

Abstract
AIMS
To investigate the pharmacokinetics and pharmacodynamics of nasal formulations containing midazolam (5–30 mg ml−1) complexed with cyclodextrin.
METHODS
An open-label sequential trial was conducted in eight healthy subjects receiving single doses of 1 mg and 3 mg intranasally and 1 mg midazolam intravenously. Pharmacokinetic parameters were obtained by non-compartmental and two-compartmental models. Pharmacodynamic effects of midazolam were assessed using VAS and a reaction time test.
RESULTS
Mean bioavailability of midazolam after nasal administration ranged from 76 ± 12% to 92 ± 15%. With formulations delivering 1 mg midazolam, mean Cmax values between 28.1 ± 9.1 and 30.1 ± 6.6 ng ml−1 were reached after 9.4 ± 3.2–11.3 ± 4.4 min. With formulations delivering 3 mg midazolam, mean Cmax values were between 68.9 ± 19.8 and 80.6 ± 15.2 ng ml−1 after 7.2 ± 0.7–13.0 ± 4.3 min. Chitosan significantly increased Cmax and reduced tmax of midazolam in the high-dose formulation. Mean ratios of dose-adjusted AUC after intranasal and intravenous application for 1′-hydroxymidazolam were between 0.97 ± 0.15 and 1.06 ± 0.24, excluding relevant gastrointestinal absorption of intranasal midazolam. The pharmacodynamic effects after the low-dose nasal formulations were comparable with those after 1 mg intravenous midazolam. The maximum increase in reaction time by the chitosan-containing formulation delivering 3 mg midazolam was greater compared with 1 mg midazolam i.v. (95 ± 78 ms and 19 ± 22 ms, mean difference 75.5 ms, 95% CI 15.5, 135.5, P < 0.01). Intranasal midazolam was well tolerated but caused reversible irritation of the nasal mucosa.
CONCLUSIONS
Effective midazolam serum concentrations were reached within less than 10 min after nasal application of a highly concentrated midazolam formulation containing an equimolar amount of the solubilizer RMβCD combined with the absorption enhancer chitosan.
Keywords: chitosan, cyclodextrin, 1′-hydroxymidazolam, midazolam, nasal application, pharmacokinetics
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Nasal application of midazolam has been studied for a variety of indications. Due to the limited application volume, highly concentrated formulations are required to reach clinically relevant concentrations in adult patients. No data on the pharmacokinetics and pharmacodynamics of nasal midazolam formulations based on cyclodextrin and chitosan are available.
WHAT THIS STUDY ADDS
Clinically effective midazolam concentrations can be reached within less than 10 min after nasal administration of highly concentrated formulations containing an equimolar amount of the solubilizer randomly methylated-β-cyclodextrin combined with the absorption enhancer chitosan. Immediate non-invasive application of such formulations in emergency treatment of seizure patients by lay persons could offer clinical benefits in situations where intravenous access cannot be quickly established.
Introduction
Nasal application of midazolam (MDZ) has been studied for sedation before surgical, dental or diagnostic procedures, and for treatment of seizures in children and adult patients. Since no nasal preparation is commercially available, MDZ solution for injection has been used in many studies for nasal delivery in paediatric and adult patients [1–7]. For adequate dosing of adult patients often large application volumes exceeding nasal capacity had to be used due to the low concentration of the intravenous formulation (5 mg ml−1). To avoid swallowing and gastrointestinal absorption of excess fluid reaching the oropharynx, the maximal volume for nasal application is restricted to approximately 0.1 ml [8]. Therefore, highly concentrated solutions with a high bioavailability are required to achieve clinically relevant serum concentrations, especially in adult patients. Availability of such highly concentrated nasal MDZ formulations would offer clinical benefits, e.g. in lay treatment of patients with generalized seizures, saving important time until i.v. access can be established.
The water solubility of MDZ depends on pH; at physiological pH MDZ exists in the lipophilic ring-closed form, in an acidic environment at pH < 4 the ring structure opens and water soluble salts (e.g. MDZ hydrochloride) can be formed. However, even in the open-ring state, solubility in water-based formulations is too low to reach sufficiently high concentrations for nasal application. Different additives such as propylene glycol [9], propylene glycol combined with polyethylene glycol [10, 11] or complexation with cyclodextrins [12–15] have been used to increase water solubility. Nasal formulations containing the highest MDZ concentrations (25–27.8 mg ml−1) so far published are based on polyethylene and/or propylene glycol. However, high concentrations of polyethylene or propylene glycol increase the osmolarity of these solutions far above physiological levels causing irritation of the nasal mucosa with possible negative effects on absorption processes due to the osmotic gradient.
An alternative approach uses complexation with cyclodextrins (CDs) to enhance solubility [14]. CDs are cyclic oligosaccharides with a hydrophilic surface and a lipophilic cavity. CDs increase apparent water solubility of small lipophilic molecules by forming non-covalent inclusion complexes. CDs are biocompatible, non-immunogenic and several chemically modified CDs are approved as pharmaceutical excipients for human use [16]. However, the highest reported MDZ concentration in CD-based formulations (17 mg ml−1) [12–14] was considerably lower compared with polyethylene/propylene based formulations and the impact of CDs on permeation processes on the nasal mucosa is controversial. Decreased drug permeability due to impaired release of drug from the CD-drug complex has been reported, especially if high CD : drug molar ratios are used [17]. In vitro data, on the other hand, suggest no negative effect on drug permeability if CDs are used at concentrations just sufficient to solvate the drug [14, 18].
Chitosan, a derivative from chitin, is another polysaccharide used to promote transmucosal absorption [19]. Chitosan has mucoadhesive properties, decreases clearance from the nasal cavity and facilitates paracellular transport of hydrophilic drugs by transient opening of inter-cellular tight junctions [20, 21]. Chitosan has been used to improve nasal absorption of peptides, vaccines and low-molecular-weight drugs such as morphine [22, 23].
