

Abstract
A variety of genetic lesions, including chromosomal translocations, internal tandem duplications and mutations have been described in acute myeloid leukaemia (AML). Expression profiling has shown that chromosomal translocations, in particular, are associated with distinctive patterns of gene expression. AML exhibiting the translocation t(8;21), which fuses the AML1 and ETO genes, has such a characteristic expression profile. One gene whose expression is highly correlated with the presence of the AML1/ETO fusion is POU4F1, which encodes the POU homeodomain transcription factor BRN3A. Here we demonstrate using specific siRNA in t(8;21) cells and overexpression studies in progenitor cells that AML1/ETO promotes expression of POU4F1/BRN3A. This effect requires DNA-binding function of AML1/ETO, and accordingly AML1/ETO is bound to the POU4F1 locus in t(8;21) cells. Functionally, while over-expression of Brn3a in murine haematopoietic progenitor cells induces terminal myeloid differentiation, co-expression of AML1/ETO or AML/ETO9a blocks this effect. Furthermore, Brn3a reduction by shRNA impairs AML1/ETO-induced immortalisation of murine progenitors. In summary, we identify POU4F1/BRN3A as a novel potential up-regulated AML1/ETO target gene whose dramatically high expression may co-operate with AML1/ETO in t(8;21) cells.
Keywords: AML1/ETO, BRN3A, AML, transcription, myeloid
Introduction
Gene expression profiling by microarray has identified many genes whose unusually high or low expression shows specificity for a particular cancer type or sub-type.(1-3) In acute myeloid leukaemias, the common chromosome translocations have each been associated with distinctive gene expression patterns. However, the specific relationships between particular gene fusions and many of their associated genes remain obscure.
The t(8;21) translocation fuses coding sequence of the gene AML1 gene to that of ETO producing a chimaeric transcription factor AML1/ETO, also called AML1/MTG8 or RUNX1/RUNX1T1.(4;5) Over-expression of AML1/ETO is sufficient to promote self-renewal of both murine and human myeloid progenitor cells in vitro(6-9) and siRNA studies demonstrate a continuing requirement for AML1/ETO in maintenance of proliferation and inhibition of terminal differentiation.(10-12) Expression of AML1/ETO is not sufficient to generate overt leukaemia in vivo,(6;8;13;14) suggesting that t(8;21) leukaemia requires ‘second hit’ alterations in proliferation-associated pathways such as those of the c-KIT and FLT3 growth factor receptors.(5;15;16) Additional less well-characterised splice variants of AML1/ETO such as the oncogenic AML/ETO9a may also play a role in t(8;21) AML.(17)
AML1/ETO retains the AML1 DNA-binding domain and the repressor domain of ETO, thus has some function by aberrantly repressing transcription.(5) Indeed AML1/ETO binds directly to transcriptional repression machinery and represses expression of genes such as CSF1R (also known as MCSFR or c-FMS) and p14ARF.(18;19) However, mostly as a result of gene expression profiling, it is now becoming clear that whilst AML1/ETO can repress gene expression it is perhaps also capable of activating gene expression.(11;20-23)
Expression profiling in AML has identified POU4F1 to be highly expressed in t(8;21) samples.(1-3) Specificity for t(8;21) is such that high POU4F1 expression can assist positive identification of those carrying this translocation.(1) However, functions of the murine Pou4f1 gene product Brn3a have been studied almost exclusively in the nervous system where it promotes neuronal development and survival.(24-27) Potential function of Brn3a/BRN3A in haematopoietic cells is presently unknown.
Here we demonstrate that expression of POU4F1 is promoted by AML1/ETO proteins, and that DNA binding of AML1/ETO, which occurs at AML1 binding sites in the POU4F1 locus, is required for this effect. Functionally, over-expression of Brn3a in murine haematopoietic progenitor cells results almost exclusively in terminal macrophage differentiation. In contrast, AML1/ETO proteins inhibit and possibly even reverse this effect, with Brn3a being required for some AML1/ETO dependent growth. Thus AML1/ETO and BRN3A appear to co-operatively inhibit differentiation and promote growth in t(8;21) AML.
Materials and methods
Patient samples and cell culture
Leukaemia samples at presentation were obtained with informed consent from adult patients at St Bartholomew's Hospital, and mononuclear cells purified by standard techniques. Primary AML blasts and t(8;21) positive Kasumi-1 cells (DSMZ ACC 220)(28) were cultured as described previously.(11)
Plasmid construction
To generate pMSCV-3a-neo, murine Brn3a(L) cDNA was inserted into pMSCVneo (Clontech) between Eco RI and Bgl II sites. Brn3a(L) was inserted between Eco RI and and Not I sites of a modified pMSCV-hCD2tailless vector(29) to generate pMSCV-3a-hCD2. To generate Brn3ashRNA plasmids, double-stranded oligonucleotides were cloned into pMSCV-hCD2tailless-miR30 vector pM2miR(30) generating a fused hCD2-miR transcript. Targeting sequence for sh24 is 5′-CGCATTGAAACTGAGCACTAAA-3′ and for sh43 5′-AGCCGAGAAACTGGACCTCAAA-3′. pMiG-AML1/ETO, pMiG-AML1/ETO9a and derived mutant plasmids(17;31) were a kind gift of Dong-Er Zhang (Dept. of Pathology, UCSD, LaJolla, CA).
Retroviral infection and culture of haematopoietic progenitors
Progenitor cells were purified from day E12.5 or E13.5 murine foetal livers by Ter119 depletion and c-Kit positive selection as per manufacturer's instructions (Miltenyi). Cells were cultured in DMEM containing 10% FCS (Stem Cell Technologies) supplemented with 100 ng/ml SCF, 10 ng/ml IL-3 and IL-6, and infected by spinfection in the presence of polybrene 24 or 48 hours post-isolation with concentrated retroviral supernatants generated by lipofectamine transfection of packaging cell line LinXE.(32) Cells were transferred to methylcellulose (M3434 Stem Cell Technologies, with 10 ng/ml GM-CSF) 72 hours post-isolation according to manufacturers protocols and replatings performed at 6/7-day intervals. At third replating cells were transferred to liquid culture conditions as above and growth monitored.
siRNA transfection
Cells were electroporated with 100 nM siRNA oligonucleotides as described previously.(10;11) Transfection efficiency was greater than 95% as assessed by electroporation of a Cy3-labelled siLaminA oligonucleotide pair into control cells. Oligonucleotide pairs or commercial reagents used for targeting are described in Supplementary Table 1.
RNA and protein isolation
Total RNA was isolated from cultured cells using RNeasy columns (Qiagen). Protein was isolated from flow-through fractions as previously.(11) RNA, DNA and protein were extracted from patient samples using TRIzol (Invitrogen) as per manufacturer's instructions. Protein samples were dissolved in 9 M urea, 1% (w/w) dithiothreitol.
