

Abstract
A specific irreversible inhibitor of both cathepsins B and L, Fmoc-Tyr-Ala-CHN2 (FYAD) induced apoptosis of neuroblastoma cells but not other tumor cells. Cysteine protease inhibitors that were not efficient inhibitors of both proteases did not cause death of any cell line tested. Apoptosis was preceded by accumulation of large electron dense vesicles and multivesicular bodies in the cytoplasm. Exposure of cells to the cathepsin D inhibitor, pepstatin, failed to rescue cells from FYAD-induced death. These results indicate that inhibition of cathepsins B and L may provide a unique mechanism for selectively inducing death of neuroblastoma with limited toxicity to normal cells and tissues.
Keywords: cathepsin, neuroblastoma, apoptosis, protease, inhibition
1. Introduction
Neuroblastoma is the major solid tumor found in infants and is a unique cancer of children. Most infants diagnosed with neuroblastoma receive either limited surgery or no treatment, and the disease is monitored until it regresses [1]. Unfortunately if the disease is not detected until the patient is older (above 2 years), the disease often presents as stage 3 or 4 and the prognosis for the patient is poor. These patients are typically treated with more aggressive surgery, radiation and chemotherapy in attempts to eradicate the cancer. However, side effects of general cytotoxic chemotherapy are particularly severe for children, and the success rate for treatment of these patients remains poor. Alternative approaches to treatment of neuroblastoma are clearly needed.
Inhibitors of lysosomal proteases block both the growth and invasive properties of many different tumor cells, including neuroblastoma, and in vivo aggressiveness of numerous tumors correlates with expression of lysosomal proteases [2; 3; 4; 5; 6]. Within the cell, release of lysosomal proteases into the cytosol is proposed to induce apoptosis [7; 8; 9; 10; 11; 12], indicating that cathepsin inhibition might prevent cell death and thus have a negative impact on cancer treatment. Conversely, broad based inhibitors such as E-64 and Z-Phe-Gly-NHO-Bz have been shown to induce apoptosis in a variety of cell types, indicating general cytotoxic effects of these compounds [5; 13].
Peptidyl diazomethylketones have been found to be remarkably specific inhibitors of cathepsins [14]. The diazomethylketone moiety allows covalent modification of the active-site cysteine of cathepsins. When a radio-iodinated form of Z-Tyr-Ala-CHN2 was incubated with live cells, the only reactive proteins identified were cathepsins B and L [15]; other cellular and extracellular proteins were not labeled. Treatment of a range of breast cancer cells with this specific inhibitor of cathepsins B and L was shown to effectively inactivate both enzymes and impair cell division [16; 17]. The inhibitor was shown to be cytostatic but not cytotoxic, and upon removal of inhibitor cells continued to divide. A recent study has shown that i.v. injection of a simple iodinated diazomethylketone inhibitor into mice labels only cathepsins B, L and S in whole tissue extracts [18]. This remarkable specificity and selectivity of the diazomethylketone inhibitors for cathepsins make them ideal tools to define biological functions of cathepsins. Treatment of rodents with inhibitors of cathepsins B and L does not have any significant toxic effects [19; 20], although inhibitors are teratogenic when administered to pregnant animals [21; 22; 23]. Major drawbacks of therapies to treat cancers arise when drug targets also perform critical functional roles in non-cancerous cells, so lack of general toxicity is a desirable feature of cathepsin inhibition. In this study we discovered that neuroblastoma cells are uniquely sensitive to inhibition of both cathepsins B and L, causing apoptotic cell death.
2. Materials and Methods
2.1 Neuroblastoma cell lines
Four different neuroblastoma cell lines were chosen for this study. IMR-32, SK-N-SH and NB-1691 cells were obtained from ATCC (Manassas, VA) and GM11027 cells were from Coriell (Camden, NJ). SK-N-SH cells, representing less aggressive S-type tumors and IMR-32 cells from more aggressive N-type tumors are well-established neuroblastoma cell lines. NB-1691 cells (a gift from Peter Houghton, St Jude's Children's Hospital, Memphis, TN), like IMR-32 cells, are N-Myc amplified aggressive tumor cells. GM11027 is a less well established cell line derived from primary tumor tissue passaged in a nude mouse. These cell lines were chosen to give a wide variety of samples of neuroblastoma cells with different phenotypes.
2.2 Determination of effective targets of protease inhibitors
For clear identification of targets of each of the cathepsin inhibitors, SK-N-SH and IMR-32 cells were incubated with Fmoc-Tyr-Ala-CHN2 (FYAD), Z-Phe-Tyr(OtBu)-CHN2 (ZFYD), Ca074Me or vehicle control for up to 48 h and then incubated with Fmoc-Tyr(I-125)-Ala-CHN2 (1 μM) for an additional 2 h. Cells were then washed in serum-free medium and total proteins harvested by dissolving the cell pellets in SDS/PAGE sample buffer containing 8 M urea. Proteins were separated by SDS/PAGE and then the radioactive bands identified by phosphor image analysis of the dried gels. We have previously found that this live cell labeling technique is particularly valuable for detecting cathepsin L, an enzyme that is rapidly inactivated on cell lysis [24], as the radiolabeled inhibitor covalently binds to all active forms of cathepsins B and L prior to cell lysis. Compounds that inactivate these enzymes in live cells block this binding, providing a clear demonstration of effectiveness in inhibiting these targeted proteases [24].
2.3 Effect of cathepsin inhibitors on cell growth
IMR-32 cells were seeded onto 12 well plates and cultured in serum-containing medium alone, with addition of 0.05 to 5 μM FYAD, with addition of 0.1 to 10 μM Ca074Me, or with addition of 1 μM ZFYD and 2 – 10 μM Ca074Me. After 6 days, cells were fixed and stained with crystal violet to visualize cell colonies.
In separate experiments, SK-N-SH and IMR-32 cells were plated in 12-well dishes at 10% confluence in MEM containing 10% fetal bovine serum and cultured for 24 h to allow cells to adhere to the plates. Cells were cultured for up to an additional 6 days in serum-containing medium alone (controls, no addition), or with addition of 2 μM FYAD. Medium and inhibitor were replenished by exchange of 50% of the media every 2 days. Triplicate samples of cells were removed from the plates by harvesting both floating and adherent cells by trypsinization. Viable cells were counted by trypan blue exclusion and dead cells counted after trypan blue uptake. While this technique is labor intensive, we have found that it is more reliable for determining viable cell numbers than standard MTT assays of these inhibitor treated neuroblastoma cells. Furthermore, cells that take up trypan blue were counted to determine extent of cell death. Cell numbers at each time-point were compared to untreated controls using student t-tests.
2.4 Determining relationship between inhibitor treatment, cathepsin inhibition and cell growth
SK-N-SH cells were cultured in 24 well plates with a range of concentrations of FYAD for 3 days. Viable cells were quantified using Cell Titer Blue (Promega, Madison, WI) and residual cathepsin B + L activity was determined using Z-Phe-Arg-NHMec substrate [25]. Residual enzyme activity and viable cells were calculated using untreated cells as 100% controls.