The purpose of the present study was to develop highly concentrated nasal MDZ formulations based on complexation with CD alone or in combination with the absorption enhancer chitosan and to investigate systematically their pharmacokinetic and pharmacodynamic properties in human volunteers.
Methods
Materials
Midazolam hydrochloride was purchased from Fährhauspharma (Hamburg, Germany), randomly methylated-β-cyclodextrin (RMβCD, Cavasol® W7M Pharma), hydroxypropyl-β-cyclodextrin (HPβCD, Cavasol® W7HP Pharma), and hydroxypropyl-γ-cyclodextrin (HPγCD, Cavasol® W8HP Pharma) from Wacker Chemicals (München, Germany). Chitosan hydrochloride for preliminary testing was obtained from Kraeber GmbH & Co (Ellerbek, Germany) and chitosan hydrochloride for pharmaceutical preparations from NovaMatrix FMC BioPolymer (Oslo, Norway). All components used for the production of nasal MDZ formulations fulfilled quality requirements specified in Ph. Eur. and Ph. Helv. and are available from commercial sources. For the preliminary experiments all chemicals were of analytical reagent grade and obtained from commercial sources. Preparations for stability testing were produced with chemicals of pharmaceutical quality. Semi-permeable cellophane membranes (Spectra/Por® Dialysis Tubing, MWCO 2000 from regenerated cellulose) were obtained from Spectrum Europe (Breda, the Netherlands). Unit dose nasal sprays, delivering 0.1 ml, were obtained from Ing. Erich Pfeiffer GmbH (Radolfzell, Germany).
Development of nasal MDZ formulations
Solubility of MDZ was assessed in water and in Britton-Robinson (BR) buffer (40 mm boric acid, 40 mm acetic acid, 40 mm phosphoric acid adjusted to pH 3.0, 4.0, 5.0, or 7.4 with 1.0 m sodium hydroxide) alone or with 10% solubilizer RMβCD, HPβCD or HPγCD. MDZ hydrochloride was added in excess amount to the different solutions, suspensions were equilibrated for 3 days, then filtered (0.45 µm pore size) and the saturation concentration of MDZ assessed by HPLC.
MDZ release from RMβCD-MDZ complexes in aqueous solutions was studied using two diffusion cells separated by a semi-permeable cellophane membrane (molecular weight cut off MWCO 2000, 2.3 cm2 surface area). MDZ passage across the membrane was assessed from the following solutions: 5 mg ml−1 MDZ in water; 5 mg ml−1 and 30 mg ml−1 MDZ dissolved with RMβCD at equimolar ratio with MDZ (2% w/v and 12% w/v RMβCD, respectively); and 5 mg ml−1 and 30 mg ml−1 MDZ dissolved with the three-fold molar ratio of RMβCD (6% w/v and 36% w/v RMβCD, respectively). The receiver compartment contained 0.9% sodium chloride in water. Donor and receiver compartments were stirred and maintained at 37°C. Withdrawn samples were replaced with aliquots of 0.9% sodium chloride. MDZ passage per minute was assessed from the slope of the concentration–time plot.
Nasal MDZ formulations for the clinical study were produced at the University Hospital Pharmacy, Basel, Switzerland, according to current guidelines of good manufacturing practice (ICH-GMP). Composition and application mode (unilateral vs. bilateral) of all clinically tested nasal MDZ formulations are given in Table 1. Formulation 1 was comparable with the commercially available 5 mg ml−1 MDZ solution for intravenous use. In formulation 2 RMβCD was added to test whether equimolar complexation with cyclodextrin had negative effects on absorption in vivo. Both formulations were applied bilaterally to administer a total dose of 1 mg. In formulation 3 concentrations of MDZ and RMβCD were doubled to test equivalence of unilateral application to bilateral application of formulation 2. Formulation 4 was used to test dose-proportionality and formulation 5 to test the effect of the absorption enhancer chitosan.
Table 1.
Composition of tested nasal MDZ formulations
| Applied MDZ dose | MDZ concentration (base) | Delivered volume | RMβCD (w : v) | Chitosan HCl (w : v) | NaCl (w : v) | |
|---|---|---|---|---|---|---|
| Formulation 1 | 1 mg | 5 mg ml−1 | 2 × 0.1 ml | – | – | 0.9% |
| MDZ | ||||||
| Formulation 2 | 1 mg | 5 mg ml−1 | 2 × 0.1 ml | 2% | – | 0.8% |
| MDZ + RMβCD | ||||||
| Formulation 3 | 1 mg | 10 mg ml−1 | 0.1 ml | 4% | – | 0.8% |
| MDZ + RMβCD | ||||||
| Formulation 4 | 3 mg | 30 mg ml−1 | 0.1 ml | 12% | – | 0.2% |
| MDZ + RMβCD | ||||||
| Formulation 5 | 3 mg | 30 mg ml−1 | 0.1 ml | 12% | 0.5% | 0.1% |
| MDZ + RMβCD + Chitosan | ||||||
Composition of aqueous midazolam solution for intranasal application. MDZ, midazolam; RMβ5CD, randomly methylated-β5-cyclodextrin. Concentration of RMβ5CD is equimolar to MDZ. Osmolality adjusted to 300 mosmol kg−1 with NaCl. Formulations 1 and 2 were applied bilaterally to deliver total dose of 1 mg.
All preparations were aqueous solutions, the osmolality was adjusted with sodium chloride to 300 mosmol kg−1. First, RMβCD was dissolved in water, then MDZ, sodium chloride and chitosan hydrochloride were slowly added while the solution was stirred. The pH of the formulation was adjusted with 0.1m HCl to a pH below 4.5.