Real-time mRNA analysis
cDNA synthesized from 33 ng RNA using M-MLV Reverse Transcriptase RNase H minus point mutant (Promega) was used per reaction with either Universal or SYBR Green master mix (Applied Biosystems). Oligonucleotide sequences (Sigma-Genosys) or commercial products used are described in Supplementary Table 1. Normalized mean gene expression values ± S.D. were determined from triplicate cycle threshold (CT) values for each gene and the housekeeping gene 18S or GAPDH. Relative transcript levels were determined by the 2−ΔΔCT method.
Immunoblot analysis
Cell lysates (20-50 μg) were separated by SDS-PAGE, transferred to membranes, blocked, and subjected to overnight primary antibody incubation with anti-ETO C-20 (Santa Cruz), anti-Brn3a MAB1585 (Chemicon), anti β-actin (Sigma), anti-GAPDH, anti-βTubulin or anti-HA (Cell Signaling). Expression was quantitated using a GS800 densitometer and QuantityOne software (Biorad).
EMSA analysis
Whole cell lysates from Kasumi-1 or 293T cells were incubated with either cold oligonucleotide or specific antisera recognising AML1 (Ab-1, Calbiochem) or ETO (C-20) prior to 20 min incubation with 32P-dCTP radiolabelled double-stranded oligonucleotides. Complexes were resolved in 5% acrylamide non-denaturing gels. Oligonucleotides are detailed in Supplementary Table 1.
FACS analysis
Cells pre-blocked in normal donkey serum (human cells) or unlabeled anti-Fc{gamma} III/II receptor mAb2.4G2 (murine cells) were stained in PBS containing 2% FCS, 0.1% BSA, 0.1% sodium azide with isotype controls or specific antibodies anti-human CD117-PE 555714 (BD), anti-mouse Cd11b, CD117, F4/80, CD61, and/or anti-humanCD2 (all eBioscience). Data were acquired using LSR (Becton Dickinson) or CyAnADP (DakoCytomation) analysers. Cell sorting was performed using a MoFloXDP (Beckman Coulter).
Immunofluorescence
Cells were cytospun, paraformaldehyde fixed, rinsed twice in PBS then permeabilised with 0.1% (v/v) Triton X-100 in PBS before incubation in PBS containing 0.1% BSA and 0.1% sodium azide. After primary antibody incubation (rabbit anti-Brn3a,(33) goat anti-ETO, normal rabbit or goat IgG controls - Santa Cruz) and washing, cells were incubated with FITC-conjugated donkey anti-goat IgG and TRITC-conjugated donkey anti-rabbit IgG; (both F(ab')2 fragments Jackson Immunoresearch), and DAPI (0.1 μg/ml - Polysciences, Inc.). Washed coverslips were mounted and cells visualized by confocal laser scanning microscopy (LSM510; Zeiss).
Chromatin immunoprecipitation
This was performed by a modification of Ivins et al.(34) Cells (5×106) were incubated with 1% formaldehyde in growth medium for 10 min at 37 °C, washed and then lysed in 0.5 ml lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris pH 8.0, 1 mM PMSF, 1 μg/ml aprotinin 1 μg/ml leupeptin) for 10 min on ice. After sonication 10% lysate was removed as input and remainder diluted in IP dilution buffer (0.01% SDS, 0.01% Triton X-100, 2 mM EDTA, 150 mM NaCl, 20 mM Tris pH 8.0, plus protease inhibitors). Lysate was divided equally and incubated with 1 μg test or control antibodies: normal goat IgG, α-ETO C-20, α-AML1 (Ab-1) or α-AML1 (Ab-2, Calbiochem); the latter does not recognize AML1 when DNA-bound and is a negative control. Precipitated complexes were cross-link reversed, proteinase-K digested, and purified DNA was subjected to real-time PCR using oligonucleotide primers and probes (Supplementary Table 1). Quantitation by 2−ΔΔCT method was as per manufacturers protocol (Applied Biosystems) comparing input to test samples, results displayed as ratio of test to control antibody signals for each cell type.
Statistical analyses
Comparisons to determine statistical significance of differences were performed using two-sided Student's t-test, either paired or unpaired as appropriate.
Results
Expression of BRN3A is extremely high and nuclear in t(8;21) patient cells
Global gene expression profiling of AML has identified high expression of POU4F1/BRN3A to be correlated closely with presence of t(8;21) translocation.(1-3) To confirm high BRN3A expression in primary t(8;21) cells we performed real-time PCR, immunoblot and immunofluoresence analyses (Figures 1A-C). Real-time PCR verified that both bone marrow and peripheral blood t(8;21) samples exhibit very high BRN3A expression in comparison with other AML samples (Figure 1A). Immunoblot and immunofluoresence analyses using two independent antisera confirmed that BRN3A protein is expressed abundantly in t(8;21) primary cells (Figures 1B and 1C), with the long isoform BRN3A(L) being predominant. Importantly, expression of BRN3A is nuclear and exhibited by cells also expressing AML1/ETO as detected by α-ETO immunofluorescence (Figure 1C). In the t(8;21) cell lines Kasumi-1 and SKNO-1 there is significant BRN3A expression, however this is 50-100 fold reduced in comparison with primary t(8;21) samples and similar to that in other cell lines (Figure 1D).
Figure 1. BRN3A protein is highly expressed in nuclei of t(8;21) cells.

A) Real-time PCR analysis of BRN3A relative to 18S expression in patient blasts from bone marrow (BM) and peripheral blood (PB), expression relative to highest expressing sample in a representative experiment is shown. Non-t(8;21) (other) include both marrow and blood samples. B) Immunoblot analysis of BRN3A and GAPDH in AML patient samples. The lowest band (asterisk) represents a non-specific cross-reactive protein as BRN3A transcripts are absent from the non-t(8;21) samples shown. The 45 kDa long (L) isoform of BRN3A is predominant whilst the 32 kDa short (S) isoform is also present in t(8;21) samples. C) Indirect immunofluorescence of M2 t(8;21) and M3 t(15;17) patient blasts and Kasumi-1 cells using specific anti-sera or IgG control as indicated, nuclei visualised using DNA counterstain (DAPI). One representative experiment of three is shown. D) Real-time PCR analysis of BRN3A relative to GAPDH and 18S expression in cell lines, a representative patient sample and cord blood (CB), relative to Kasumi-1. BRN3A(L) expression in ND7 cells (neuronal positive control), haematopoietic cell lines and primary AML samples (as indicated) was determined by immunoblot (inset).
