

Abstract
Glycosyltransferases are carbohydrate-active enzymes with essential roles in numerous important biological processes. We have developed a novel donor analogue for galactosyltransferases which locks a representative target enzyme in a catalytically inactive conformation, thus almost completely abolishing sugar transfer. Results with other galactosyltransferases suggest that this novel and unique mode of glycosyltransferase inhibition is, very likely, generally applicable to other members of this very important enzyme family also.
In all domains of life, the biosynthesis of complex glycoconjugates requires the concerted action of a multitude of glycosyltransferases (GTs), enzymes that catalyse the transfer of a mono- or oligosaccharide from a glycosyl donor, e.g. a sugar-nucleotide, to a suitable acceptor, e.g. a glycan, peptide or lipid [1-4]. GTs play a key role in many fundamental biological processes underpinning human health and disease, such as cell signalling, cellular adhesion, carcinogenesis, and cell wall biosynthesis in human pathogens [5-8]. The development of small molecular GT inhibitors is therefore of considerable scientific interest in chemical glycobiology and drug discovery [9]. Most existing GT inhibitors are ground-state donor or acceptor analogues whose inhibition constants (Ki) are, at best, of a similar order of magnitude (10-1000 μM) as the Km value of the respective natural donor or acceptor substrate [9]. The rational design of potent GT inhibitors has been complicated by the complex reaction mechanism and unusual conformational plasticity of many GTs, which often undergo significant conformational rearrangement in the active site during the catalytic cycle [1-4,10]. On the other hand, it has recently been suggested that targeting these very conformational changes may be a promising route towards potent GT inhibitors [11]. However, no structural information has been provided to date that would allow the rational design of such a novel class of allosteric GT inhibitors.
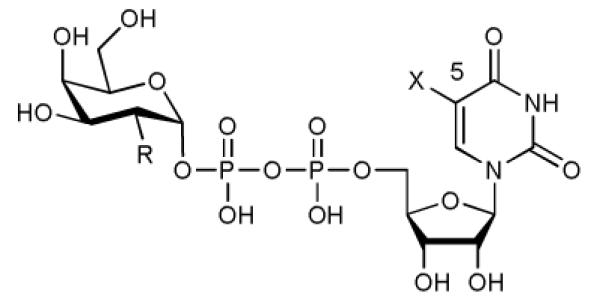
UDP-Galactose (UDP-Gal, 1, Table 1) is the general donor substrate for all Leloir-type galactosyltransferases (GalTs) [12]. Despite the considerable potential of GalTs as therapeutic targets, there is a significant lack of potent inhibitors for these enzymes. Thus, a Ki value of 1.8 μM has been reported for the best GalT inhibitor to date, against a single enzyme, bovine β-1,4-GalT [11]. Herein, we describe a new, base-modified UDP-Gal derivative (compound 2, Table 1), which blocks the closure of a flexible loop in the active site of a human blood group GalT and acts, toward five different GalTs, as an inhibitor of glycosyl transfer, with Ki values in the low micromolar to nanomolar range. This is a novel mode of inhibition for glycosyltransferases which, given the strong mechanistic similarities between many GTs [1-4,10], will very likely be applicable to other enzymes in this class also.
Table 1.
Enzymological characterisation of UDP-Gal 1, UDP-Gal derivative 2, and UDP-GalNAc 7 with AA(Gly)B.
| |||
|---|---|---|---|
|
| |||
| 1 (UDP-Gal) | 2 | 7 (UDP-GalNAc) | |
| Substituent R | OH | OH | NHAc |
| Substituent X | H | 5-formylthien-2-yl | H |
|
| |||
| Km donor [μM] | 0.7 ± 0.1 [1] 0.7 ± 0.06 [2] |
< 0.4 [2] | 1.7 ± 0.2 [1] |
|
| |||
| Km acceptor [μM] | 21 ± 2 [3] | 211 ± 24 [2] | 7.9 ± 0.6 [3] |
|
| |||
| kcat [s−1] | 0.37 ± 0.06 [1] 0.25 ± 0.01 [2] |
0.024 ± 0.001 [2] | 0.88 ± 0.07 [1] |
radiochemical assay, with 100 μM acceptor;
HPLC assay;
radiochemical assay, with 100 μM donor.