Analytical methods
In vitro samples were analyzed in duplicate using a specific HPLC method with UV-detection of MDZ at 220 nm. Aliquots (10 µl) were injected into the HPLC system (Waters Alliance, 2690 Separation Module, 996 Photodiode Array Detector, Millenium32 Software, Waters Corporation, Milford, Massachusetts, USA) and separated on a X-Terra RP18 column (3.9 × 100 mm, 3.5 µm) kept at 35 °C. Flow rate was 0.8 ml min−1, isocratic, mobile phases were phosphate buffer (pH 8) 65% and acetonitrile 35%. Quantification of MDZ, 1′-hydroxymidazolam (1′-OH-MDZ), and 4-hydroxymidazolam in human serum was performed using an adapted, validated HPLC-MS method [24]. Briefly, 1 ml serum was spiked with 200 ng of the internal standard brotizolam and extracted with 3 ml butyl chloride. The organic phase was transferred into a test tube containing 20 µl ethylene glycol, then the sample was evaporated and redissolved in 40 µl of acetonitrile. Aliquots (10 µl) were injected into the LC-MS system (Finnigan LCQ Duo, Thermo Fisher Scientific Inc., MA, USA) equipped with an APCI source and a Restek Allure C18 (150 × 3.2 mm, 5 µm) column (Restek Coorp., PA, USA). The lower limit of quantification of the adapted method was 0.2 ng ml−1 for MDZ and metabolites and the assay was linear up to 1500 ng ml−1.
Human volunteer study
The clinical protocol was approved by the local Ethics Committee (EKBB, Basel, Switzerland) and notified to the national regulatory authority (Swiss Agency for Therapeutic Products, Swissmedic). The study was carried out at the Clinical Research Centre, University Hospital Basel, Switzerland according to the Declaration of Helsinki and current national regulations. Eight non-obese, healthy, non-smoking male volunteers (age 18–45 years) were included in this open-label, sequential trial. Subjects with acute or chronic impairment of nasal function or anatomical anomalies (e.g. nasal polyps), intolerance to benzodiazepines or adjuvants (including allergy to crustaceans) or with any clinically relevant systemic disease were excluded. Before enrolment volunteers were given detailed information about the study and written informed consent was obtained. Subjects were fasted for 10 h before and until 4 h after administration of study medication. Six different treatments were administered, starting with MDZ 1 mg ml−1 i.v., followed by nasal MDZ formulations 1–5 in sequential order, with a minimum washout period of 2 days between MDZ administrations. Venous blood samples (7.5 ml) were obtained from an indwelling venous catheter placed in the non-dominant forearm predose and at 1, 2.5, 5, 10, 15, 20, 30, 45 60, 120, 180, 240, and 360 min after MDZ administration. Blood samples were obtained in serum tubes, centrifuged at 1800 g for 10 min at 4 °C and serum was stored at −20°C until analysis. Volunteers were asked to classify local irritation immediately after nasal MDZ administration and after 5, 15, 30 and 240 min. Blood pressure, heart rate and transcutaneous oxygen saturation were monitored for 30 min after MDZ administration.
Pharmacokinetic analysis
Serum concentration data (except data of formulations 4 and 5) were analyzed using non-compartmental methods. Cmax and tmax were obtained directly from observed concentration–time data. The terminal elimination rate constant (λz) was estimated by log-linear regression after semi-logarithmic transformation of the data using at least three data points. Terminal elimination half-life was calculated using λz.
AUC from time of dosing to the last observable concentration (AUC(0,last) was calculated using the trapezoidal rule. AUC from time of dosing extrapolated to infinity (AUC(0,∞)) was calculated as follows:
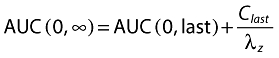 |
(1) |
To fit concentration data from formulations 4 and 5 adequately a two-compartmental model was used with first-order input, first-order output, lag-time and micro-constants as primary parameters and maximum serum concentration (Cmax), time to reach Cmax (tmax), area under concentration–time curve (AUC), and terminal elimination half-life (t1/2) as secondary parameters (WinNonlin, Pharsight Corp., Mountain View, CA, USA).
Pharmacodynamics
Visual analogue scale (subjective sedation)
Subjective sedation was rated by study subjects on a visual analogue scale (VAS) using a 100 mm non-graduated line, the left end referring to ‘no fatigue’ (no pharmacological effect) and the right end referring to ‘close to falling asleep’ (maximum pharmacological effect) at 5, 15, 30, 120 min after nasal MDZ administration. VAS scores were parameterized by calculating the area under effect curve (AUECVAS) from the first to the last recorded measurement.
Choice reaction time test (CRTT)
Sedative effects of MDZ were assessed with an adaptive choice reaction time test (CRTT) at 20, 120 and 240 min after study drug administration. During a 3 min period subjects had to respond to the presentation of coloured lights (five different colours) appearing in random sequence by pressing the button with the corresponding colour as quickly and accurately as possible. A computer-based algorithm adjusted the interstimulus interval (ISI) in steps of 50 ms according to the performance in a continuously moving window of the previous seven stimulus-response pairs. ISI decreased when four or more out of seven stimulus-response pairs were correct, otherwise ISI increased. Thus, test difficulty was continuously adjusted to individual performance resulting in a subject false response rate of approximately 50% [25]. Mean reaction time (RT) of all correct responses was calculated as a dependent variable. RTs less than 300 ms were considered accidental, not representing true cognitive-motor reactions to a stimulus and were therefore excluded. To familiarize subjects with the test procedure a training run was performed before the first baseline assessment.
Statistical analysis
Statistical analysis was performed using the SPSS statistical software package, version 15.0 (SPSS, Chicago, IL, USA). Results are presented as mean and SD unless indicated otherwise. A P value of less than 0.05 was considered to be statistically significant. Normal distribution of the data was assessed using the Shapiro-Wilk test. Parameters with normal distribution were tested using one-way repeated measures anova. Significant overall results were further analyzed with an appropriate post-hoc procedure for multiple comparisons (Bonferroni). Non-normally distributed parameters were analyzed by the non-parametric Friedman test.