AML1/ETO proteins promote BRN3A expression in t(8;21) AML cells
All primary adult t(8;21) samples studied thus far exhibit high BRN3A expression(1), and this correlation may result from direct regulation of BRN3A by AML1/ETO. To test this hypothesis, expression of AML1/ETO in primary t(8;21) patient cells was reduced by siRNA and the effect on endogenous BRN3A transcript levels determined. As observed previously,(10;11) AML1/ETO expression was reduced efficiently by this method (Figure 2A). Loss of AML1/ETO significantly reduced BRN3A expression (Figure 2A and 2B) indicating BRN3A as a potential AML1/ETO target. Similar AML1/ETO targeting in t(8;21) Kasumi-1 and SKNO-1 cells also decreased low basal BRN3A expression, but generally less than two-fold (data not shown), explaining why BRN3A was not identified as an AML1/ETO target in previous cell-line microarray screens whose cut-off requirement was two-fold regulation(11).
Figure 2. AML1/ETO promotes BRN3A/Brn3a expression.
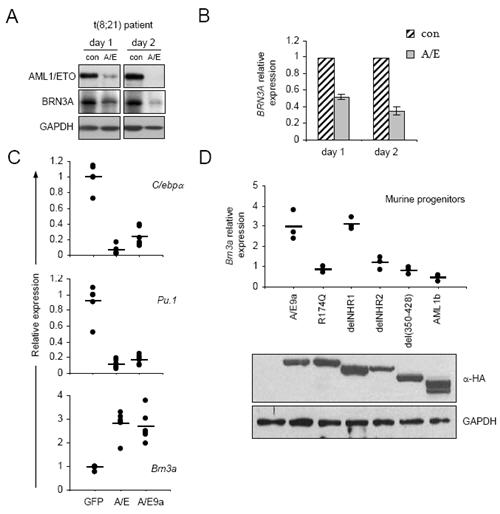
A) Immunoblot analysis of AML1/ETO, BRN3A and GAPDH in Patient M2 t(8;21) blasts electroporated on day 0 with siRNAs targeting AML1/ETO (A/E) or control (con) and harvested at indicated timepoints. BRN3A immunoblots show BRN3A(L). An additional independent t(8;21) patient showed similar results. B) Densitometric analysis of immunoblots as in A, mean ± S.E. of two experiments C) Real-time PCR analyses of transcripts as indicated relative to 18s in murine progenitor cells transduced with retroviruses encoding GFP (GFP) or AML1/ETO (A/E) or AML1/ETO9a (9a), and sorted for GFP positivity prior to lysis 48 hrs later. Data are pooled from three separate experiments using cells from both day E12.5 (2 expts) and E13.5 (1 expt) foetal liver, each data point represents one of two experimental replicates, relative to the first GFP sample in each experiment. Comparisons with Actin and Gapdh gave similar results (data not shown). D) In similar experiments to those described in C, GFP, AML1/ETO9a, AML1b and the indicated AML1/ETO9a mutants were expressed in progenitors and Brn3a expression monitored in sorted cells harvested at 48 hrs (graph). Expression of AML1 proteins was confirmed by anti-HA immunoblotting of producer cell lysates (bottom), leftmost lane from GFP alone transfected.
The relatively rapid reduction in BRN3A expression following AML1/ETO siRNA treatment suggested a direct relationship between the fusion protein and BRN3A. To determine whether AML1/ETO is sufficient to increase Brn3a expression, AML1/ETO or its shorter variant AML1/ETO9a(17) were introduced into primary murine haematopoietic progenitor cells. Significant down-regulation of myeloid differentiation transcripts C/ebpa and Pu.1 verified correct function of AML1/ETO (P≤0.001, Figure 2C). In contrast, Brn3a transcript levels were significantly increased approximately three-fold 48 hrs after either AML1/ETO or AML1/ETO9a transduction (P≤0.001, Figure 2C). To investigate the mechanism by which AML1/ETO up-regulates Brn3a, the effect of specific AML1/ETO9a mutants and wild-type AML upon Brn3a expression was examined (Figure 2D). AML1/ETO9a mutants R174Q, delNHR2 and del(350-428) which disrupt DNA binding, oligomerisation and NCoR interaction respectively(31) do not upregulate Brn3a expression (P=0.238, P=0.403, P=0.188) (Figure 2D). In contrast, AML1/ETO9a with delNHR1 mutation, which disrupts E-protein interaction, remains competent to upregulate Brn3a significantly (P=0.007). Thus the pattern of Brn3a regulation by AML1/ETO9a mutants parallels their ability to induce rapid leukaemia in vivo.(31) In an opposite manner AML1b significantly represses Brn3a expression in this assay (P=0.036) further supporting direct regulation of Brn3a by AML1-containing proteins.
The AML1 DNA-binding domain is intact in both AML1/ETO and AML1/ETO9a,(17) thus both are likely to bind DNA in a similar if not identical manner to AML1. MatInspector screening(35) of the entire BRN3A locus identified three potential sites with varying homology to the AML1 binding consensus(4) in both upstream and downstream regulatory regions (Figures 3A and 3B). Although these AML1 binding sites are not globally conserved in the mouse, all are proximal to validated(36) and predicted BRN3A binding sites. Initially, EMSA analyses showed that. all three sites can bind to a specific factor present in 293T cells transfected with AML1/ETO but not GFP (Figure 3C upper panel). Mutation of AML1 sites in competing unlabelled oligonucleotides prevented competition for binding, demonstrating interaction specificity. Presence of AML1/ETO in these complexes and similar endogenous complexes in Kasumi-1 cells was confirmed by supershift analysis using both anti-ETO and anti-AML antisera (Figure 3C lower panel). An additional complex present in all extracts bound specifically to site 2 in a manner dependent upon an intact AML1 site but was not supershifted by either antisera.
Figure 3. AML1/ETO can bind to the POU4F1/BRN3A locus via AML1 sites.