2.5 Co-treatment of cells with FYAD and caspase and cathepsin D inhibitors
IMR-32 cells were treated with 5 μM FYAD, 10 μM pepstatin, or both compounds for 48 h. Similarly, cells were treated with the pan-caspase inhibitor, Z-Val-Ala-Asp (OMe)-fluoromethylketone (ZVAD) with or without FYAD. The effect of inhibitor treatment on PARP cleavage was determined by western blotting.
2.6 Other cell types
MCF-7, BT474 and MDA-MB-231, MDA-MB-435S and SK-BR-3 breast cancer cells, 3T3 mouse fibroblasts and HT-1080 fibrosarcoma cells were obtained from ATCC and cultured in 10% serum using recommended media. These cells were treated with FYAD for 3 days to determine effect on cell growth and viability.
2.7 Western blotting
Equal quantities (10 μg) of total cell protein were loaded to SDS/PAGE (7% for poly (ADP-ribose) polymerase (PARP), 12% for caspase 3), separated by electrophoresis, transferred to PVDF membrane and then detected with commercially available antibodies for PARP and caspase 3. β-actin was used as a control for loading of cellular proteins.
2.8 Flow cytometry
Cells were harvested and thoroughly resuspended in PBS. Cells were then fixed in 70% ethanol. Prior to analysis, cells were transferred to PBS and then DNA stained by resuspending cells in PBS containing 0.1% Triton-X-100, propidium iodide (20 μg/ml) and RNase A (200 μg/ml). DNA content was analyzed using the 488 nm argon ion laser on Beckman Coulter flow cytometer, model EPICS XL/MCL.
2.9 Ultrastructural analysis of neuroblastoma cells
Transmission electron microscopy of cultured cells was performed using a modified protocol, previously described [26]. Media and detached cells were removed from culture plates and remaining cells washed with PBS. Cells were then incubated in fixative (2% glutaraldehyde in 0.1M sodium cacodylate buffer, pH 7.4) for 45 min at room temperature. Following fixation, cell monolayers were scraped and harvested by centrifugation at 600 g for 3.5 min at 5°C. The cell pellets were resuspended in 1 ml 0.1 M sodium cacodylate buffer. Subsequently, cells were embedded in 4% agar and cut into 1×1 mm cubes, washed in cacodylate buffer, and fixed for 2 h in 1% osmium tetroxide in buffer. Following secondary fixation, cells were washed twice in buffer, rinsed in double de-ionized water and dehydrated in an ascending acetone series. The samples were infiltrated in Spurr's resin and polymerized at 60° C for 24 h.
Ultrathin sections were cut on a Reichert-Jung Ultracut E ultramicrotome, collected on formvar-carbon coated copper grids, and stained with saturated methanolic uranyl acetate and Reynolds' lead citrate [27]. All images were acquired with a Zeiss CEM 902 TEM at 80kV equipped with an Olympus Soft Imaging System GmbH Megaview II digital camera.
3. Results
3.1 Protease inhibitors have varying specificities in live cells
A range of cysteine protease inhibitors have been reported to induce cell death of many cancer cells, including neuroblastoma cells. In this study, we chose FYAD, an inhibitor of both cathepsins B and L; ZFYD, a specific inhibitor of cathepsin L and Ca074Me, a specific inhibitor of cathepsin B [14; 17; 28]. Fmoc-Tyr(I-125)-Ala-CHN2 enters cells and binds covalently to the catalytic site of active cathepsins B and L. The covalent bond is stable under conditions used for SDS/PAGE and consequently active enzymes can be visualized directly by autoradiography. Inactive enzymes do not react with these inhibitors [15]. Reactivity with the labeled inhibitor shows that SK-N-SH cells contain more active cathepsin B than active cathepsin L, whereas IMR-32 cells contain more active cathepsin L than cathepsin B (fig 1a). Pre-treatment of cells with 5 μM unlabeled FYAD for 2 h completely blocked labeling with the radioiodinated inhibitor, showing that this compound efficiently inhibits both enzymes in cells (fig 1a). Complete inhibition was maintained for the duration of the experiment (48 h). These results are similar to findings using the closely related compound, Z-Phe-Ala-CHN2, confirming the high specificity of diazomethanes for efficient inhibition of cathepsins in cellular systems [24]. Using a similar technique, ZFYD was shown to specifically block reactivity of cathepsin L within 2 h and inhibition of this enzyme was also maintained for at least 3 days (fig 1b). However, cathepsin B remained active and was subsequently labeled with Fmoc-Tyr(I-125)-Ala-CHN2. Ca074Me blocked labeling of both cathepsins B and L within 2 h, but after 3 days active cathepsin B was restored in these cells. Cathepsin B activity was also restored when both ZFYD and Ca074Me were added to cells. Ca074 is a very specific inhibitor of cathepsin B, partly mediated by a carboxyl group that specifically reacts with two histidine residues in the active site cleft of the enzyme [29]. This charged group is neutralized as a methyl ester in Ca074Me to allow the inhibitor to enter the cell. The neutralized inhibitor can react with cathepsin L, making it less specific [30] and the ester is readily hydrolyzed in water making it less effective with time. Thus although Ca074 is one of the best known specific inhibitors of cathepsin B [28], the results of this study show that protease inhibition with Ca074Me is not specific or efficient in living cells.
Figure 1.
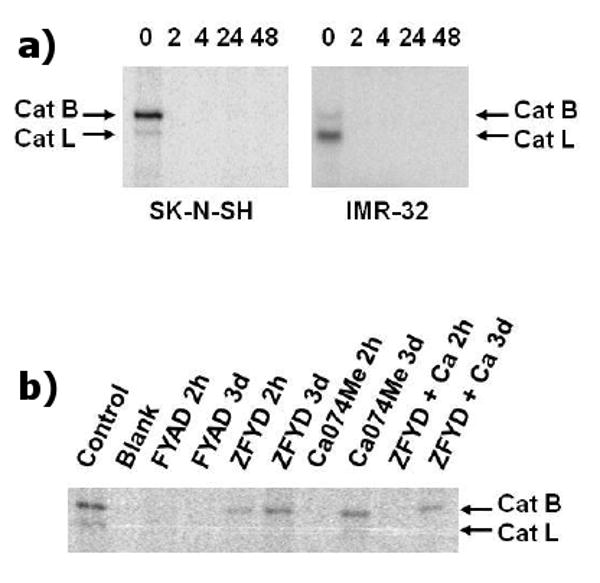
Specific inhibition of cathepsins B and L. These images are phosphor images of SDS gels of proteins extracted from cells treated with Fmoc-Tyr(I-125)-Ala-CHN2 for 2 h. Cells in the upper panels (a) were pre-treated with 5 μM unlabeled FYAD for 2, 4, 24 or 48 h prior to addition of the labeled compound. Zero lanes correspond to cell extracts treated with radiolabeled inhibitor alone. The labeled band of approx. 32 kDa is cathepsin B and the labeled band of approx. 25 kDa is cathepsin L. In the lower panel (b), SK-N-SH cells were treated with either 2 μM FYAD, 5 μM ZFYD, 5 μM Ca074Me or both (ZFYD + Ca) for 2 h or 3 days prior to labeling of residual active proteases with Fmoc-Tyr(I-125)-Ala-CHN2 for 2 h.