Results
Pharmaceutical preparation
MDZ solubility in water alone or in aqueous solutions containing 10% HPβCD, 10% RMβCD or 10% HPγCD was 9.1 mg ml−1, 26.2 mg ml−1, 33.1 mg ml−1 and 40.8 mg ml−1, respectively. For the complexes of HPβCD, RMβCD or HPγCD with MDZ the calculated molar ratios of cyclodextrin to MDZ were 1:1.13, 1:1.33, and 1:1.97, respectively. Figure 1 shows the pH-dependent solubility of MDZ in BR buffer alone or with addition of 10% HPβCD, 10% RMβCD or 10% HPγCD. All tested CD derivatives increased the solubility of MDZ to a similar degree compared with the solubility in BR buffer alone. Above pH 5 solubility of MDZ was low (<10 mg ml−1) for all preparations. At a pH < 5 solubility rose rapidly for all CD-containing preparations reaching approximately 70 mg ml−1 at pH 3.0.
Figure 1.
pH-dependent solubility of MDZ. Saturation concentration of MDZ in Britton-Robinson (BR) buffer alone and in BR buffer combined with 10% solubilizer hydroxypropyl-β-cyclodextrin (HPβCD), randomly methylated-β-cyclodextrin (RMβCD), or hydroxypropyl-γ-cyclodextrin (HPγCD) depended on pH. At a pH < 5 all tested CD derivatives increased the solubility of MDZ to a similar degree compared with the solubility in BR buffer alone. Above pH 5 solubility of MDZ was below 10 mg ml−1. Britton-Robinson (BR) buffer ( ); BR buffer + 10% RMβCD (
); BR buffer + 10% RMβCD ( ); BR buffer + 10% HPβCD (
); BR buffer + 10% HPβCD ( ); BR buffer + 10% HPγCD (
); BR buffer + 10% HPγCD ( )
)
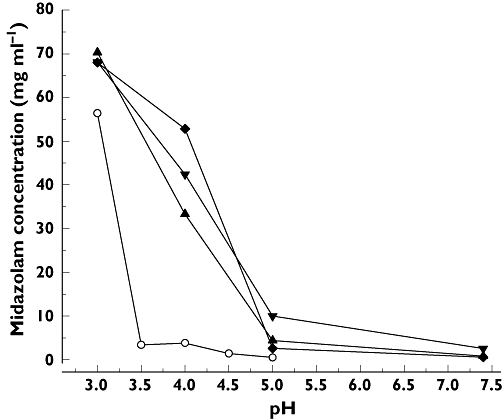
MDZ passage through semi-permeable cellophane membranes is shown in Figure 2. At a concentration of 5 mg ml−1, MDZ passage significantly decreased with increasing concentration of RMβCD (P= 0.027). At 30 mg ml−1, MDZ passage was higher than for 5 mg ml−1 but not proportional to the MDZ concentration and decrease of MDZ passage at thre-fold molar RMβCD to MDZ ratio was not statistically significant (P= 0.058).
Figure 2.
MDZ release from RMβCD-MDZ complexes was estimated by assessing MDZ passage through semi-permeable cellophane membranes. Total MDZ concentrations were 5 and 30 mg ml−1. RMbCD was added at equimolar (2% and 12% w/v) and threefold (6% and 36% w/v) molar ratio compared with MDZ base. RMβCD added at three-fold molar ratio significantly decreased MDZ passage at 5 mg ml−1 (P= 0.027) but not at 30 mg ml−1 (P= 0.058) MDZ concentration. Data are presented as mean ± SD (n= 3). Midazolam 5 mg ml−1 ( ); Midazolam 30 mg ml−1 (
); Midazolam 30 mg ml−1 ( )
)
Human volunteer study
Eight subjects completed the study; one subject who experienced a vasovagal reaction after insertion of the i.v. catheter and withdrew consent after the first study day was replaced. Data of the drop-out subject were not analyzed. Age (mean ± SD) was 24 ± 3 years (range 20–31 years) and weight 74 ± 5 kg (range 66–84 kg).
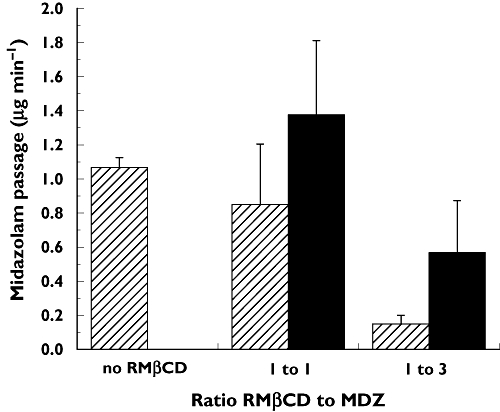
After intravenous and intranasal application, MDZ and its major metabolite 1′-OH-MDZ were detected and quantified in serum. The concentrations of 4-hydroxymidazolam were too low to allow reliable quantification. Pharmacokinetic parameters of MDZ after intravenous and intranasal application are shown in Table 2 and serum concentration–time profiles of MDZ and 1′-OH-MDZ are given in Figure 3A, B. Mean bioavailability of formulations 1, 2, and 3 containing 1 mg MDZ was in the range of 88–93%, mean time to reach maximal serum concentrations ranged from 9.4 min to 11.3 min. There were no statistically significant pharmacokinetic differences between these low dose formulations. The addition of the solubilizer RMβCD at a concentration of 2% (formulation 2) had no influence on peak exposure (Cmax) or total exposure (AUC) compared with formulation 1 (standard 5 mg ml−1 MDZ solution). Doubling the concentration of MDZ and RMβCD (formulation 3) allowed a reduction of the applied volume and a decrease of the absorption surface of 50% (application of 100 µl into one nasal cavity instead of 100 µl on each side) without impact on exposure measures. Dose-proportionality was tested with formulation 4, which contained three times higher concentrations of MDZ and RMβCD than formulation 3. For the ratio of the dose-adjusted AUC of formulation 4 to AUC of formulation 3 the 90% confidence interval was 0.86, 1.05 and was within the range for bioequivalence (0.8, 1.25). However, for the comparison of dose-normalized Cmax of formulation 4 to Cmax of formulation 3 the 90% confidence interval for the ratio was 0.67, 92 and dose-proportionality was not met. Addition of the absorption enhancer chitosan (formulation 