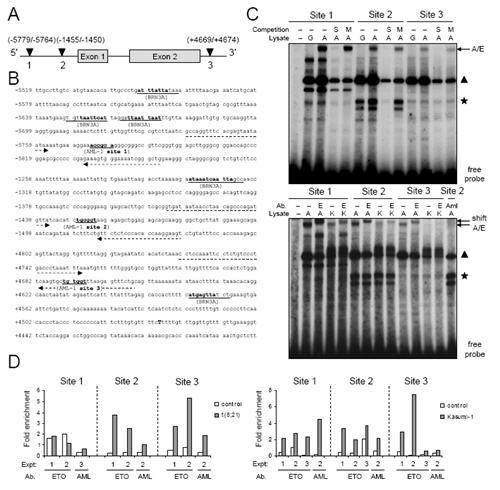
A) A schematic of AML1 sites identified close to the BRN3A exons by screening the entire human POU4F1/BRN3A locus (Accession AL445209) for potential AML1 and BRN3A binding sites using MatInspector software.(35) B) Detail of potential binding sites in the BRN3A locus. Numbering relative to translation start is indicated left, potential binding sites are underlined, dotted arrows indicate sites of primers used for chromatin immunoprecipitation (ChIP) analysis, end of exon 2 is indicated in bold capital. C) EMSA analysis of AML1/ETO binding to AML1 sites 1, 2 and 3 in the POU4F1/BRN3A locus. Radiolabelled site 1, 2 and 3 probes as indicated were incubated without lysate (−) or with lysate from either GFP-transfected 293T cells (G) or AML1/ETO-transfected 293T cells (A) or Kasumi-1 cells (K). Upper panel, Reactions were subject to either no competition (−) or competition with a 50-fold excess of either unlabelled same oligonucleotide (S) or same oligonucelotide with mutant AML1 binding site (M). A potential AML1/ETO complex migrated above a non-specific complex (triangle) and a specific complex formed only with site 2 probe (asterisk). Lower panel, Identity of the potential AML1/ETO complex was confirmed by supershift with either an anti-ETO antibody (E) or anti-AML1 antibody (Aml). The faster-migrating site 2-specific complex (asterisk) was unaffected by either antibody. D Left panel) ChIP analysis of AML1/ETO binding to potential BRN3A sites in t(8;21) patient blasts (Expt1 M2, expt2 M1) cultured for 24 hr. Negative controls were non-t(8;21) blasts (M4 NK - white bars). Specific signals relative to input are expressed as fold enrichment over signal obtained with total IgG (for anti-ETO) or anti-AML-RHD (for anti-AML). AML1/ETO-specific versus control precipitations did not enrich for fragments of the β−2M locus (data not shown). AML1/ETO binding determined by anti-ETO and anti-AML precipitation data combined was significant at site 3 (P=0.046), borderline significant at site 2 (P=0.058) and not significant at site 1 (P=0.361). D right panel) Kasumi-1 cells were subjected to ChIP analysis as in A to measure binding of AML1/ETO to the BRN3A locus (grey bars). AML1/ETO negative U937 or Bristol-8 cells were used as negative controls (white bars). AML1/ETO binding determined by anti-ETO and anti-AML precipitation data combined was significant at sites 1 and 2 (P=0.012 and 0.004 respectively), and not significant at site 3 (P=0.103). Experiments were performed at least three times for each site in each cell type with either ETO or AML antibodies.
To examine AML1/ETO binding to the endogenous POU4F1/BRN3A gene, chromatin immunoprecpitation (ChIP) assays were performed using both t(8;21) patient and Kasumi-1 cells. Importantly, both anti-ETO and anti-AML anti-sera co-precipitate two of the predicted AML1 binding sites in BRN3A, sites 2 and 3, from patient t(8;21) cells (Figure 3D left panel). Binding to the BRN3A locus occurs also in Kasumi-1 cells, although only significantly at sites 1 and 2 (Figure 3D right panel). This altered binding correlates with reduced BRN3A expression (Figure 1D) and its reduced dependency upon AML1/ETO in Kasumi-1 compared with primary t(8;21) AML. In summary, siRNA, EMSA and ChIP studies of AML1/ETO in t(8;21) cells alongside AML1/ETO overexpression studies in primary cells combine to suggest that BRN3A is a novel direct target of AML1/ETO.
High Brn3a expression in murine progenitor cells promotes myeloid differentiation
AML1/ETO increases Brn3a expression in murine progenitors in vitro, however resulting expression levels are modest when compared to those in patient samples. Therefore to investigate the function of high BRN3A expression in haematopoietic cells we over-expressed Brn3a in multi-lineage progenitors, and monitored lineage phenotypes and methylcellulose replating capacity. Progenitors were transduced with retroviruses encoding Brn3a cDNA under the control of MSCV promoter (Figure 4A) and infection monitored using either selection for neomycin resistance or electronic gating for humanCD2tailless (hCD2) marker gene expression. Background expression of Brn3a was low in purified progenitor populations (Figure 2C,D and data not shown), and expression of Brn3a protein from the viruses was confirmed in the packaging cell line LinXE (Figure 4A).
Figure 4. High Brn3a expression promotes myeloid differentiation.
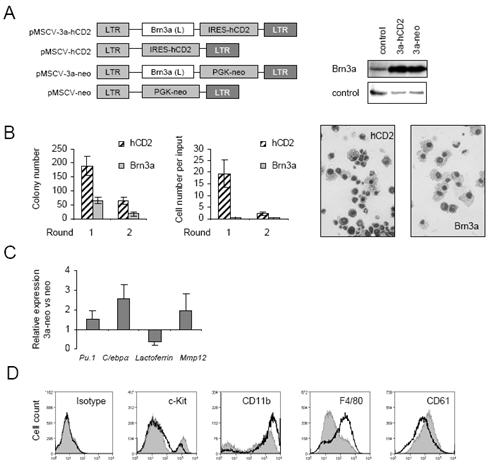
A, left panel) Retroviral plasmids encoding Brn3a(L) cDNAs under the control of MSCV promoter and co-expressing tailless human CD2 (hCD2) or neomycin resistance marker genes were generated as described in Materials and Methods. A, right panel) Brn3a overexpression from retroviruses in comparison with pMSCV-neo (control) was confirmed by immunoblotting of packaging cells. B) Semi-solid colony-forming assays of murine haematopoietic progenitor cells from day E13.5 foetal liver transduced with pMSCV-hCD2 (hCD2) or pMSCV-3a-hCD2 (Brn3a-hCD2) viruses, 1 × 104 FACS-sorted hCD2+ cells plated into each culture 2 days after infection. Colony number per plate and cell number were monitored at rounds 1 and 2, cell morphology (× 40 magnification) at round 1. C) Real-time PCR analysis of gene expression as indicated relative to that of 18s in progenitor cells transduced with pMSCV-neo (control) or pMSCV-3a-neo (Brn3a) and cultured for 6 days under G418 selection. D) Cell-surface FACS analysis of cells transduced with pMSCV-neo (grey filled histogram) or pMSCV-3a-neo (black line histogram) viruses as in C performed at day 9 post-infection, data are representative of three independent experiments
In contrast to AML1/ETO, whose expression is sufficient to immortalise murine progenitor cells in methylcellulose replating assays,(6) over-expression of Brn3a alone inhibited replating capacity (Figure 4B). While control cultures contained multiple myeloid lineages at the first round of replating, Brn3a-hCD2 transduced cultures comprised almost exclusively macrophages, with occasional neutrophils (Figure 4B). Similarly Brn3a-neo infected cultures failed to replate and exhibited macrophage morphology (data not shown). Furthermore, real-time PCR analyses showed that ectopic Brn3a increases expression of myeloid genes Pu.1, Cebpα, and macrophage-specific Mmp12 (Figure 4C), while decreasing expression of the neutrophil marker gene Lactoferrin. FACS analysis showed that ectopic Brn3a decreases CD117/c-Kit and CD61 expression and increases CD11b and F4/80 expression on the cell surface (Figure 4D). Thus combined data from replating assays and morphology, gene expression, and surface marker analyses indicate that ectopic Brn3a induces monocyte/macrophage differentiation at the expense of neutrophilic and erythroid differentiation.