3.2 Fmoc-Tyr-Ala-CHN2 inhibits cell growth of neuroblastoma cells in a colony-forming assay
FYAD inhibited growth of IMR-32 cells in a colony-forming assay with an IC-50 of approximately 100 nM, and growth inhibition was detectable at concentrations as low as 50 nM (fig 2a). By comparison, ZFYD and Fmoc-Tyr-Ala-OH, an inactive peptidyl homolog, failed to inhibit growth of these cells at concentrations as high as 10 μM (results not shown). Ca074Me inhibited colony formation with an IC-50 of 5 μM (fig 2b). Addition of 1 μM ZFYD to the Ca074Me had a small additional effect on colony formation, but growth inhibition was not as efficient as with the bifunctional inhibitor, FYAD.
Figure 2.
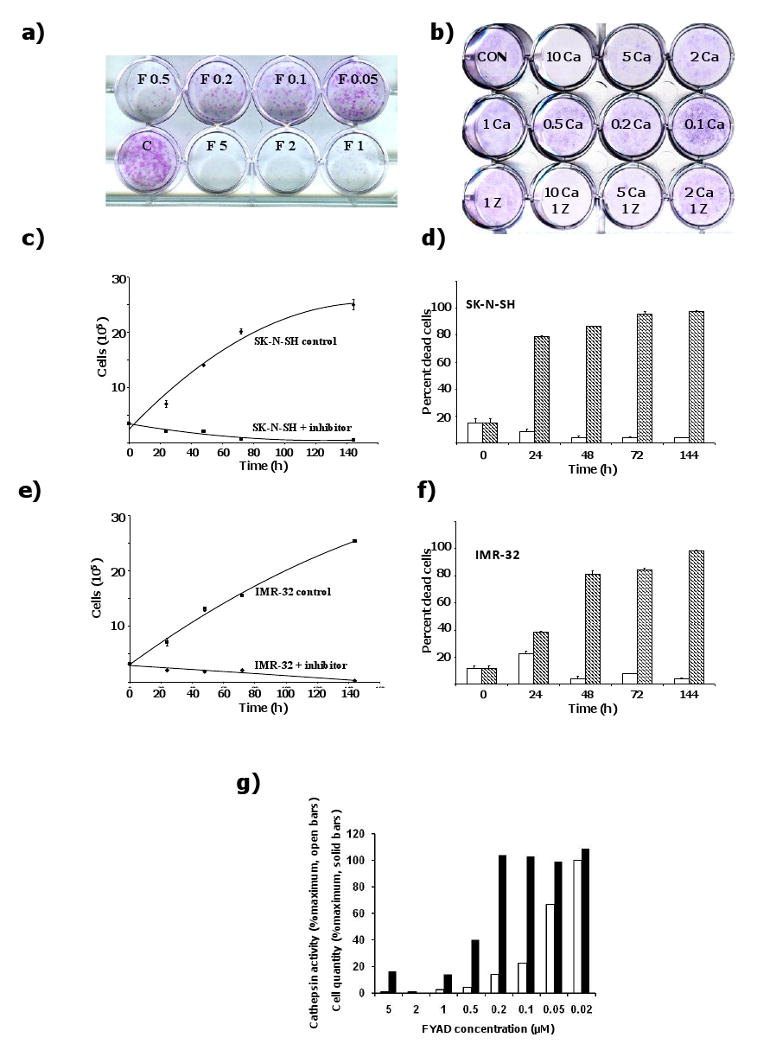
Fmoc-Tyr-Ala-CHN2 inhibits of growth of IMR-32 cells. a) IMR-32 cells were seeded onto 12 well plates and cultured for 6 days in serum-containing medium alone (controls, C), or with addition of 0.05 to 5 μM FYAD (F 0.05 to F 5). Cells were then fixed and stained with crystal violet. In a separate experiment (b), IMR-32 cells were incubated with 0.1 to 10 μM Ca074Me (0.1 Ca to 10 Ca) or 1 μM ZFYD + 2 – 10 μM Ca074Me (Z + Ca). Representative images of 4 separate experiments are shown. SK-N-SH (c,d) or IMR-32 (e,f) neuroblastoma cells were seeded onto 12 well plates and cultured for up to 6 days in serum-containing medium alone (controls), or with addition of 2 μM FYAD (+ inhibitor). At the times indicated, triplicate samples of cells were removed from the plates by harvesting both floating and adherent cells by trypsinization. Viable cells were counted by trypan blue exclusion and dead cells counted after trypan blue uptake. Values are presented as mean +/- standard deviation. Medium and inhibitor were replenished by exchange of 50% of the media every 2 days. The graphs (c, e) show numbers of live cells at each time point. The bar charts (d, f) show percentage dead cells at each time point with (shaded) and without (open) FYAD. Residual cathepsin B + L activity and viable cell numbers were determined after 3 days of treatment using the substrate Z-Phe-Arg-NHMec (open bars) and a Cell Titer Blue assay (solid bars, g). Values are means of triplicate samples that had standard deviations of 10 – 20%.
3.3 Fmoc-Tyr-Ala-CHN2 causes cell death of neuroblastoma cells
IMR-32 and SK-N-SH cells were cultured in the presence or absence of 2 μM FYAD for up to 6 days. Live and dead cells were counted at timed intervals. After 6 days of treatment, the vast majority of cells were dead, being detached and unable to exclude trypan blue (fig 2 c - f). No live IMR-32 cells remained and the few remaining SK-N-SH cells were large S-type flat cells. The remaining SK-N-SH cells may be a sub-population of the SK-N-SH cell line although they may be differentiated forms of the more aggressive N-type cells [31; 32]. This induction of death of neuroblastoma cells by FYAD contrasts with a range of breast cancer cells and transformed fibroblasts which have previously been shown to grow more slowly in the presence of FYAD and recover on removal of inhibitor [16]. Inhibition of growth of breast cancer cells without causing cell death was confirmed in this study (fig S1).
3.4 Cathepsins B and L must be inhibited by greater than 90% to affect cell growth
When cells are incubated for 3 days with 5 μM FYAD, cathepsins B and L are efficiently inhibited (fig 1). However at concentrations of 1 μM and lower some activity can be measured (fig 2g). Cell viability assays show that at FYAD concentrations of 0.2 μM and lower, cell growth was not impaired, even though enzyme activity was reduced by as much as 90%.