The characteristic structural feature of the new UDP-Gal derivative 2 is an additional formylthienyl substituent in position 5 of the uracil base (Table 1). While sugar-nucleotide analogues modified at the sugar have been commonly used for the investigation of glycosyltransferases, examples for base-modified sugar-nucleotides are extremely rare [13]. Our initial interest in 5-substituted UDP-Gal derivatives was prompted by the analysis of different GalT structures [14,15], which suggested that these enzymes might be able to accommodate donor analogues with an additional substituent in this position (Supplementary Figure S1). We reasoned that such novel UDP-Gal derivatives might be useful as GalT inhibitor candidates or, in view of the strong fluorescence emission reported for structurally related, 5-substituted uridine nucleosides [16], as fluorescent probes for assay development. For the synthesis of the representative UDP-Gal derivative 2 we used Suzuki-Miyaura chemistry previously developed in our group for the direct modification of unprotected sugar-nucleotides [17,18]. This synthetic strategy allowed the efficient preparation of 2, in five synthetic steps, from uridine (Supplementary Methods). In the key step of our synthesis, 2 was obtained in 56% isolated yield from the cross-coupling of 6 with (5-formylthien-2-yl)boronic acid under mild aqueous conditions.
In order to assess the effect of the additional substituent at the uracil base on the binding affinity and biological activity of this novel UDP-Gal derivative, we carried out enzymological studies with 5 and the representative human blood group GalT AA(Gly)B (see Supplementary Methods for terminology). This dual specificity enzyme can utilise either UDP-Gal 1 or UDP-GalNAc 7 as a donor substrate, and can transfer to the H-antigen with equal efficiency, producing both blood group A and B structures (Supplementary Methods, Scheme 2). In an HPLC-based assay of galactosylation, we determined a Km (donor) for 2 similar to that of the natural donor 1 (Table 1). The kcat of 2, on the other hand, was considerably lower than that of 1. These results suggested that the additional substituent at the uracil base was tolerated by the enzyme, and that binding of 2 occurred with relatively high affinity, but also that 2 was a much poorer donor substrate for AA(Gly)B than 1. Moreover, when we investigated the acceptor kinetics for the AA(Gly)B mutant, we found that for 2, Km (acceptor) was increased by ca. 10-fold compared to the natural donor. Consequently, the specificity constant, kcat/Km, of 2 is only 1% that of 1.
Prompted by this unusual and unexpected enzymological profile, we further investigated the behaviour of 2 in a second, radiochemical assay. The kinetic profile of the natural donor 1 was near identical in both the HPLC and radiochemical assay (Table 1). As was expected for the dual specificity enzyme AA(Gly)B, the alternative donor UDP-GalNAc 7 also showed similar Km (donor) and kcat values as 1. When we adapted the radiochemical assay format for competition studies, co-incubating AA(Gly)B with donor, donor analogue 2 and acceptor, 2 efficiently inhibited the transfer of both [3H]-Gal and [3H]-GalNAc in a competitive manner, with Ki values similar to the Km values of the respective donor (Table 2). Moreover, when we extended these experiments to a broader range of wild-type GalTs, in order to assess the generality of this inhibitory activity, 2 acted as an inhibitor of glycosyl transfer toward all five enzymes included in these experiments (Table 2). Notably, for individual GalTs the Ki values of 2 were up to 10-fold lower than the Km of the natural donor. At the same time, turnover numbers, as determined in the HPLC assay, were up to 100-fold lower for 2 than for 1 (Supplementary Information).
Table 2.