AML1/ETO proteins alter Brn3a function in myeloid differentiation
High BRN3A levels in AML occur only in the context of t(8;21)-dependent AML1/ETO expression. Therefore AML1/ETO was co-expressed alongside Brn3a in murine progenitors by dual infection using GFP and hCD2 markers respectively. As previously described,(6;8) AML1/ETO alone impaired myeloid differentiation as determined by reduced CD11b expression at the first round of replating as did the splice variant AML1/ETO9a (Figure 5A upper panels and 5B). AML1/ETO reduced cell number immediately after transduction consistent with growth arrest observed in previous studies,(8) whilst either AML1/ETO or AML1/ETO9a alone enabled continued long-term replating and immortalisation (data not shown). Loss of CD117/c-Kit and gain of CD11b induced by ectopic Brn3a (Figure 4D) was not affected by co-transduction with GFP control virus (Figure 5A left panels and 5B). Importantly, co-expression of AML1/ETO or AML1/ETO9a respectively reduced and abolished the ability of Brn3a to induce differentiation. In fact, Brn3a over-expression in the presence of AML1/ETO9a appeared to inhibit differentiation as indicated by increased percentage c-Kit positivity (Figure 5b). Morphological analyses of FACS-sorted Brn3a-hCD2+/GFP+ and Brn3a-hCD2+/AML1/ETO9a+ infected populations at round 1 of replating (Figure 5C) confirmed impairment of Brn3a-dependent differentiation by AML1/ETO9a. Furthermore, gene expression analyses 48 hr post-transduction (Figure 5D) showed that AML1/ETO9a inhibits Brn3a-dependent activation of Pu.1 and C/ebpa but may co-operate with Brn3a to repress Lactoferrin and activate Mmp12.
Figure 5. AML1/ETO proteins inhibit Brn3a function in myeloid differentiation.
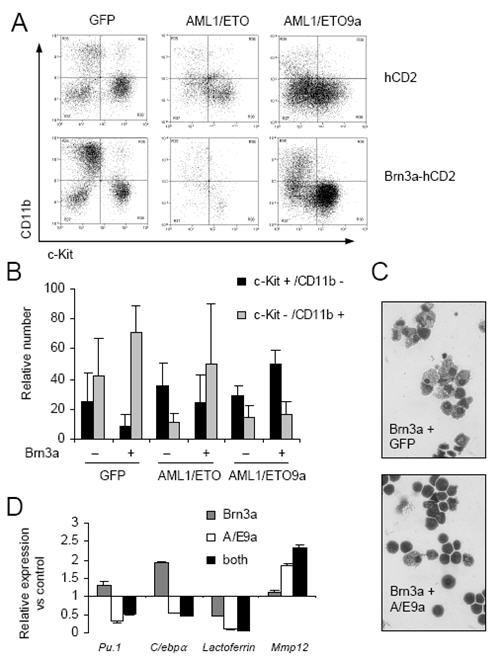
A, B) FACS analysis of CD11b and CD117 expression at day 3 on progenitors infected at day 0 with pMSCV-GFP (GFP) or pMiG-AML1/ETO (AML1/ETO) or pMiG-AML1/ETO9a (AML1/ETO9a) and at day 1 with either pMSCV-hCD2 (hCD2 control) or pMSCV-3a-hCD2 (Brn3a). Graph represents mean percentage of total viable population ± S.D. from three experiments. Differentiated cell number (c-Kit−/CD11b+) relative to progenitor number (c-Kit+/CD11b−) was increased in Brn3a/GFP cultures (P=0.013), not significantly different in Brn3a/AML1ETO cultures (P=0.386), and decreased in Brn3a/AML1ETO9a cultures (P=0.010). C) Morphology of FACS-sorted GFP+/hCD2+ cells. D) Real-time PCR analysis of gene expression relative to that of 18s in hCD2+/GFP+ sorted progenitors 48 hr after dual-transduction, relative to hCD2/GFP control.
Brn3a contributes to AML1/ETO-dependent immortalisation
As upregulation of Brn3a expression closely parallels leukaemogenicity of AML1/ETO9a proteins and AML1/ETO9a plus Brn3a co-expression produces high c-Kit positivity, we considered that endogenous Brn3a might play a role in AML1/ETO-dependent immortalisation. To test this, several Brn3a-targeting retroviral shRNA plasmids containing the hCD2 marker gene were generated and tested (Figure 6A). The most efficient of these, sh24 and sh43, or a non-targeting control (shcon), were co-transduced into progenitors alongside either AML1/ETO or an unrelated oncogene also capable of myeloid immortalisation, E2A/HLF.(37) ShRNA effects on immortalisation were determined by measuring hCD2 positivity over time. Strikingly, sh24- or sh43-transduced cells were lost from AML1/ETO but not E2A/HLF cultures (Figures 6B and 6C). Absolute growth curves in liquid culture confirmed that AML1/ETO-dependent growth can be dramatically reduced by presence of either Brn3a-targeting plasmid, consistent with Brn3a having an important role in immortalisation driven by AML1/ETO.
Figure 6. Brn3a is required for AML1-ETO dependent immortalisation of murine progenitors.
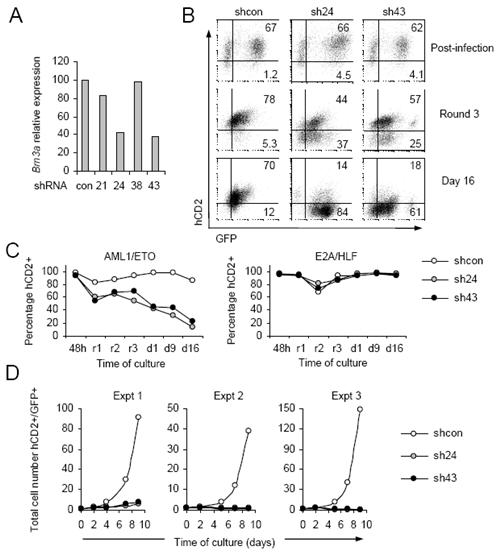
A) Real-time PCR analysis of Brn3a expression relative to that of 18s in unsorted progenitors 48 hr after transduction to approximately 80% efficiency with non-targeting pM2miR-based retrovirus (con) or viruses designed to target Brn3a (21, 24, 38 and 43). B) Representative FACS analysis of hCD2 and GFP positivity in murine progenitors 48 hrs (post-infection) after simultaneous transduction with pM2miR (shcon, sh24 or sh43) alongside pMIG-AML1/ETO (GFP), to 60-70 % double-positive efficiency. Positivity was monitored during subsequent semi-solid methylcellulose culture (Round 3) and liquid culture (Day 16). C) Representative hCD2+ in AML1/ETO and E2A/HLF cultures co-transduced with pM2miR viruses, monitored 48 hr after transduction (48h) at rounds 1-3 of semi-solid culture replating (r1-r3) and at days 1, 9 and 16 of subsequent liquid culture (d1,9,16). D) Numbers of viable hCD2+/GFP+ cells during suspension culture.