3.5 Fmoc-Tyr-Ala-CHN2 induces apoptosis of neuroblastoma cells
Treatment of SK-N-SH and IMR-32 cells with FYAD for 3 days resulted cleavage of the DNA repair enzyme PARP (fig 3a). Treatment with ZFYD or Ca074Me failed to cause cell death or PARP cleavage. FYAD treatment also induced cleavage of caspase 3, another hallmark of apoptosis (fig 3b). The majority of the apoptotic cells were detached from the plates and consequently both detached and adherent cells had to be harvested to see the processed proteins. Flow cytometry analysis of treated cells showed time-dependent appearance of sub-G1 DNA containing particles after FYAD treatment, characteristic of apoptotic bodies (fig 3c). After 24 h of treatment, low levels of apoptotic fragments were seen by flow cytometry and very low levels of PARP cleavage could be detected by western blotting (fig 3f). Remaining live cells appeared to be shifted towards G1/G0, consistent with cell cycle arrest as seen for other cell types treated with FYAD [16]. Two other neuroblastoma cell lines, NB1691 and GM11027, were also sensitive to FYAD, showing evidence of apoptosis after 3 days (fig 3g). The GM11027 cells were more resistant to FYAD than the other three cell lines and did not show signs of apoptosis until 3 days after treatment.
Figure 3.
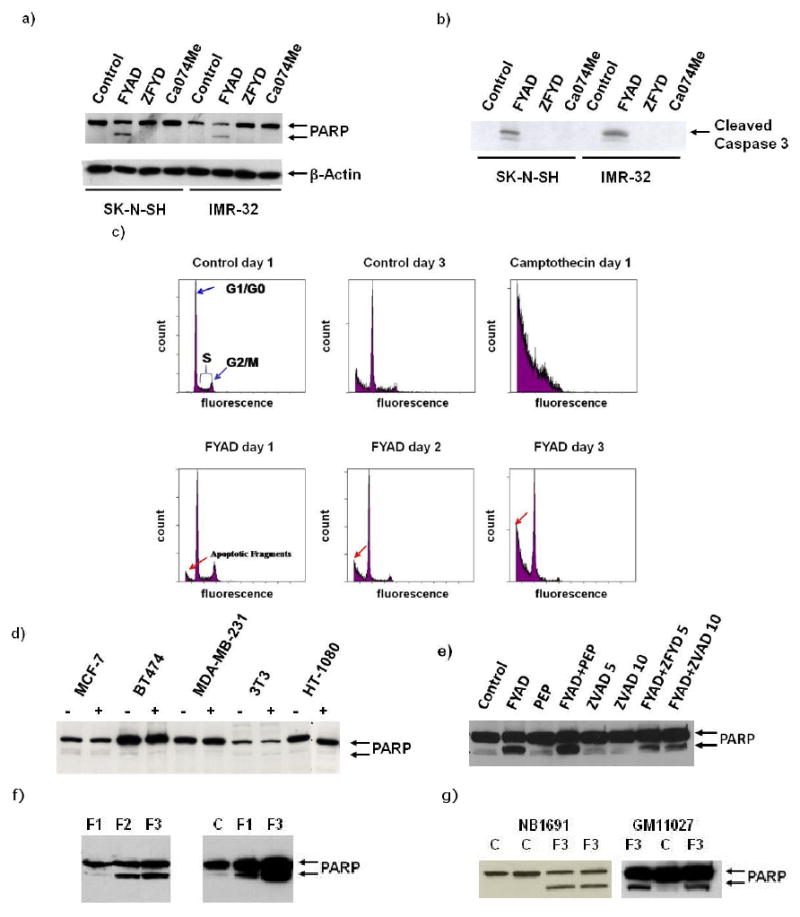
FYAD induces apoptosis of neuroblastoma cells. Proteins were extracted from neuroblastoma cells treated with different inhibitors of cathepsins for 3 days (a). Western blotting was performed using antibodies to PARP and β-actin. Inhibitor concentrations were 5 μM for FYAD, Ca074Me, and ZFYD. FYAD consistently induces PARP cleavage in these cells and can be detected as early as 24 h after treatment. Similarly treated cells were also western blotted for cleaved caspase 3, another hallmark of apoptosis (b). FYAD treated and untreated SK-N-SH cells were fixed, permeabilized and stained with propidium iodide for analysis by flow cytometry (c). In control cells the characteristic pattern of cells in G1/G0, S and G2/M phase are shown. Cells treated with camptothecin are shown as a control for detection of apoptotic bodies. Increases in apoptotic fragments are shown in cells treated with 5 μM for FYAD for 1 - 3 days. The breast cancer cell lines MCF7, BT474 and MDA-MB-231, and fibroblastic cell lines 3T3 and HT1080 were cultured in the presence (+) or absence (-) of 5 μM FYAD for 3 days. Cell proteins were then extracted and western blotted for PARP (d). SK-N-SH cells were cultured in the presence of FYAD with addition of ZVAD (5 or 10 μM) to inhibit caspases or pepstatin (PEP) to inhibit cathepsin D and after 3 days protein extracts were western blotted for PARP (e). Similar results were obtained at least three times for each of these analyses. IMR-32 cells were treated with 5 μM for 1,2 or 3 days (F1, F2, F3) and then total samples harvested and western blotted for PARP. Low levels of cleaved PARP could be seen after 24 h, and were higher than in controls (C) in an over-exposed blot (right panel, f). Cleavage of PARP is also seen in NB1691 and GM11027 cells after 3 days of treatment with 5 μM FYAD (g).
3.6 Fmoc-Tyr-Ala-CHN2 does not induce apoptosis of other cell types
We have examined the effect of FYAD on growth of a range of cells including breast cancer cell lines, a fibrosarcoma cell line, and mouse fibroblasts. Although growth of these cells was impaired (as noted previously, [16]), no significant cell death occurred during 3 days of treatment and there was no evidence of PARP cleavage (fig 3d). These results indicate that induction of cell death shows specificity towards neuroblastoma.
3.7 Inhibition of cathepsin D does not rescue cells from FYAD-induced death
It has been proposed that release of cathepsins B or D from lysosomes into the cytosolic compartment can induce cell death in a variety of cell types [11; 33]. When SK-N-SH or IMR-32 cells were treated simultaneously with 5 μM FYAD and 10 μM pepstatin, a membrane permeant inhibitor of aspartic proteases, cell death was not reduced compared to cells treated with FYAD alone, showing that cathepsin D inhibition does not rescue cells from FYAD induced cell death. The cell death kinetics induced by treatment with both pepstatin and FYAD were not significantly affected compared to treatment with FYAD alone. Pepstatin did not induce PARP cleavage by itself but slightly increased PARP cleavage induced by FYAD (fig 3e). By contrast, the pan-caspase inhibitor, ZVAD, reduced FYAD-induced PARP cleavage.