Inhibition of AA(Gly)B and four other GalTs by UDP-Gal derivative 2 [1].
| AA(Gly)B | GTB H. sapiens |
α-1,3-GalT B. taurus |
α-1,4-GalT N. meningitidis |
β-1,4-GalT B. taurus |
|
|---|---|---|---|---|---|
| Ki [μM] | 0.53 [2] 0.52 [3] |
2.4 | 9.8 [4] | 0.45 | 38.8 [4] |
|
| |||||
| Km UDP-Gal [μM] | 0.7 ± 0.1 | 27 [5] | 77 ± 14 | 0.5 ± 0.05 | 22 ± 9 |
Ki and Km values were determined in the radiochemical assay. Ki values were obtained from Dixon plots with inhibitor 2 at three different concentrations (n = 3), unless otherwise stated;
donor: UDP-Gal (2 μM);
donor: UDP-GalNAc (2 μM);
n = 4;
Reference 19.
To understand the molecular basis for the surprising enzymological behaviour of 2, we solved the high-resolution crystal structure of 2 bound to AA(Gly)B and, for direct comparison, structures of the enzyme in its unliganded apo form and in complex with UDP (see Supplementary Table 1 for data collection and refinement statistics). The AA(Gly)B apo structure was solved to near atomic resolution (1.25 Å, Figure 1a) and reveals an overall conformation isomorphous to the previously described wild-type GTB apo structure [15]. Similar to the wt-GTB, AA(Gly)B-apo has adopted an “open” conformation of the flexible internal loop (residues 176-199). However, this loop is significantly more ordered and the α-helix (α3) of the internal loop has a slightly different conformation around residues 194-199 compared to wt-GTB. The structure of the AA(Gly)B-UDP complex was solved to 1.65 Å resolution and has both a UDP molecule and a Mn2+ ion bound in the active site (Figure 1b). The structure shows excellent density for the entire nucleotide, as well as for a glycerol molecule bound in the acceptor binding site (Supplementary Figure S2a). While the C-terminus remains partly disordered, the entire internal loop is completely ordered and adopts a “semi-closed” conformation [15] where the loop has moved toward the UDP ligand to partially occlude the active site.
Figure 1. Galactosyltransferase (GalT) structures (continues overleaf).



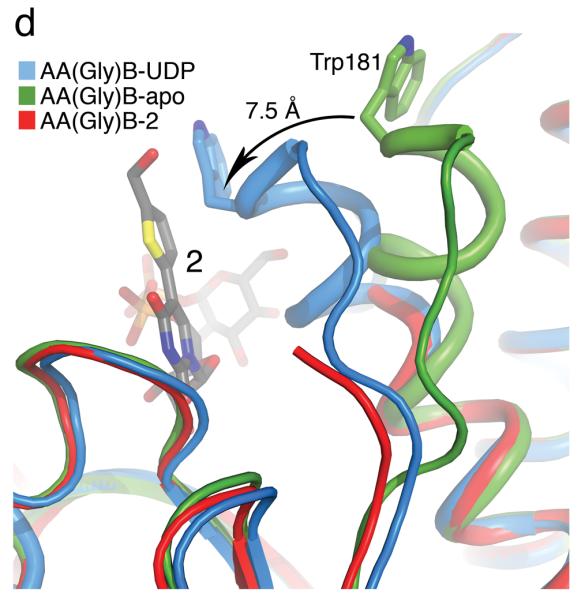
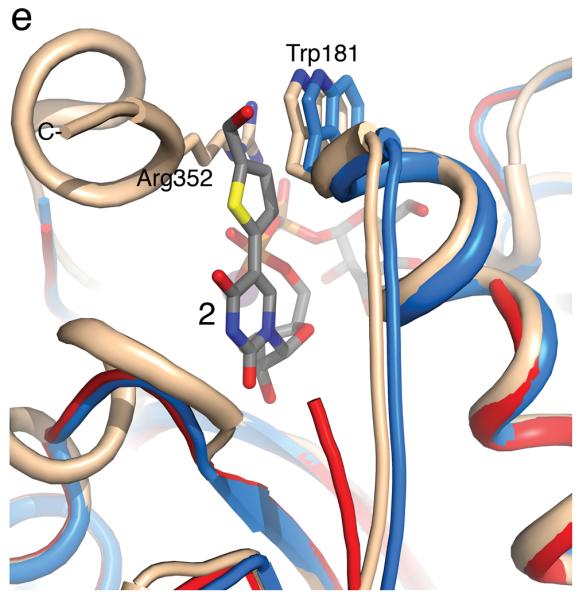


(a) Cartoon representation of the 1.25 Å structure of the AA(Gly)B GalT. Helices are green, strands are brown and the DXD motif is shown in yellow. (b) The 1.65 Å structure of the AA(Gly)B-UDP complex, oriented and colored as in (a). The internal loop is shown in light blue whereas the UDP is illustrated in dark grey sticks, and the Mn2+ by a violet sphere. A glycerol molecule in the acceptor binding site is omitted for clarity. (c) The 1.45 Å structure of AA(Gly)B with 2 bound in the active site, oriented and colored as in (a). The inhibitor is shown in light grey sticks and the Mn2+ is shown as in (b). The beginning and end of the flexible internal loop is shown in