B. The when and wheres of CDC25 phosphatases. Curr Opin Cell Biol. 2006;18:185–91. doi: 10.1016/j.ceb.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Busino L, Chiesa M, Draetta GF, Donzelli M. Cdc25A phosphatase: combinatorial phosphorylation, ubiquitylation and proteolysis. Oncogene. 2004;23:2050–6. doi: 10.1038/sj.onc.1207394. [DOI] [PubMed] [Google Scholar]
- Busino L, Donzelli M, Chiesa M, Guardavaccaro D, Ganoth D, Dorrello NV, et al. Degradation of Cdc25A by beta-TrCP during S phase and in response to DNA damage. Nature. 2003;426:87–91. doi: 10.1038/nature02082. [DOI] [PubMed] [Google Scholar]
- Chen L, Li C, Pan Y, Chen J. Regulation of p53-MDMX interaction by casein kinase 1 alpha. Mol Cell Biol. 2005;25:6509–20. doi: 10.1128/MCB.25.15.6509-6520.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MS, Ryan CE, Piwnica-Worms H. Chk1 Kinase Negatively Regulates Mitotic Function of Cdc25A Phosphatase through 14-3-3 Binding. Mol Cell Biol. 2003;23:7488–7497. doi: 10.1128/MCB.23.21.7488-7497.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P, Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol. 2001;2:769–76. doi: 10.1038/35096075. [DOI] [PubMed] [Google Scholar]
- Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–86. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donzelli M, Busino L, Chiesa M, Ganoth D, Hershko A, Draetta GF. Hierarchical order of phosphorylation events commits Cdc25A to betaTrCP-dependent degradation. Cell Cycle. 2004;3:469–71. [PubMed] [Google Scholar]
- Donzelli M, Squatrito M, Ganoth D, Hershko A, Pagano M, Draetta GF. Dual mode of degradation of Cdc25 A phosphatase. Embo J. 2002;21:4875–84. doi: 10.1093/emboj/cdf491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falck J, Mailand N, Syljuasen RG, Bartek J, Lukas J. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature. 2001;410:842–847. doi: 10.1038/35071124. [DOI] [PubMed] [Google Scholar]
- Flotow H, Graves PR, Wang AQ, Fiol CJ, Roeske RW, Roach PJ. Phosphate groups as substrate determinants for casein kinase I action. J Biol Chem. 1990;265:14264–9. [PubMed] [Google Scholar]
- Galaktionov K, Chen X, Beach D. Cdc25 cell-cycle phosphatase as a target of c-myc. Nature. 1996;382:511–517. doi: 10.1038/382511a0. [DOI] [PubMed] [Google Scholar]
- Goloudina A, Yamaguchi H, Chervyakova DB, Appella E, Fornace AJ, Jr, Bulavin DV. Regulation of human Cdc25A stability by serine 75 phosphorylation is not sufficient to activate a S-phase checkpoint. Cell Cycle. 2003;2:473–478. [PubMed] [Google Scholar]
- Gross SD, Anderson RA. Casein kinase I: spatial organization and positioning of a multifunctional protein kinase family. Cell Signal. 1998;10:699–711. doi: 10.1016/s0898-6568(98)00042-4. [DOI] [PubMed] [Google Scholar]
- Hassepass I, Voit R, Hoffmann I. Phosphorylation at serine-75 is required for UV-mediated degradation of human Cdc25A phosphatase at the S-phase checkpoint. J Biol Chem. 2003;278:29824–29829. doi: 10.1074/jbc.M302704200. [DOI] [PubMed] [Google Scholar]
- Jin J, Ang XL, Ye X, Livingstone M, Harper JW. Differential roles for checkpoint kinases in DNA damage-dependent degradation of the Cdc25A protein phosphatase. J Biol Chem. 2008;283:19322–8. doi: 10.1074/jbc.M802474200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Shirogane T, Xu L, Nalepa G, Qin J, Elledge SJ, et al. SCFbeta-TRCP links Chk1 signaling to degradation of the Cdc25A protein phosphatase. Genes Dev. 2003;17:3062–74. doi: 10.1101/gad.1157503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanemori Y, Uto K, Sagata N. Beta-TrCP recognizes a previously undescribed nonphosphorylated destruction motif in Cdc25A and Cdc25B phosphatases. Proc Natl Acad Sci U S A. 2005;102:6279–84. doi: 10.1073/pnas.0501873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang T, Wei Y, Honaker Y, Yamaguchi H, Appella E, Hung MC, et al. GSK-3beta Targets Cdc25A for Ubiquitin-Mediated Proteolysis, and GSK-3beta Inactivation Correlates with Cdc25A Overproduction in Human Cancers. Cancer Cell. 2008;13:36–47. doi: 10.1016/j.ccr.