5) significantly increased maximum serum concentrations (80.6 ± 15.2 and 68.9 ± 19.8 ng ml−1, mean difference 11.7 ng ml−1, 95% CI 5.0, 18.5, P= 0.005) and significantly decreased time to reach peak concentrations (7.2 ± 0.7 and 13.0 ± 4.3 min, respectively, mean difference 5.8 min, 95% CI 2.4, 9.1, P= 0.005) compared with formulation 4. In contrast to formulation 4, dose-normalized Cmax of formulation 5 met dose-proportionality criteria compared with formulation 3 (90% confidence interval for ratio 0.81, 1.09). However, chitosan decreased total exposure. Dose-normalized AUC of formulation 5 did not meet bioequivalence criteria compared with AUC of formulation 3 (90% confidence interval for ratio 0.78, 0.90) and mean bioavailability of formulation 5 was significantly lower compared with formulation 4 (76 ± 12% and 85 ± 8%, respectively, P < 0.01). Partial AUCs from 0 to 120 min (AUC(0,120 min)) were significantly higher for the two high dose nasal formulations compared with the low dose nasal formulations and the intravenous administration (P < 0.001). In contrast to AUC(0,∞) there was no significant difference between AUC(0,120 min) of formulation 5 compared with AUC(0,120 min) of formulation 4 (Table 2). To visualize the overall effect of chitosan on absorption, fitting curves were generated by a two-compartmental pharmacokinetic model applied to the individual concentration–time data of formulations 4 and 5 (Figure 3C).
Table 2.
Pharmacokinetic parameters of MDZ 1 mg i.v. (Dormicum®, Roche) and the 5 nasal MDZ formulations tested in eight healthy volunteers
| tmax | Cmax | AUC(0,∞) | AUC(0,120 min) | t1/2 | ||
|---|---|---|---|---|---|---|
| Formulation | (min) | (ng ml−1) | (ng ml−1min) | (ng ml−1min) | (min) | F |
| Midazolam | 2.1 ± 0.8 | 87.6 ± 58.7 | 2799 ± 509 | 1796 ± 418 | 113.5 ± 25.9 | N.A. |
| 1 mg. i.v. | ||||||
| Formulation 1 | 10.6 ± 5.0 | 28.1 ± 9.1 | 2461 ± 628 | 1424 ± 399 | 114.2 ± 23.0 | 88 ± 17% |
| 0.5 mg i.n. both sides | ||||||
| Formulation 2 | 9.4 ± 3.2 | 30.1 ± 6.6 | 2596 ± 680 | 1535 ± 370 | 113.0 ± 22.8 | 92 ± 15% |
| 0.5 mg i.n. both sides | ||||||
| Formulation 3 | 11.3 ± 4.4 | 28.9 ± 5.4 | 2511 ± 541 | 1496 ± 269 | 105.4 ± 17.7 | 90 ± 16% |
| 1 mg i.n. one side | ||||||
| Formulation 4 | 13.0 ± 4.3 | 68.9 ± 19.8 | 7143 ± 1568 | 4117 ± 798 | 117.5 ± 32.0 | 85 ± 8% |
| 3 mg i.n. one side | ||||||
| Formulation 5 | 7.2 ± 0.7** | 80.6 ± 15.2** | 6320 ± 1458** | 3741 ± 717ns | 111.4 ± 20.6 | 76 ± 12%* |
| 3 mg i.n. one side | ||||||
Values are mean ± SD. i.n., intranasal application. Cmax, maximum serum concentration; tmax, time to maximum serum concentration; AUC(0,∞), area under concentration–time curve extrapolated to infinity; t1/2, elimination half-life; F, bioavailability.
Significantly different (P < 0.05) from formulation 4.
Significantly different (P < 0.005) from formulation 4.
Figure 3.
Kinetics of MDZ (A) and 1′-OH-MDZ (B) after intravenous administration of 1 mg MDZ and intranasal administration of 1 mg MDZ (formulations 1–3) and 3 mg MDZ (formulations 4 and 5). Fitting curves (C) generated by a two-compartmental pharmacokinetic model applied to the individual concentration–time data of the 3 mg nasal formulation without (formulation 4) and with chitosan (formulation 5). The absorption enhancer chitosan decreases variability in the early absorption phase, and significantly reduces tmax and increases Cmax
The concentration–time profiles of 1′-OH-MDZ after administration of the low dose nasal formulations 1, 2 and 3 were comparable with the profile obtained after intravenous administration of 1 mg MDZ (Figure 3B). Accordingly mean ratios of dose-adjusted AUCin : AUCiv for 1′-OH-MDZ, a measure for the relative amount of metabolite formed, were between 0.97 (formulation 5) and 1.06 (formulations 2 and 3, Table 3), suggesting that there was no evidence for relevant gastrointestinal absorption of intranasally applied MDZ.
Table 3.
Comparison of areas under concentration–time curve of the major midazolam metabolite 1′-hydroxymidazolam after intranasal and intravenous application of midazolam as indicator of oral absorption of midazolam after intranasal administration
| AUC(0,∞) (ng ml−1min) | ||
|---|---|---|
| Formulation | 1′-hydroxymidazolam | AUCin : AUC iv |
| Midazolam 1 mg i.v. | 294 ± 73 | NA |
| Formulation 1 | 305 ± 67 | 1.05 ± 0.09 |
| Formulation 2 | 317 ± 130 | 1.06 ± 0.24 |
| Formulation 3 | 311 ± 82 | 1.06 ± 0.11 |
| Formulation 4 | 293 ± 83* | 1.00 ± 0.18 |
| Formulation 5 | 280 ± 53* | 0.97 ± 0.15 |
Values are mean ± SD. i.n., intranasal application.