3.8 Fmoc-Tyr-Ala-CHN2 causes the accumulation of electron dense vesicles in neuroblastoma cells
Inhibition of cathepsins B and L is likely to impair lysosomal proteolysis. SK-N-SH and IMR-32 cells treated for 1 and 3 days with 5 μM FYAD were processed for ultrastructural analysis. Accumulations of electron dense vesicles were apparent in treated cells after 24 h and these increased in size and number with prolonged exposure to the inhibitor (fig. 4). Multivesicular bodies were also evident in treated cells. Mitochondria decreased in size and number with treatment whereas the Golgi apparatus became more prominent. Condensed chromatin detached from the inner nuclear membrane was also observed in FYAD-treated cells.
Figure 4.
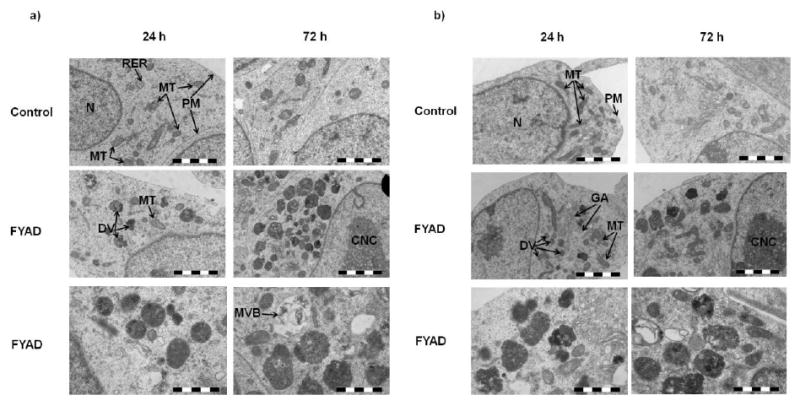
Ultrastructural changes caused by FYAD treatment. IMR-32 (a) and SK-N-SH (b) cells were examined by electron microscopy. The upper 2 panels for each cell line are control cells after 24 h and 72 h and the lower 4 panels are cells treated with 5 μM FYAD for 24 h and 72 h. Scale bar is 2 μm in the upper 4 panels and 1 μm in the lower 2 panels. In control cells, euchromatic nuclei (N), mitochondria (MT), rough endoplasmic reticulum (RER) and plasmamembrane (PM) are identified. In treated cells, accumulating dense vesicles (DV), reduced numbers of mitochondria (MT), prominent Golgi apparatus (GA), condensed nuclear chromatin (CNC) detached from the nuclear envelope, and multivesicular bodies (MVB) are indicated. These images are representative of multiple fields of view from three separate samples.
4. Discussion
In this study we showed that FYAD binds to both cathepsins B and L and that complete inhibition of both of these enzymes is maintained in cultured neuroblastoma cells, creating a chemical knockout of both enzyme activities. The chemically related compound, ZFYD, bound only to cathepsin L and inhibition of this enzyme was also shown to be maintained in cultured neuroblastoma cells. The ability of the diazomethanes to penetrate cell membranes and to selectively bind only to active cathepsins in cells makes them unique tools for the complete inhibition of cellular cathepsins [17]. The more widely used cathepsin B inhibitor, Ca074Me, was shown to be less effective at inhibiting cathepsin B and was not specific for this enzyme in cells. The un-esterified inhibitor, Ca074, has been shown to inhibit cathepsin B and not cathepsin L or calpain 1 [28], but the charged carboxyl group of this compound impairs cellular penetration. While esterification enables the compound to enter cells as Ca074Me, this modified compound reacts with both cathepsins B and L and could react with additional enzymes and proteins [30; 34].
In our studies, complete inhibition of both cathepsins B and L induced cell death in the neuroblastoma cell lines (SK-N-SH and IMR-32) but not other tumor cells. Inhibitors that affected only one enzyme or failed to maintain inhibition of both cathepsins B and L did not induce apoptosis of these cells. This contrasts with other reports of inhibitors of either single or multiple cathepsins that cause cell death. Z-Phe-Gly-NHO-Bz is an inhibitor of both cathepsins B and L that causes death of many cell types [13; 35]. The specificity of this inhibitor in cellular systems is not well-established and in aqueous solution it hydrolyzes to an hydroxamate that can react with other enzymes [36]. Inhibition of cathepsin B alone with Ca074Me has previously been proposed as a potential treatment for neuroblastoma, although the efficiency and specificity of cathepsin inhibition was not examined in this study [5]. The specificity of Ca074Me in cellular systems has not been shown by direct protease labeling and it has been shown to have cellular effects unrelated to cathepsin B inhibition, making it less useful for in vivo studies of cathepsin function [37]. Although many cathepsin inhibitors are commercially available, few have been shown to be specific in cellular systems.
Low-level inhibition of the proteases did not affect growth of SK-N-SH cells and 95 -100% enzyme inhibition was required to cause cell death. Lysosomal concentrations of cathepsins can be as high as 1 mM [17], and consequently partial enzyme inhibition is unlikely to have a major impact on cellular proteolysis. We have not been able to generate siRNA techniques that stably knock-down expression of cathepsins B or L in neuroblastoma cells by more than 70% so are unable to confirm our inhibitor studies with this technology. The major targets of FYAD are clearly shown to be cathepsins B and L (fig 1) and a related inhibitor that does not inhibit both cathepsins B and L does not cause cell death indicating that efficient inhibition of cathepsins B and L is most likely the cause of neuroblastoma cell death.
A direct role in apoptosis is becoming established as an important physiological role for cathepsin B, and stimulation of cathepsin B mediated apoptosis by lysosomal membrane disruption is proposed for cancer therapy [8; 38]. Cell death can also be stimulated by lysosomal rupture and release of cathepsin D [39; 40; 41]. There was little evidence of lysosomal rupture in FYAD treated neuroblastoma cells and addition of pepstatin to treated cells did not prevent cell death in our studies. Pepstatin alone had little effect on growth of IMR-32 and SK-N-SH neuroblastoma cells and did not impair cell death induced by FYAD treatment. This indicates that it is lysosomal protease inhibition that initiates the cell death process and that release of other lysosomal enzymes such as cathepsin D into cytosol is not a significant cause of cell death after FYAD treatment of these neuroblastoma cells.
The mechanism by which cathepsin inhibition leads to cell death is not clear, but is likely to be due to accumulation of one or more proteins that are normally degraded by the concerted action of cathepsins B and L. Electron microscopy clearly shows that inhibition of cathepsins B and L caused major accumulation of dense granules in both IMR-32 and SK-N-SH neuroblastoma cell lines within 24 h. Although there is some evidence of apoptosis 24 h after treatment, it is likely that accumulation of undigested proteins precedes induction of apoptosis. Accumulation of autophagic-like vacuoles are seen in double knockout mice lacking both cathepsins B and L [42; 43], indicating that autophagy is probably induced prior to apoptosis. More detailed studies are required to elucidate the critical steps that lead to death of neuronal cells in the knock-out animal studies and death of neuroblastoma cells in our inhibitor studies.