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knippschild U, Gocht A, Wolff S, Huber N, Lohler J, Stoter M. The casein kinase 1 family: participation in multiple cellular processes in eukaryotes. Cell Signal. 2005;17:675–89. doi: 10.1016/j.cellsig.2004.12.011. [DOI] [PubMed] [Google Scholar]
- Kristjansdottir K, Rudolph J. Cdc25 phosphatases and cancer. Chem Biol. 2004;11:1043–51. doi: 10.1016/j.chembiol.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Loffler H, Syljuasen RG, Bartkova J, Worm J, Lukas J, Bartek J. Distinct modes of deregulation of the proto-oncogenic Cdc25A phosphatase in human breast cancer cell lines. Oncogene. 2003;22:8063–71. doi: 10.1038/sj.onc.1206976. [DOI] [PubMed] [Google Scholar]
- Mailand N, Falck J, Lukas C, Syljuasen RG, Welcker M, Bartek J, et al. Rapid destruction of human Cdc25A in response to DNA damage. Science. 2000;288:1425–1429. doi: 10.1126/science.288.5470.1425. [DOI] [PubMed] [Google Scholar]
- Melixetian M, Klein DK, Sorensen CS, Helin K. NEK11 regulates CDC25A degradation and the IR-induced G2/M checkpoint. Nat Cell Biol. 2009;11:1247–53. doi: 10.1038/ncb1969. [DOI] [PubMed] [Google Scholar]
- Molinari M, Mercurio C, Dominguez J, Goubin F, Draetta GF. Human Cdc25A inactivation in response to S phase inhibition and its role in preventing premature mitosis. EMBO Reports. 2000;1:71–79. doi: 10.1093/embo-reports/kvd018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash P, Tang X, Orlicky S, Chen Q, Gertler FB, Mendenhall MD, et al. Multisite phosphorylation of a CDK inhibitor sets a threshold for the onset of DNA replication. Nature. 2001;414:514–21. doi: 10.1038/35107009. [DOI] [PubMed] [Google Scholar]
- Ray D, Kiyokawa H. CDC25A phosphatase: a rate-limiting oncogene that determines genomic stability. Cancer Res. 2008;68:1251–3. doi: 10.1158/0008-5472.CAN-07-5983. [DOI] [PubMed] [Google Scholar]
- Ray D, Terao Y, Nimbalkar D, Chu LH, Donzelli M, Tsutsui T, et al. Transforming growth factor beta facilitates beta-TrCP-mediated degradation of Cdc25A in a Smad3-dependent manner. Mol Cell Biol. 2005;25:3338–47. doi: 10.1128/MCB.25.8.3338-3347.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rena G, Bain J, Elliott M, Cohen P. D4476, a cell-permeant inhibitor of CK1, suppresses the site-specific phosphorylation and nuclear exclusion of FOXO1a. EMBO Rep. 2004;5:60–5. doi: 10.1038/sj.embor.7400048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigo E, Muller H, Properini E, Hatevoer G, Cartwright P, Moroni MC, et al. CDC25A phosphatase is a target of E2F and is required for efficient E2F-induced S phase. Mol Cell Biol. 1999;19:6379–6395. doi: 10.1128/mcb.19.9.6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabludoff SD, Deng C, Grondine MR, Sheehy AM, Ashwell S, Caleb BL, et al. AZD7762, a novel checkpoint kinase inhibitor, drives checkpoint abrogation and potentiates DNA-targeted therapies. Mol Cancer Ther. 2008;7:2955–66. doi: 10.1158/1535-7163.MCT-08-0492. [DOI] [PubMed] [Google Scholar]
- Zhao H, Watkins JL, Piwnica-Worms H. Disruption of the checkpoint kinase 1/cell division cycle 25A pathway abrogates ionizing radiation-induced S and G2 checkpoints. Proc NatlAcad Sci USA. 2002;99:14795–14800. doi: 10.1073/pnas.182557299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M, et al. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol. 2004;6:931–40. doi: 10.1038/ncb1173. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.