Discussion
The most widely described mechanism of action for AML1/ETO is one of DNA-binding dependent transcriptional repression at AML1 binding sites in genes whose expression is activated by normal AML1.(4) Recent gene expression profiling experiments indicate that AML1/ETO may not only repress a specific set of target genes but also increase the expression of others.(11;20-23) However, whilst mechanisms for AML1/ETO dependent transcriptional inhibition have been relatively well characterised,(4;18;19) the means by which AML1/ETO might directly activate transcription are unclear.
We show that BRN3A expression in t(8;21) cells is maintained by AML1/ETO, that AML1/ETO can up-regulate Brn3a expression in a DNA-binding dependent manner, and that AML1/ETO can bind to consensus AML1 TGT/CGGT sites in the BRN3A gene. These findings therefore add BRN3A to the short list of genes including TRKA whose expression is up-regulated by AML1/ETO via potentially direct means.(22) It is most likely that BRN3A has not previously been identified as a potential AML1/ETO target gene due to moderate degree of regulation by AML1/ETO in cell lines and potential tissue-specific requirements for this effect. Previous studies have demonstrated negative autoregulation by Brn3a at its own locus,(36) and binding of AML1/ETO adjacent to BRN3A may activate BRN3A expression via a de-repressive effect.(38;39) Our results from AML1/ETO9a mutant studies do suggest that corepressor recruitment to AML1/ETO9a may be important for Brn3a control. Additionally, upregulation of BRN3A by AML1/ETO in t(8;21) AML may be enhanced by either in vivo factors or possible second-hit pathways(5;16;40) in this disease.
Brn3a is sufficient to induce terminal neuronal differentiation when over-expressed in neuronal precursor type cells.(24;27;41) The present study provides evidence that in an analogous manner Brn3a over-expression in haematopoietic progenitors (in the absence of AML1/ETO) promotes terminal monocyte/macrophage differentiation, associated with Pu.1, C/ebpa and Mmp12 activation and Lactoferrin repression. Candidate Brn3a binding sites in the activated promoters hint at some direct Brn3a-dependent regulation of these important haematopoietic genes, while potential inhibition of Brn3a expression by exogenous AML1 further suggests a role for Brn3a in haematopoiesis.
In the presence of AML1/ETO, Brn3a appears to support immortalisation. Previously the Brn3a long isoform predominant in t(8;21) cells has been described to co-operate with Ras to transform rat fibroblasts,(42) and we show here it may co-operate with AML1/ETO but not E2A/HLF to inhibit myeloid differentiation and promote growth of murine progenitor cells. This AML1/ETO-dependent switch of Brn3a function, from inducer to inhibitor of differentiation provides a parallel with the inhibition of Brn3a neuronal differentiation function by EWS/FLI1 fusions proteins present in Ewing's sarcoma.(41)
Importantly, AML1/ETO prevents Brn3a-induced Pu.1 and C/ebpa activation while potentially co-activating Mmp12. There are several possibilities regarding the mechanisms for these phenomena. Firstly, AML1/ETO could bind Brn3a directly, but despite interaction in pulldown assays (D.M.G. unpublished data) the two proteins do not extensively co-localise in t(8;21) cells. Possibly adjacent AML1 and Brn3a binding sites in Mmp12 facilitate interaction and co-operative target regulation. In contrast, AML1/ETO is likely to control Brn3a function at Pu.1 and C/ebpa promoters (where adjacent binding sites appear to be absent) via altered cofactor recruitment or changes in promoter structure. Inhibitory interaction of Brn3a with HIPK2,(43) which is mutated in some AML,(44) may also modulate Brn3a function in t(8;21) cells. The ability of both AML1/ETO and Brn3a to upregulate TrkA growth factor expression(22;45) may also be relevant in t(8;21) AML pathogenesis.
In summary, AML1/ETO contributes to both high expression and aberrant function of BRN3A in t(8;21) cells. Identification of specific pathways and target genes regulated by the AML1/ETO and BRN3A combination, and additional factors controlling BRN3A expression should provide insight into t(8;21) AML pathogenesis. Reactivation of BRN3A pro-differentiation function by inhibition of AML1/ETO, and/or inhibition of BRN3A expression directly may prove therapeutically beneficial in t(8;21) AML.
Supplementary Material
Acknowledgements
The authors thank patients who contributed to this study, D.E. Zhang for AML1/ETO plasmids, E. Turner (Dept. Psychiatry, UCSD, LaJolla, CA) for Brn3a antiserum, G. Molloy for patient samples, A. Tonks and S. Debernardi for sharing data, J. Marshall, A. Eddaoudi and CRUK FACS facility for excellent technical assistance, and O. Williams for AML1b plasmid and useful discussions.
Funding: Cancer Research UK (J.D and B.Y.), Children With Leukaemia (D.M.G. and H.B.) and the Deutsche Jose Carreras Leukaemie-Stiftung (O.H.)
Footnotes
No potential conflicts of interest were disclosed.