Data from genetic deletion of cathepsins B and L in mice provide some support for the selective induction of apoptosis that we see for neuroblastoma cells. Deletion of cathepsin L in mice shows that this enzyme plays a role in endosomal processing of the MHC II invariant chain and turnover of EGF and its receptor but there is no evidence that deletion of this enzyme induces cellular apoptosis [44; 45]. A major phenotype is actually uncontrolled proliferation of keratinocytes, resulting in periodic hair loss [44]. Apoptosis is not induced by genetic deletion of cathepsin B either, but TNF-induced apoptosis (a process mediated by cathepsin B activity) is blocked in these mice [46; 47]. However, double knockout mice lacking both cathepsins B and L show significant evidence of apoptosis in neuronal cells, with particularly significant loss of Purkinje cells [42; 43]. Other cell types are not significantly affected, supporting our observation that both enzymes must be inhibited and that apoptosis may be specifically induced in proliferating cells of neuronal origin. Current cancer therapies target abnormal growth characteristics of tumor cells while cathepsin inhibition may be targeting a normal function of proliferating cells of neuronal origin. The common defect of all neuroblastoma tumors is a failure in differentiation and they continue to proliferate after most normal cells have differentiated into cells of the peripheral nervous system.
Degeneration of the central nervous system by genetic deletion of both cathepsins B and L raises some concerns about potential use of inhibitors of these enzymes therapeutically. However, inhibitors of cathepsin B (and L) have been shown to have a neuroprotective effect in animal models of ischemia, preventing apoptosis [48; 49; 50; 51; 52] and prolonged treatment of animals with cathepsin inhibitors do not produce any significant neurological toxic effects [19; 20]. The deleterious effects of protease loss in the cathepsin B and L double knockout mice are primarily due to early post-natal effects on rapidly proliferating neuronal cells whereas non-proliferating cells of the central nervous system of adult mice appears to be refractory to protease inhibition. Thus in children, particularly those over two years old, post-mitotic neuronal cells may not be affected by treatment with protease inhibitors, making such compounds potentially useful for development of treatments to eradicate neuroblastoma. Nevertheless, careful monitoring of potential adverse neurological effects of inhibitor treatment would need to be monitored and drugs that do not cross the blood-brain barrier may provide a less toxic treatment of this cancer of the peripheral nervous system. The unusual sensitivity of neuroblastoma cells to inhibitors of cathepsins B and L may provide a novel therapeutic approach to treat this particularly intractable childhood cancer.
Supplementary Material
FYAD inhibits growth of breast cancer cell lines but does not cause cell death. MDA-MB-231, MDA-MB-435S, SK-BR-3 breast cancer cells were seeded onto 12 well plates and cultured for up to 3 days in serum-containing medium alone (controls), or with addition of 5 μM FYAD. At the times indicated, triplicate samples of cells were removed from the plates by harvesting both floating and adherent cells by trypsinization. Viable cells were counted by trypan blue exclusion and dead cells counted after trypan blue uptake. Values are presented as mean +/- standard deviation. Medium and inhibitor were replenished by exchange of 50% of the media at 2 days. The graphs show numbers of live cells at each time point. Cells treated with the non-inhibitory peptide, Fmoc-Tyr-Ala-OH, gave values similar to those of controls. The bar charts show percentage dead cells at each time point with (shaded) and without (open) FYAD.
Acknowledgments
Nemours Research Programs, the Center for Pediatric Research NIH NCRR Grant P20RR20173, the Delaware INBRE, P20RR016472 and Alex's Lemonade Stand Foundation have provided financial support for this research. Particular thanks go to Shannon Modla and Kirk Czymmek for assistance with electron microscopy and Victoria Maduskuie for assistance with flow cytometry.
Footnotes
Conflict of interest: The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Castel V, Canete A. A comparison of current neuroblastoma chemotherapeutics. Expert Opin Pharmacother. 2004;5:71–80. doi: 10.1517/14656566.5.1.71. [DOI] [PubMed] [Google Scholar]
- 2.Szpaderska AM, Frankfater A. An intracellular form of cathepsin B contributes to invasiveness in cancer. Cancer Res. 2001;61:3493–3500. [PubMed] [Google Scholar]
- 3.Mohanam S, Jasti SL, Kondraganti SR, Chandrasekar N, Lakka SS, Kin Y, Fuller GN, Yung AW, Kyritsis AP, Dinh DH, Olivero WC, Gujrati M, Ali-Osman F, Rao JS. Down-regulation of cathepsin B expression impairs the invasive and tumorigenic potential of human glioblastoma cells. Oncogene. 2001;20:3665–3673. doi: 10.1038/sj.onc.1204480. [DOI] [PubMed] [Google Scholar]
- 4.Strojnik T, Zidanik B, Kos J, Lah TT. Cathepsins B and L are markers for clinically invasive types of meningiomas. Neurosurgery. 2001;48:598–605. doi: 10.1097/00006123-200103000-00029. [DOI] [PubMed] [Google Scholar]
- 5.Castino R, Pace D, Demoz M, Gargiulo M, Ariatta C, Raiteri E, Isidoro C. Lysosomal proteases as potential targets for the induction of apoptotic cell death in human neuroblastomas. Int J Cancer. 2002;97:775–779. doi: 10.1002/ijc.10139. [DOI] [PubMed] [Google Scholar]
- 6.Levicar N, Dewey RA, Daley E, Bates TE, Davies D, Kos J, Pilkington GJ, Lah TT. Selective suppression of cathepsin L by antisense cDNA impairs human brain tumor cell invasion in vitro and promotes apoptosis. Cancer Gene Ther. 2003;10:141–151. doi: 10.1038/sj.cgt.7700546. [DOI] [PubMed] [Google Scholar]
- 7.Nagaraj NS, Vigneswaran N, Zacharias W. Cathepsin B mediates TRAIL-induced apoptosis in oral cancer cells. J Cancer Res Clin Oncol. 2006;132:171–183. doi: 10.1007/s00432-005-0053-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chwieralski CE, Welte T, Buhling F. Cathepsin-regulated apoptosis. Apoptosis. 2006;11:143–149. doi: 10.1007/s10495-006-3486-y. [DOI] [PubMed] [Google Scholar]