AUCs of the 3 mg formulations are dose-normalized.
AUECVAS was higher after application of the 3 mg formulations compared with the 1 mg formulations (Figure 4) without reaching statistical significance due to high variability (P= 0.06). Results of the adaptive choice reaction time test are summarized in Table 4. There was a significant training effect (P= 0.005) over all study periods, evident by decreasing mean baseline RT in subsequent study periods. Therefore, increases in RT were baseline-corrected (ΔRT) for further analysis. Maximal increase of reaction time (ΔRTmax) after nasal application of 1 mg MDZ was comparable with the effect of 1 mg MDZ given intravenously. The 3 mg nasal MDZ formulations resulted in a larger effect. After application of formulation 5, ΔRTmax increased four-fold (95 ± 78 ms vs. 19 ± 22 ms, mean difference 75.5 ms, 95% CI 15.5, 135.5, P < 0.01) compared with 1 mg MDZ given intravenously. ΔRTmax and the area under effect curve from 0 to 120 min (AUECRT(0,120 min) as well as AUEC over the whole observation period (AUECRT) were significantly larger (P < 0.01) compared with 1 mg MDZ given intranasally.
Figure 4.
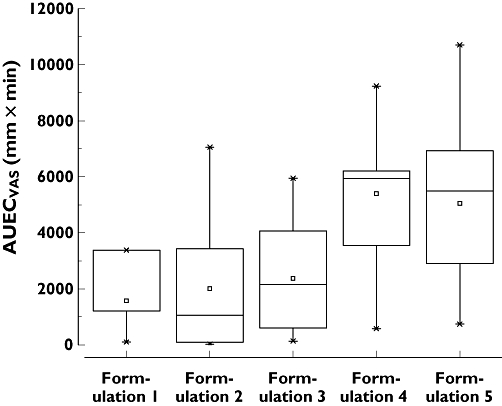
MDZ induced sedative effects assessed by VAS. AUECVAS was higher after application of the 3 mg formulations compared with the 1 mg formulations without reaching statistical significance
Table 4.
Adaptive choice reaction time test
| Reaction time (RT) at baseline (ms) | Maximum increase(ΔRTmax) of baseline-corrected RT (ms) | Time to reach ΔRTmax (min) | Area under effect curve (AUECRT) (ms × h) | Partial area under effect curve (AUECRT(0,120 min)) (ms × h) | |
|---|---|---|---|---|---|
| Midazolam i.v. | 575 ± 44 | 19 ± 22 | 120 (20–120) | 43 ± 84 | 22 ± 42 |
| Formulation 1 | 564 ± 45 | 16 ± 18 | 120 (20–240) | 4 ± 47 | 3 ± 31 |
| Formulation 2 | 548 ± 40 | 15 ± 16 | 70 (20–120) | 5 ± 77 | 15 ± 32 |
| Formulation 3 | 528 ± 32 | 19 ± 15 | 120 (20–120) | 38 ± 55 | 26 ± 25 |
| Formulation 4 | 519 ± 36 | 70 ± 39 | 20 (20–20) | 106 ± 81 | 85 ± 49 |
| Formulation 5 | 502 ± 42 | 95 ± 78* | 20 (20–120) | 183 ± 160** | 128 ± 102* |
Data are mean ± SD, except time to reach ΔRTmax which is given as median (range).
Significantly different (P < 0.01) from midazolam 1 mg i.v. and nasal 1 mg formulations 1, 2, and 3.
Significantly different (P < 0.05) from nasal 1 mg formulations 1, 2 and 3.
No relevant changes in blood pressure, heart rate or transcutaneous oxygen saturation were observed after intravenous or nasal administration of MDZ. Intranasal administration of MDZ caused mild to moderate local irritation of the nasal mucosa and occasionally in the upper throat area immediately after application. Application was described as tolerable (65%), unpleasant (25%) or indifferent (10%) by study subjects. Administration of the chitosan containing formulation was associated with a delayed stinging sensation (starting about 30–60 s after application) and tearing of the ipsilateral eye (63%) and volunteers classified administration more often as unpleasant (50%) than administration of the formulations not containing chitosan. Fifteen minutes after application local irritation was reported as mild by most study subjects and all adverse effects were completely reversible within 30 min.
Discussion
In our study we systematically investigated the pharmacokinetics and pharmacodynamics of five different nasal MDZ formulations using 1 mg intravenous MDZ as a reference. Mean bioavailability of all tested MDZ formulations was high, allowing clinically effective concentrations to be reached within a few minutes after nasal application of the high-dose formulations. Absorption of MDZ was essentially limited to the nasal mucosa as demonstrated by the comparable concentration–time profiles of 1′-hydroxymidazolam after intranasal and intravenous administration. Using RMβCD as solubilizer allowed an increase in concentration of MDZ in aqueous solution by six-fold compared with the commercially available intravenous MDZ solution. In vitro, equimolar RMβCD reduced MDZ passage across a cellophane membrane by about 20% compared with MDZ without RMβCD, but in vivo we did not observe a negative effect of RMβCD on exposure measures of MDZ. Our finding that RMβCD added at three-fold molar ratio reduced MDZ release in vitro by more than 85% could explain the clinical observation that RMβCD added in five-fold excess concentration (20% w/v for a 10 mg ml−1 MDZ nasal formulation) reduced MDZ exposure after nasal application of 7.5 mg MDZ (Cmax < 40 ng ml−1 and tmax > 30 min) [15].