red, with a broken line illustrating the disordered part (residues 178-185). (d) Superposition of AA(Gly)B-2 (red); AA(Gly)B-UDP (light blue) and AA(Gly)B-apo (green) using the SSM tool in COOT. The position of 2 is shown in grey sticks. (e) Superposition of the human blood group GT AABB mutant, in the “fully-closed” conformation (light brown, PDB entry 2RJ7, UDP-Gal and acceptor removed for clarity), AA(Gly)B-UDP, in the “semi-closed” conformation” (light blue, UDP removed for clarity), and AA(Gly)B-5 (red). Trp181 in the internal loop, stacking to Arg352 in the C-terminus, and ligand 2 are shown in sticks. (f) Superposition of various GTs. The C-terminus and internal loop of GT AABB (residues 176-195) and the structurally similar bovine α-1,3-GalT (residues 187-205) (PDB entry 2RJ7 and 1K4V) are shown in light brown and dark blue, respectively. The flexible loops of human β-1,4-GalT (residues 340-358) and LgtC (residues 70-90) from N. meningitidis (PDB entry 3EE5 and 1GA8) are shown in green and pink, respectively. UDP, UDP-Gal and other donor derivatives bound to the structures are represented in sticks with matching color. The position of 2 is shown in white ball and sticks.
The structure of AA(Gly)B in complex with compound 2, solved to 1.45 Å resolution, shows an intact compound 2 molecule as well as a Mn2+ ion bound to the active site (Figure 1c). The electron density is well defined for the entire UDP moiety, whereas the density for the Gal and the formylthienyl substituent in position 5 is slightly less ordered (Supplementary Figure S2b). Although the formylthienyl substituent seems to have some degree of rotational freedom, a strong electron density peak places the ring sulfur in a position facing the O4 on the uracil. Compound 2 adopts a classic “folded back” conformation and binds to the donor-binding site in the same orientation as previously described for 1 [15], with the Gal moiety interacting with Arg188, Asp211 and Asp302. Despite the higher resolution of AA(Gly)B-2 compared to the AA(Gly)B-UDP structure, the overall internal loop is noticeably less ordered, and is completely disordered from residue 178 to 185.
In the superposition of the three AA(Gly)B structures, it can be seen that the ordered part of the internal loop in AA(Gly)B-2 is aligning with the “semi-closed” conformation in AA(Gly)B-UDP (Figure 1d). Despite the missing residues in the internal loop, it is clear that the enzyme is attempting to adopt the “semi-closed” conformation as a response to the binding of 2. Importantly, the overall conformation of the protein in the AA(Gly)B-2 complex is in excellent agreement with comparable previous GalT structures [15], confirming the reliability of the new AA(Gly)B-2 structure (see Figure 1e for superposition with the human blood group GT AABB).