- 9.Ichinose S, Usuda J, Hirata T, Inoue T, Ohtani K, Maehara S, Kubota M, Imai K, Tsunoda Y, Kuroiwa Y, Yamada K, Tsutsui H, Furukawa K, Okunaka T, Oleinick NL, Kato H. Lysosomal cathepsin initiates apoptosis, which is regulated by photodamage to Bcl-2 at mitochondria in photodynamic therapy using a novel photosensitizer, ATX-s10 (Na) Int J Oncol. 2006;29:349–355. [PubMed] [Google Scholar]
- 10.Kagedal K, Johansson AC, Johansson U, Heimlich G, Roberg K, Wang NS, Jurgensmeier JM, Ollinger K. Lysosomal membrane permeabilization during apoptosis-- involvement of Bax? Int J Exp Pathol. 2005;86:309–321. doi: 10.1111/j.0959-9673.2005.00442.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cirman T, Oresic K, Droga Mazovec G, Turk V, Reed JC, Myers RM, Salvesen GS, Turk B. Selective disruption of lysosomes in HeLa cells triggers apoptosis, mediated by cleavage of Bid by multiple papain-like lysosomal cathepsins. J Biol Chem. 2004;279:3578–3587. doi: 10.1074/jbc.M308347200. [DOI] [PubMed] [Google Scholar]
- 12.Guicciardi ME, Leist M, Gores GJ. Lysosomes in cell death. Oncogene. 2004;23:2881–2890. doi: 10.1038/sj.onc.1207512. [DOI] [PubMed] [Google Scholar]
- 13.Zhu DM, Uckun FM. Z-Phe-Gly-NHO-Bz, an inhibitor of cysteine cathepsins, induces apoptosis in human cancer cells. Clin Cancer Res. 2000;6:2064–2069. [PubMed] [Google Scholar]
- 14.Shaw E. Peptidyl diazomethanes as inhibitors of cysteine proteinases. Meth Enzymol. 1994;244:649–656. doi: 10.1016/0076-6879(94)44048-4. [DOI] [PubMed] [Google Scholar]
- 15.Mason RW, Wilcox D, Wikstrom P, Shaw EN. The identification of active forms of cysteine protienases in Kirsten-virus-transformed mouse fibroblasts by use of a specific radiolabelled inhibitor. Biochem J. 1989;257:125–129. doi: 10.1042/bj2570125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xing RY, Wu F, Mason RW. Control of breast tumor cell growth using a targeted cysteine protease inhibitor. Cancer Res. 1998;58:904–909. [PubMed] [Google Scholar]
- 17.Xing RY, Addington AK, Mason RW. Quantification of cathepsins b and L in cells. Biochem J. 1998;332:499–505. doi: 10.1042/bj3320499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Desmarais S, Black WC, Oballa R, Lamontagne S, Riendeau D, Tawa P, Duong LT, Pickarski M, Percival MD. Effect of cathepsin K inhibitor basicity on in vivo off-target activities. Mol Pharmacol. 2007 doi: 10.1124/mol.107.039511. [DOI] [PubMed] [Google Scholar]
- 19.Sugita H, Kimura M, Tarumoto Y, Tamai M, Hanada K, Ishiura S, Nonaka I, Ohzeki M, Imahori K. In vivo administration of a thiol protease inhibitor, E-64-C, to hereditary dystrophic chicken. Muscle & Nerve. 1982;5:738–744. [Google Scholar]
- 20.Komatsu K, Inazuki K, Hosoya J, Satoh S. Beneficial effect of new thiol protease inhibitors, epoxide derivatives, on dystrophic mice. Exp Neurol. 1986;91:23–29. doi: 10.1016/0014-4886(86)90022-1. [DOI] [PubMed] [Google Scholar]
- 21.Afonso S, Romagnano L, Babiarz B. The expression and function of cystatin C and cathepsin B and cathepsin L during mouse embryo implantation and placentation. Development. 1997;124:3415–3425. doi: 10.1242/dev.124.17.3415. [DOI] [PubMed] [Google Scholar]
- 22.Freeman SJ, Lloyd JB. Inhibition of proteolysis in rat yolk sac as a cause of teratogenesis. Effects of leupeptin in vitro and in vivo. J Embryol Exp Morph. 1983;78:183–193. [PubMed] [Google Scholar]
- 23.Ambroso JL, Harris C. In vitro embryotoxicity of the cysteine proteinase inhibitors benzyloxycarbonyl-phenylalanine-alanine-diazomethane(Z-Phe-Ala-CHN2) and benzyloxycarbonyl-phenylalanine-phenylalanine-diazomethane (Z-Phe-Phe-CHN2) Teratology. 1994;50:214–228. doi: 10.1002/tera.1420500307. [DOI] [PubMed] [Google Scholar]
- 24.Wilcox D, Mason RW. Inhibition of cysteine proteinases in lysosomes and whole cells. Biochem J. 1992;285:495–502. doi: 10.1042/bj2850495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barrett AJ, Kirschke H. Cathepsin B, cathepsin H and cathepsin L. Meth Enzymol. 1981;80:535–561. doi: 10.1016/s0076-6879(81)80043-2. [DOI] [PubMed] [Google Scholar]
- 26.Graham L, Orenstein JM. Processing tissue and cells for transmission electron microscopy in diagnostic pathology and research. Nat Protoc. 2007;2:2439–2450. doi: 10.1038/nprot.2007.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reynolds ES. The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. J Cell Biol. 1963;17:208–212. doi: 10.1083/jcb.17.1.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buttle DJ, Murata M, Knight CG, Barrett AJ. CA074 Methyl Ester - A Proinhibitor for Intracellular Cathepsin-B. Arch Biochem Biophys. 1992;299:377–380. doi: 10.1016/0003-9861(92)90290-d. [DOI] [PubMed] [Google Scholar]
- 29.Krupa J, Hasnain SC, Nagler DK, Menard R, Mort JS. S'2 substrate specificity and the role of His110 and His111 in the exopeptidaseactivity of human cathepsin B. Biochem J. 2002;361:613–619. doi: 10.1042/0264-6021:3610613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Montaser M, Lalmanach G, Mach L. CA-074, but not its methyl ester CA-074Me, is a selective inhibitor of cathepsin B within living cells. Biol Chem. 2002;383:1305–1308. doi: 10.1515/BC.2002.147. [DOI] [PubMed] [Google Scholar]
- 31.Cohen N, Betts DR, Rechavi G, Amariglio N, Trakhtenbrot L. Clonal expansion and not cell interconversion is the basis for the neuroblast and nonneuronal types of the SK-N-SH neuroblastoma cell line. Cancer Genet Cytogenet. 2003;143:80–84. doi: 10.1016/s0165-4608(02)00835-x. [DOI] [PubMed] [Google Scholar]
- 32.Wainwright LJ, Lasorella A, Iavarone A. Distinct mechanisms of cell cycle arrest control the decision between differentiation and senescence in human neuroblastoma cells. Proc Natl Acad Sci U S A. 2001;98:9396–9400. doi: 10.1073/pnas.161288698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Erdal H, Berndtsson M, Castro J, Brunk U, Shoshan MC, Linder S. Induction of lysosomal membrane permeabilization by compounds that activate p53-independent apoptosis. Proc Natl Acad Sci U S A. 2005;102:192–197. doi: 10.1073/pnas.0408592102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bogyo M, Verhelst S, Bellingard-Dubouchaud V, Toba S, Greenbaum D. Selective targeting of lysosomal cysteine proteases with radiolabeled electrophilic substrate analogs. Chemistry & Biology. 2000;7:27–38. doi: 10.1016/s1074-5521(00)00061-2. [DOI] [PubMed] [Google Scholar]