Reference List
- 1.Schoch C, Kohlmann A, Schnittger S, Brors B, Dugas M, Mergenthaler S, et al. Acute myeloid leukemias with reciprocal rearrangements can be distinguished by specific gene expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(15):10008–13. doi: 10.1073/pnas.142103599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Debernardi S, Lillington DM, Chaplin T, Tomlinson S, Amess J, Rohatiner A, et al. Genome-wide analysis of acute myeloid leukemia with normal karyotype reveals a unique pattern of homeobox gene expression distinct from those with translocation-mediated fusion events. Genes Chromosomes & Cancer. 2003;37(2):149–58. doi: 10.1002/gcc.10198. [DOI] [PubMed] [Google Scholar]
- 3.Ross ME, Mahfouz R, Onciu M, Liu HC, Zhou XD, Song GC, et al. Gene expression profiling of pediatric acute myelogenous leukemia. Blood. 2004;104(12):3679–87. doi: 10.1182/blood-2004-03-1154. [DOI] [PubMed] [Google Scholar]
- 4.Licht JD. AML1 and the AML1-ETO fusion protein in the pathogenesis of t(8;21) AML. Oncogene. 2001;20(40):5660–79. doi: 10.1038/sj.onc.1204593. [DOI] [PubMed] [Google Scholar]
- 5.Peterson LF, Boyapati A, Ahn EY, Biggs JR, Okumura AJ, Lo MC, et al. Acute myeloid leukemia with the 8q22;21q22 translocation: secondary mutational events and alternative t(8;21) transcripts. Blood. 2007;110(3):799–805. doi: 10.1182/blood-2006-11-019265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rhoades KL, Hetherington CJ, Harakawa N, Yergeau DA, Zhou LM, Liu LQ, et al. Analysis of the role of AML1-ETO in leukemogenesis, using an Inducible transgenic mouse model. Blood. 2000;96(6):2108–15. [PubMed] [Google Scholar]
- 7.Mulloy JC, Cammenga J, MacKenzie KL, Berguido FJ, Moore MAS, Nimer SD. The AML1-ETO fusion protein promotes the expansion of human hematopoietic stem cells. Blood. 2002;99(1):15–23. doi: 10.1182/blood.v99.1.15. [DOI] [PubMed] [Google Scholar]
- 8.de Guzman CG, Warren AJ, Zhang Z, Gartland L, Erickson P, Drabkin H, et al. Hematopoietic stem cell expansion and distinct myeloid developmental abnormalities in a murine model of the AML1-ETO translocation. Molecular and Cellular Biology. 2002;22(15):5506–17. doi: 10.1128/MCB.22.15.5506-5517.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tonks A, Tonks AJ, Pearn L, Pearce L, Hoy T, Couzens S, et al. Expression of AML1-ETO in human myelomonocytic cells selectively inhibits granulocytic differentiation and promotes their self-renewal. Leukemia. 2004;18(7):1238–45. doi: 10.1038/sj.leu.2403396. [DOI] [PubMed] [Google Scholar]
- 10.Heidenreich O, Krauter J, Riehle H, Hadwiger P, John M, Heil G, et al. AML1/MTG8 oncogene suppression by small interfering RNAs supports myeloid differentiation of t(8;21)-positive leukemic cells. Blood. 2003;101(8):3157–63. doi: 10.1182/blood-2002-05-1589. [DOI] [PubMed] [Google Scholar]
- 11.Dunne J, Cullmann C, Ritter M, Soria NM, Drescher B, Debernardi S, et al. siRNA-mediated AML1/MTG8 depletion affects differentiation and proliferation-associated gene expression in t(8 ; 21)-positive cell lines and primary AML blasts. Oncogene. 2006;25(45):6067–78. doi: 10.1038/sj.onc.1209638. [DOI] [PubMed] [Google Scholar]
- 12.Fazi F, Zardo G, Gelmetti V, Travaglini L, Ciolfi A, Di Croce L, et al. Heterochromatic gene repression of the retinoic acid pathway in acute myeloid leukemia. Blood. 2007;109(10):4432–40. doi: 10.1182/blood-2006-09-045781. [DOI] [PubMed] [Google Scholar]
- 13.Yergeau DA, Hetherington CJ, Wang Q, Zhang P, Sharpe AH, Binder M, et al. Embryonic lethality and impairment of haematopoiesis in mice heterozygous for an AML1-ETO fusion gene. Nature Genetics. 1997;15(3):303–6. doi: 10.1038/ng0397-303. [DOI] [PubMed] [Google Scholar]
- 14.Fenske TS, Pengue G, Mathews V, Hanson PT, Hamm SE, Riaz N, et al. Stem cell expression of the AML1/ETO fusion protein induces a myeloproliferative disorder in mice. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(42):15184–9. doi: 10.1073/pnas.0400751101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang YY, Zhou GB, Yin T, Chen B, Shi JY, Liang WX, et al. AML1-ETO and C-KIT mutation/overexpression in t(8;21) leukemia: Implication in stepwise leukemogenesis and response to Gleevec. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(4):1104–9. doi: 10.1073/pnas.0408831102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schessl C, Rawat VPS, Cusan M, Deshpande A, Kohl TM, Rosten PM, et al. The AML1-ETO fusion gene and the FLT3 length mutation collaborate in inducing acute leukemia in mice. Journal of Clinical Investigation. 2005;115(8):2159–68. doi: 10.1172/JCI24225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yan M, Kanbe E, Peterson LF, Boyapati A, Miao Y, Wang Y, et al. A previously unidentified alternatively spliced isoform of t(8;21) transcript promotes leukemogenesis. Nature Medicine. 2006;12(8):945–9. doi: 10.1038/nm1443. [DOI] [PubMed] [Google Scholar]
- 18.Linggi B, Muller-Tidow C, van de Locht L, Hu M, Nip J, Serve H, et al. The t(8;21) fusion protein, AML1-ETO, specifically represses the transcription of the p14(ARF) tumor suppressor in acute myeloid leukemia. Nature Medicine. 2002;8(7):743–50. doi: 10.1038/nm726. [DOI] [PubMed] [Google Scholar]
- 19.Follows GA, Tagoh H, Lefevre P, Hodge D, Morgan GJ, Bonifer C. Epigenetic consequences of AML1-ETO action at the human c-FMS locus. Embo Journal. 2003;22(11):2798–809. doi: 10.1093/emboj/cdg250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alcalay M, Meani N, Gelmetti V, Fantozzi A, Fagioli M, Orleth A, et al. Acute myeloid leukemia fusion proteins deregulate genes involved in stem cell maintenance and DNA repair. Journal of Clinical Investigation. 2003;112(11):1751–61. doi: 10.1172/JCI17595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fliegauf M, Stock M, Berg T, Lubbert M. Williams-Beuren syndrome critical region-5/non-T-cell activation linker: a novel target gene of AML1/ETO. Oncogene. 2004;23(56):9070–81. doi: 10.1038/sj.onc.1208042. [DOI] [PubMed] [Google Scholar]