Addition of 0.5% (w/v) chitosan had a positive effect on the early phase of MDZ absorption with a significant increase in Cmax and a significant reduction in tmax. However, total exposure as reflected by AUC and bioavailability was reduced compared with RMβCD alone.
To increase solubility of MDZ in aqueous solutions, either co-solvents such as polyethylene glycol and propylene glycol [9–12], or molecular complexation with cyclodextrins [12–14, 26] have been used in experimental nasal formulations. The highest MDZ concentrations reported in nasal formulations are 27.8 mg ml−1 for propylene glycol [9] and 25 mg ml−1 for a combination of propylene glycol and polyethylene glycol 400 as co-solvents [10, 11]. In cyclodextrin-based formulations maximal MDZ concentrations used so far were considerably lower, 17 mg ml−1 in a formulation containing sulfobutylether-β-cyclodextrin (14% w/v) [12–14] and 10 mg ml−1 in a formulation using (20% w/v) RMβCD [15]. With a concentration of 30 mg ml−1, our formulation was thus comparable with the best co-solvent-based rather than the other cyclodextrin-based formulations. This was true also for the pharmacokinetic properties. While bioavailability of the sulfobutylether-β-cyclodextrin containing formulations was in the range of 64–77%, mean bioavailability of our equimolar RMβCD-MDZ formulations without chitosan (formulations 2–4) was in the range of 85–93% and thus comparable with the highest reported bioavailability (83%) reached by a propylene glycol based formulation [9]. Compared with this propylene glycol based formulation containing 2.5 mg MDZ, mean peak serum concentrations of our 3 mg formulation without chitosan (formulation 4) was similar (69 and 71 ng ml−1, respectively). However, the addition of chitosan increased Cmax significantly to 81 ng ml−1, which is close to the peak serum concentration of 84 ng ml−1 reached after application of a 66% higher dose (5 mg) of MDZ with a formulation based on the combination of propylene and polyethylene-glycol [10]. The other main effect of chitosan was a significant decrease in the time needed to reach maximal serum concentrations. For most reported nasal MDZ formulations, values for tmax are in the range of 10–15 min, which is in line with the values we obtained with our non-chitosan containing formulations. As illustrated by the modeled PK curves (Figure 3C), chitosan significantly enhanced the early phase of absorption and significantly increased Cmax and reduced tmax to 7.2 min. In view of a possible application of nasal MDZ in the emergency treatment of patients with seizures these properties may be essential.
The strength of our study is that we systematically investigated the effects of cyclodextrin and chitosan on the pharmacokinetics and pharmacodynamics of MDZ after nasal application. Due to lack of a commercially available nasal formulation the intravenous MDZ solution is often used for intranasal application. In a first step we demonstrated that equimolar complexation with the solubilizer RMβCD (formulation 2) did not change the absorption characteristics in vivo compared with the nasal application of the standard MDZ solution for intravenous use (formulation 1).
Next we showed that doubling the concentration of MDZ and RMβCD and concurrent reduction of the absorption surface by 50% (one-sided nasal application of the 10 mg ml−1 formulation 3) was equivalent to both-sided application of the same dose using the 5 mg ml−1 formulation 2. We then evaluated dose-proportionality by tripling the concentration of MDZ and RMβCD to 30 mg ml−1 and 12% (w/v), respectively (formulation 4). These experiments showed that A) dose-proportionality was only met for AUC but not for Cmax compared with formulation 3 and that B) the addition of chitosan (formulation 5) reversed the situation with dose-proportionality now met for Cmax but not for AUC. The reason for this finding remains unclear.
One of the limitations of our study is that we did not use pretreatment with activated charcoal to prevent gastrointestinal absorption of any MDZ that might be swallowed after nasal application. However, by limiting the application volume to 100 µl we did not exceed the accepted retention capacity of the nasal cavity which ranges from 100 to 200 µl [8]. Accordingly, neither the concentration–time profile of MDZ, nor the ratio of 1′-OH-MDZ AUC after nasal administration compared with 1′-OH-MDZ AUC after intravenous administration, showed evidence for oral absorption. A further limitation is that our study design was not blinded, without a placebo control and that we did not use an EEG to measure the effects of MDZ. These points would have strengthened our pharmacodynamic assessment. The primary goal of the study was, however, to identify the formulation with the most promising PK profile for use in subsequent studies with more elaborate PD assessment, such as quantitative EEG.
Regarding implications of our study results for clinical practice it is interesting to note that intranasal application of 6 mg MDZ (3 mg simultaneously into each nasal cavity) with a mean bioavailability of 75% corresponds to an intravenous dose of 4.5 mg, which is in the dose-range used for conscious sedation. Although not tested in this study, it can be expected that peak serum concentrations after intranasal application of 6 mg MDZ would exceed 150 ng ml−1 and thus reach a concentration range where patients experience pronounced sedation and amnesia but are still arousable [27]. For the treatment of status epilepticus the recommended bolus dose of MDZ is 0.15–0.2 mg kg−1 body weight, resulting in 10–14 mg for a person weighing 70 kg. At least 200 µl per nasal cavity would therefore have to be applied, which is still within the accepted retention capacity. This approach needs to be tested in a separate study. The chitosan containing formulation was associated with reversible local irritation of the nasal mucosa classified as unpleasant by half of the volunteers. While this observation is irrelevant for emergency applications, for sedative use, especially in the paediatric field, this is a disadvantage which might be overcome by pre-treating the nasal mucosa with a local anaesthetic.
In conclusion we demonstrated that clinically effective concentrations can be reached within less than 10 min after nasal application of a highly concentrated MDZ formulation containing an equimolar amount of the solubilizer RMβCD combined with the absorption enhancer chitosan. Although maximal serum concentrations after nasal application are not reached as quickly as after intravenous application, immediate non-invasive application of such formulations in emergency treatment of patients with generalized seizures by lay persons might offer clinical benefits and should first be tested in a controlled clinical setting.