- 35.Zhu DM, Uckun FM. Cathepsin inhibition induces apoptotic death in human leukemia and lymphoma cells. Leuk Lymphoma. 2000;39:343–354. doi: 10.3109/10428190009065834. [DOI] [PubMed] [Google Scholar]
- 36.Demuth HU, Schierhorn A, Bryan P, Hofke R, Kirschke H, Bromme D. N-peptidyl, O-acyl hydroxamates: comparison of the selective inhibition of serine and cysteine proteinases. Biochim Biophys Acta. 1996;1295:179–186. doi: 10.1016/0167-4838(96)00038-6. [DOI] [PubMed] [Google Scholar]
- 37.Mihalik R, Imre G, Petak I, Szende B, Kopper L. Cathepsin B-independent abrogation of cell death by CA-074-OMe upstream of lysosomal breakdown. Cell Death Differ. 2004;11:1357–1360. doi: 10.1038/sj.cdd.4401493. [DOI] [PubMed] [Google Scholar]
- 38.Fehrenbacher N, Jaattela M. Lysosomes as targets for cancer therapy. Cancer Res. 2005;65:2993–2995. doi: 10.1158/0008-5472.CAN-05-0476. [DOI] [PubMed] [Google Scholar]
- 39.Yu Z, Li W, Hillman J, Brunk UT. Human neuroblastoma (SH-SY5Y) cells are highly sensitive to the lysosomotropic aldehyde 3-aminopropanal. Brain Res. 2004;1016:163–169. doi: 10.1016/j.brainres.2004.04.075. [DOI] [PubMed] [Google Scholar]
- 40.Isahara K, Ohsawa Y, Kanamori S, Shibata M, Waguri S, Sato N, Gotow T, Watanabe T, Momoi T, Urase K, Kominami E, Uchiyama Y. Regulation of a novel pathway for cell death by lysosomal aspartic and cysteine proteinases. Neuroscience. 1999;91:233–249. doi: 10.1016/s0306-4522(98)00566-1. [DOI] [PubMed] [Google Scholar]
- 41.Castino R, Bellio N, Nicotra G, Follo C, Trincheri NF, Isidoro C. Cathepsin D-Bax death pathway in oxidative stressed neuroblastoma cells. Free Radic Biol Med. 2007;42:1305–1316. doi: 10.1016/j.freeradbiomed.2006.12.030. [DOI] [PubMed] [Google Scholar]
- 42.Stahl S, Reinders Y, Asan E, Mothes W, Conzelmann E, Sickmann A, Felbor U. Proteomic analysis of cathepsin B and L-deficient mouse brain lysosomes. Biochim Biophys Acta. 2007 doi: 10.1016/j.bbapap.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Felbor U, Kessler B, Mothes W, Goebel HH, Ploegh HL, Bronson RT, Olsen BR. Neuronal loss and brain atrophy in mice lacking cathepsins B and L. Proc Natl Acad Sci U S A. 2002;99:7883–7888. doi: 10.1073/pnas.112632299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reinheckel T, Hagemann S, Dollwet-Mack S, Martinez E, Lohmuller T, Zlatkovic G, Tobin DJ, Maas-Szabowski N, Peters C. The lysosomal cysteine protease cathepsin L regulates keratinocyte proliferation by control of growth factor recycling. J Cell Sci. 2005;118:3387–3395. doi: 10.1242/jcs.02469. [DOI] [PubMed] [Google Scholar]
- 45.Nakagawa T, Roth W, Wong P, Nelson A, Farr A, Deussing J, Villadangos JA, Ploegh H, Peters C, Rudensky AY. Cathepsin L: critical role in Ii degradation and CD4 T cell selection in the thymus. Science. 1998;280:450–453. doi: 10.1126/science.280.5362.450. [DOI] [PubMed] [Google Scholar]
- 46.Foghsgaard L, Lademann U, Wissing D, Poulsen B, Jaattela M. Cathepsin B mediates tumor necrosis factor-induced arachidonic acid release in tumor cells. J Biol Chem. 2002;277:39499–39506. doi: 10.1074/jbc.M206669200. [DOI] [PubMed] [Google Scholar]
- 47.Taha TA, Kitatani K, Bielawski J, Cho W, Hannun YA, Obeid LM. Tumor Necrosis Factor Induces the Loss of Sphingosine Kinase-1 by a Cathepsin B-dependent Mechanism. J Biol Chem. 2005;280:17196–17202. doi: 10.1074/jbc.M413744200. [DOI] [PubMed] [Google Scholar]
- 48.Seyfried DM, Veyna R, Han Y, Li K, Tang N, Betts RL, Weinsheimer S, Chopp M, Anagli J. A selective cysteine protease inhibitor is non-toxic and cerebroprotective in rats undergoing transient middle cerebral artery ischemia. Brain Res. 2001;901:94–101. doi: 10.1016/s0006-8993(01)02289-2. [DOI] [PubMed] [Google Scholar]
- 49.Anagli J, Abounit K, Stemmer P, Han Y, Allred L, Weinsheimer S, Movsisyan A, Seyfried D. Effects of cathepsins B and L inhibition on postischemic protein alterations in the brain. Biochem Biophys Res Commun. 2008;366:86–91. doi: 10.1016/j.bbrc.2007.11.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yoshida M, Yamashima T, Zhao L, Tsuchiya K, Kohda Y, Tonchev AB, Matsuda M, Kominami E. Primate neurons show different vulnerability to transient ischemia and response to cathepsin inhibition. Acta Neuropathol (Berl) 2002;104:267–272. doi: 10.1007/s00401-002-0554-4. [DOI] [PubMed] [Google Scholar]
- 51.Yamashima T, Kohda Y, Tsuchiya K, Ueno T, Yamashita J, Yoshioka T, Kominami E. Inhibition of ischaemic hippocampal neuronal death in primates with cathepsin b inhibitor ca-074 - a novel strategy for neuroprotection based on calpain-cathepsin hypothesis. Eur J Neurosci. 1998;10:1723–1733. doi: 10.1046/j.1460-9568.1998.00184.x. [DOI] [PubMed] [Google Scholar]
- 52.Bednarski E, Ribak CE, Lynch G. Suppression of cathepsins B and L causes a proliferation of lysosomes and the formation of meganeurites in hippocampus. J Neurosci. 1997;17:4006–4021. doi: 10.1523/JNEUROSCI.17-11-04006.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FYAD inhibits growth of breast cancer cell lines but does not cause cell death. MDA-MB-231, MDA-MB-435S, SK-BR-3 breast cancer cells were seeded onto 12 well plates and cultured for up to 3 days in serum-containing medium alone (controls), or with addition of 5 μM FYAD. At the times indicated, triplicate samples of cells were removed from the plates by harvesting both floating and adherent cells by trypsinization. Viable cells were counted by trypan blue exclusion and dead cells counted after trypan blue uptake. Values are presented as mean +/- standard deviation. Medium and inhibitor were replenished by exchange of 50% of the media at 2 days. The graphs show numbers of live cells at each time point. Cells treated with the non-inhibitory peptide, Fmoc-Tyr-Ala-OH, gave values similar to those of controls. The bar charts show percentage dead cells at each time point with (shaded) and without (open) FYAD.