The results from our structural studies provide a clear rationale for the unusual, and unexpected, enzymological profile of the novel UDP-Gal derivative 2. The catalytic mechanism of AA(Gly)B, representative for that of many other GTs [1-4,10], involves significant conformational movement in the active site prior to the glycosyl transfer reaction. Upon binding of the metal cofactor and sugar-nucleotide donor, a flexible internal loop and the C-terminus normally move from an “open” to a “semi-closed”, and then a “fully-closed”, conformation, which is required for catalytic activity. During the transition from the “semi-closed” to the “fully-closed” conformation, the last nine residues of the C-terminus become fully ordered, in response to binding of the acceptor to the active site [15]. The “fully-closed” conformation is stabilised by a stacking interaction, above the uracil moiety of the donor 1, between Trp181 in the internal loop and Arg352 in the C-terminus [15].
In the AA(Gly)B-2 complex, while a portion of the enzyme internal loop remains disordered, the internal loop has clearly undergone movement towards a “closed” conformation (Figure 1d). However, the transition to the “fully-closed” conformation is blocked by the formylthienyl substituent in 2. The superposition of the AA(Gly)B-2, AA(Gly)B-UDP and AABB structures (Figure 1e) illustrates how the formylthienyl substituent in 2 is positioned exactly where, in the “fully-closed” conformation, Trp181 forms a stacking interaction with Arg353. The substituent on compound 2 appears to interfere with this crucial interaction, preventing the enzyme from adopting a “fully-closed” conformation. Thus, 2 locks the enzyme in an unproductive conformation and effectively inhibits glycosyl transfer. The inability of the C-terminus, in the AA(Gly)B-2 complex, to fold over the active site also provides a structural explanation for the increase in Km (acceptor) observed for 2. In the “fully-closed” conformation of AABB, His348 in the C-terminus forms two hydrogen bonds with the fucose residue of the acceptor [15]. These important interactions, which likely contribute to the stabilisation of the donor-enzyme-acceptor complex, seem to be prevented in the AA(Gly)B-2 complex.
The superposition of different retaining and inverting GTs raises the intriguing possibility that this novel mode of GT inhibition is general and not limited to AA(Gly)B (Figure 1f). Despite significant structural differences between these enzymes, all of them have flexible active site loops folding over the donor binding site upon binding of the sugar-nucleotide donor. The superposition suggests that in a broad range of GTs, this flexible loop movement would be hindered by the substituent on the uracil ring of 2. This notion is strongly supported experimentally by the broad inhibitory activity we have observed for 2 toward five different GalTs. While it remains to be seen whether the residual donor substrate activity will limit cellular applications of 2, the favorable kcat/Km ratio suggests that it may serve as an inhibitor at the sub-saturating donor concentrations that are often found in cells (see Supplementary Methods).
In summary, we have developed a novel, base-modified UDP-Gal derivative which inhibits galactosyl transfer by several different GalTs. Results from structural and enzymological studies provide evidence that the inhibitory activity of 2 is due to interference with the catalytic mechanism of these enzymes. In light of the mechanistic similarities between many GTs, this novel mode of inhibition, which is reminiscent of the mode of action of allosteric protein kinase inhibitors [19], very likely represents a generally applicable strategy for the development of a novel class of potent, allosteric GT inhibitors.
Supplementary Material
Acknowledgements
We thank D. D. Djurhuus, Dr. D. Adlercreutz and M. H. Bien for excellent technical assistance during this research. The plasmid for the N. meningitidis α-1,4-GalT was a generous gift from Dr. W. W. Wakarchuk. AA(Gly)B was cloned by Dr. H. J. Lee and Dr. N. O. L. Seto. We also thank Dr. A. Henriksen for supplying equipment and tools for protein crystallography and for helpful discussions. We are grateful for the help with X-ray data collection provided by the beamline staff at I-911 at MAX-lab, Lund, Sweden and at X12 at Deutsches Elektronen-Synchrotron, EMBL-Hamburg, Germany. This work was supported by the EPSRC (First Grant EP/D059186/1, to GKW), the MRC (Discipline Hopping Award G0701861, to GKW) and the Danish Natural Science Research Council (FNU, to MMP). GKW is a Leverhulme Trust Research Fellow (RF/4/RFG/2008/0544). We thank the EPSRC National Mass Spectrometry Service Centre, Swansea, for the recording of mass spectra.