- 22.Mulloy JC, Jankovic V, Wunderlich M, Delwel R, Cammenga J, Krejci O, et al. AML1-ETO fusion protein up-regulates TRKA mRNA expression in human CD34(+) cells, allowing nerve growth factor-induced expansion. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(11):4016–21. doi: 10.1073/pnas.0404701102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tonks A, Pearn L, Musson M, Gilkes A, Mills KI, Burnett AK, et al. Transcriptional dysregulation mediated by RUNX1-RUNX1T1 in normal human progenitor cells and in acute myeloid leukaemia. Leukemia. 2007;21(12):2495–505. doi: 10.1038/sj.leu.2404961. [DOI] [PubMed] [Google Scholar]
- 24.Lakin ND, Morris PJ, Theil T, Sato TN, Moroy T, Wilson MC, et al. Regulation of Neurite Outgrowth and Snap-25 Gene-Expression by the Brn-3A Transcription Factor. Journal of Biological Chemistry. 1995;270(26):15858–63. doi: 10.1074/jbc.270.26.15858. [DOI] [PubMed] [Google Scholar]
- 25.McEvilly RJ, Erkman L, Luo L, Sawchenko PE, Ryan AF, Rosenfeld MG. Requirement for Brn-3.0 In differentiation and survival of sensory and motor neurons. Nature. 1996;384(6609):574–7. doi: 10.1038/384574a0. [DOI] [PubMed] [Google Scholar]
- 26.Xiang MQ, Gan L, Zhou LJ, Klein WH, Nathans J. Targeted deletion of the mouse POU domain gene Brn-3a causes a selective loss of neurons in the brainstem and trigeminal ganglion, uncoordinated limb movement, and impaired suckling. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(21):11950–5. doi: 10.1073/pnas.93.21.11950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ensor E, Smith MD, Latchman DS. The BRN-3A transcription factor protects sensory but not sympathetic neurons from programmed cell death/apoptosis. Journal of Biological Chemistry. 2001;276(7):5204–12. doi: 10.1074/jbc.M007068200. [DOI] [PubMed] [Google Scholar]
- 28.Asou H, Tashiro S, Hamamoto K, Otsuji A, Kita K, Kamada N. Establishment of A Human Acute Myeloid-Leukemia Cell-Line (Kasumi-1) with 8-21 Chromosome-Translocation. Blood. 1991;77(9):2031–6. [PubMed] [Google Scholar]
- 29.Ruiz A, Williams O, Brady HJM. The Ikaros splice isoform, Ikaros 6, immortalizes murine haematopoietic progenitor cells. International Journal of Cancer. 2008;123(6):1240–5. doi: 10.1002/ijc.23706. [DOI] [PubMed] [Google Scholar]
- 30.Gascoyne DM, Long E, Veiga-Fernandes H, de Boer J, Williams O, Seddon B, et al. The basic leucine zipper transcription factor E4BP4 is essential for natural killer cell development. Nature Immunology. 2009;10(10):1118–U99. doi: 10.1038/ni.1787. [DOI] [PubMed] [Google Scholar]
- 31.Yan M, Ahn EY, Hiebert SW, Zhang DE. Runx1/Aml1 Dna-Binding Domain and Eto/Mtg8 Nhr2-Dimerization Domain Are Critical to Aml1-Eto9A Leukemogenesis. Blood. 2009;113(4):883–6. doi: 10.1182/blood-2008-04-153742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hannon GJ, Sun PQ, Carnero A, Xie LY, Maestro R, Conklin DS, et al. Molecular genetics - MaRX: An approach to genetics in mammalian cells. Science. 1999;283(5405):1129–30. doi: 10.1126/science.283.5405.1129. [DOI] [PubMed] [Google Scholar]
- 33.Fedtsova NG, Turner EE. Brn-3.0 Expression Identifies Early Postmitotic Cns Neurons and Sensory Neural Precursors. Mechanisms of Development. 1995;53(3):291–304. doi: 10.1016/0925-4773(95)00435-1. [DOI] [PubMed] [Google Scholar]
- 34.Ivins S, Pemberton K, Guidez F, Howell L, Krumlauf R, Zelent A. Regulation of Hoxb2 by APL-associated PLZF protein. Oncogene. 2003;22(24):3685–97. doi: 10.1038/sj.onc.1206328. [DOI] [PubMed] [Google Scholar]
- 35.Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, et al. Matlnspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics. 2005;21(13):2933–42. doi: 10.1093/bioinformatics/bti473. [DOI] [PubMed] [Google Scholar]
- 36.Trieu M, Ma A, Eng SR, Fedtsova N, Turner EE. Direct autoregulation and gene dosage compensation by POU-domain transcription factor Brn3a. Development. 2003;130(1):111–21. doi: 10.1242/dev.00194. [DOI] [PubMed] [Google Scholar]
- 37.Ayton PM, Cleary ML. Transformation of myeloid progenitors by MLL oncoproteins is dependent on Hoxa7 and Hoxa9. Genes & Development. 2003;17(18):2298–307. doi: 10.1101/gad.1111603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Melnick A, Carlile GW, McConnell MJ, Polinger A, Hiebert SW, Licht JD. AML-1/ETO fusion protein is a dominant negative inhibitor of transcriptional repression by the promyelocptic leukemia zinc finger protein. Blood. 2000;96(12):3939–47. [PubMed] [Google Scholar]
- 39.Salat D, Liefke R, Wiedenmann J, Borggrefe T, Oswald F. ETO, but not leukemogenic fusion protein AML1/ETO, augments RBP-J kappa/SHARP-mediated repression of Notch target genes. Molecular and Cellular Biology. 2008;28(10):3502–12. doi: 10.1128/MCB.01966-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith ML, Arch R, Smith LL, Bainton N, Neat M, Taylor C, et al. Development of a human acute myeloid leukaemia screening panel and consequent identification of novel gene mutation in FLT3 and CCND3. British Journal of Haematology. 2005;128(3):318–23. doi: 10.1111/j.1365-2141.2004.05324.x. [DOI] [PubMed] [Google Scholar]
- 41.Gascoyne DM, Thomas GR, Latchman DS. The effects of Brn-3a on neuronal differentiation and apoptosis are differentially modulated by EWS and its oncogenic derivative EWS/Fli-1. Oncogene. 2004;23(21):3830–40. doi: 10.1038/sj.onc.1207497. [DOI] [PubMed] [Google Scholar]
- 42.Theil T, Mcleanhunter S, Zornig M, Moroy T. Mouse Brn-3 Family of Pou Transcription Factors - A New Aminoterminal Domain Is Crucial for the Oncogenic Activity of Brn-3A. Nucleic Acids Research. 1993;21(25):5921–9. doi: 10.1093/nar/21.25.5921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wiggins AK, Wei GW, Doxakis E, Wong C, Tang AA, Zang K, et al. Interaction of Brn3a and HIPK2 mediates transcriptional repression of sensory neuron survival. Journal of Cell Biology. 2004;167(2):257–67. doi: 10.1083/jcb.200406131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li XL, Arai Y, Harada H, Shima Y, Yoshida H, Rokudai S, et al. Mutations of the HIPK2 gene in acute myeloid leukemia and myelodysplastic syndrome impair AML1-and p53-mediated transcription. Oncogene. 2007;26(51):7231–9. doi: 10.1038/sj.onc.1210523. [DOI] [PubMed] [Google Scholar]
- 45.Ma L, Lei L, Eng R, Turner E, Parada LF. Brn3a regulation of TrkA/NGF receptor expression in developing sensory neurons. Development. 2003;130(15):3525–34. doi: 10.1242/dev.00582. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.