Footnotes
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Weadge JT, Palcic MM. Wiley Encyclopedia of Chemical Biology. 2008:1–13. DOI 10.1002/9780470048672.wecb213. [Google Scholar]
- 2.Lairson LL, Henrissat B, Davies GJ, Withers SG. Annu. Rev. Biochem. 2008;77:521–555. doi: 10.1146/annurev.biochem.76.061005.092322. [DOI] [PubMed] [Google Scholar]
- 3.Schuman B, Alfaro JA, Evans SV. Top. Curr. Chem. 2008;272:217–257. [Google Scholar]
- 4.Breton C, Snajdrova L, Jeanneau C, Koca J, Imberty A. Glycobiology. 2006;16:29R–37R. doi: 10.1093/glycob/cwj016. [DOI] [PubMed] [Google Scholar]
- 5.Marth JD, Grewal PK. Nat. Rev. Immunol. 2008;8:874–887. doi: 10.1038/nri2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rexach JE, Clark PM, Hsieh-Wilson LC. Nat. Chem. Biol. 2008;4:97–106. doi: 10.1038/nchembio.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dube DH, Bertozzi CR. Nat. Rev. Drug. Discov. 2005;4:477–488. doi: 10.1038/nrd1751. [DOI] [PubMed] [Google Scholar]
- 8.Berg S, Kaur D, Jackson M, Brennan PJ. Glycobiology. 2007;17:35R–56R. doi: 10.1093/glycob/cwm010. [DOI] [PubMed] [Google Scholar]
- 9.Qian X, Palcic MM. Glycosyltransferase Inhibitors. In: Ernst B, Hart G, Sinaÿ P, editors. Carbohydrates in Chemistry & Biology. Wiley-VCH; Weinheim: 2000. pp. 293–328. [Google Scholar]
- 10.Qasba PK, Ramakrishnan B, Boeggeman E. Trends Biochem. Sci. 2005;30:53–62. doi: 10.1016/j.tibs.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 11.Takaya K, Nagahori N, Kurogochi M, Furuike T, Miura N, Monde K, Lee YC, Nishimura S-I. J. Med. Chem. 2005;48:6054–6065. doi: 10.1021/jm0504297. [DOI] [PubMed] [Google Scholar]
- 12.Hennet T. Cell. Mol. Life Sci. 2002;59:1081–1095. doi: 10.1007/s00018-002-8489-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wagner GK, Pesnot T, Field RA. Nat. Prod. Rep. 2009;26:1172–1194. doi: 10.1039/b909621n. [DOI] [PubMed] [Google Scholar]
- 14.Gastinel LN, Cambillau C, Bourne Y. Embo J. 1999;18:3546–3557. doi: 10.1093/emboj/18.13.3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alfaro JA, Zheng RB, Persson M, Letts JA, Polakowski R, Bai Y, Borisova SN, Seto NOL, Lowary TL, Palcic MM, Evans SV. J. Biol. Chem. 2008;283:10097–10108. doi: 10.1074/jbc.M708669200. [DOI] [PubMed] [Google Scholar]
- 16.Greco NJ, Tor YZ. Nature Protocols. 2007;2:305–316. doi: 10.1038/nprot.2006.464. [DOI] [PubMed] [Google Scholar]
- 17.Pesnot T, Wagner GK. Org. Biomol. Chem. 2008;6:2884–2891. doi: 10.1039/b805216f. [DOI] [PubMed] [Google Scholar]
- 18.Collier A, Wagner GK. Chem. Commun. 2008:178–180. doi: 10.1039/b714379f. [DOI] [PubMed] [Google Scholar]
- 19.Liu Y, Gray NS. Nat. Chem. Biol. 2006;2:358–364. doi: 10.1038/nchembio799. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.