Acknowledgments
Pharmaceutical development of nasal MDZ formulations was partially financed by Akroswiss AG, Zurich, Switzerland. We thank Mrs Cornelia Hamberg Stäubli for her careful technical assistance in performing the quantitative analyses.
Competing interests
J.F. owns shares of Akroswiss AG, which supported part of the work of K.S. with financial funds.
REFERENCES
- 1.Lahat E, Goldman M, Barr J, Bistritzer T, Berkovitch M. Comparison of intranasal midazolam with intravenous diazepam for treating febrile seizures in children: prospective randomised study. BMJ (Clin Res Ed) 2000;321:83–6. doi: 10.1136/bmj.321.7253.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mahmoudian T, Zadeh MM. Comparison of intranasal midazolam with intravenous diazepam for treating acute seizures in children. Epilepsy Behav. 2004;5:253–5. doi: 10.1016/j.yebeh.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 3.Harbord MG, Kyrkou NE, Kyrkou MR, Kay D, Coulthard KP. Use of intranasal midazolam to treat acute seizures in paediatric community settings. J Paediatrics Child Health. 2004;40:556–8. doi: 10.1111/j.1440-1754.2004.00463.x. [DOI] [PubMed] [Google Scholar]
- 4.Scheepers M, Scheepers B, Clarke M, Comish S, Ibitoye M. Is intranasal midazolam an effective rescue medication in adolescents and adults with severe epilepsy? Seizure. 2000;9:417–22. doi: 10.1053/seiz.2000.0425. [DOI] [PubMed] [Google Scholar]
- 5.Bjorkman S, Rigemar G, Idvall J. Pharmacokinetics of midazolam given as an intranasal spray to adult surgical patients. Br J Anaesth. 1997;79:575–80. doi: 10.1093/bja/79.5.575. [DOI] [PubMed] [Google Scholar]
- 6.Burstein AH, Modica R, Hatton M, Forrest A, Gengo FM. Pharmacokinetics and pharmacodynamics of midazolam after intranasal administration. J Clin Pharmacol. 1997;37:711–8. doi: 10.1002/j.1552-4604.1997.tb04358.x. [DOI] [PubMed] [Google Scholar]
- 7.McCormick AS, Thomas VL, Berry D, Thomas PW. Plasma concentrations and sedation scores after nebulized and intranasal midazolam in healthy volunteers. Br J Anaesth. 2008;100:631–6. doi: 10.1093/bja/aen072. [DOI] [PubMed] [Google Scholar]
- 8.Romeo VD, deMeireles J, Sileno AP, Pimplaskar HK, Behl CR. Effects of physicochemical properties and other factors on systemic nasal drug delivery. Adv Drug Deliv Rev. 1998;29:89–116. doi: 10.1016/s0169-409x(97)00063-x. [DOI] [PubMed] [Google Scholar]
- 9.Knoester PD, Jonker DM, Van Der Hoeven RT, Vermeij TA, Edelbroek PM, Brekelmans GJ, de Haan GJ. Pharmacokinetics and pharmacodynamics of midazolam administered as a concentrated intranasal spray. A study in healthy volunteers. Br J Clin Pharmacol. 2002;53:501–7. doi: 10.1046/j.1365-2125.2002.01588.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wermeling DP, Record KA, Archer SM, Rudy AC. A pharmacokinetic and pharmacodynamic study, in healthy volunteers, of a rapidly absorbed intranasal midazolam formulation. Epilepsy Res. 2009;83:124–32. doi: 10.1016/j.eplepsyres.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 11.Wermeling DP, Record KA, Kelly TH, Archer SM, Clinch T, Rudy AC. Pharmacokinetics and pharmacodynamics of a new intranasal midazolam formulation in healthy volunteers. Anesth Analg. 2006;103:344–9. doi: 10.1213/01.ane.0000226150.90317.16. [DOI] [PubMed] [Google Scholar]
- 12.Dale O, Nilsen T, Loftsson T, Hjorth Tonnesen H, Klepstad P, Kaasa S, Holand T, Djupesland PG. Intranasal midazolam: a comparison of two delivery devices in human volunteers. J Pharm Pharmacol. 2006;58:1311–8. doi: 10.1211/jpp.58.10.0003. [DOI] [PubMed] [Google Scholar]
- 13.Gudmundsdottir H, Sigurjonsdottir JF, Masson M, Fjalldal O, Stefansson E, Loftsson T. Intranasal administration of midazolam in a cyclodextrin based formulation: bioavailability and clinical evaluation in humans. Die Pharmazie. 2001;56:963–6. [PubMed] [Google Scholar]
- 14.Loftsson T, Gudmundsdottir H, Sigurjonsdottir JF, Sigurdsson HH, Sigfusson SD, Masson M, Stefansson E. Cyclodextrin solubilization of benzodiazepines: formulation of midazolam nasal spray. Int J Pharm. 2001;212:29–40. doi: 10.1016/s0378-5173(00)00580-9. [DOI] [PubMed] [Google Scholar]
- 15.Roelofse JA, Payne PK, Morkel JA, Penkler L, Malan J. Intranasal midazolam spray in adults-pharmacokinetics and clinical use. S Afr J Anesth Analg. 2000;6:16–19. [Google Scholar]
- 16.Davis ME, Brewster ME. Cyclodextrin-based pharmaceutics: past, present and future. Nat Rev. 2004;3:1023–35. doi: 10.1038/nrd1576. [DOI] [PubMed] [Google Scholar]
- 17.Carrier RL, Miller LA, Ahmed I. The utility of cyclodextrins for enhancing oral bioavailability. J Control Release. 2007;123:78–99. doi: 10.1016/j.jconrel.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 18.Masson M, Loftsson T, Masson G, Stefansson E. Cyclodextrins as permeation enhancers: some theoretical evaluations and in vitro testing. J Control Release. 1999;59:107–18. doi: 10.1016/s0168-3659(98)00182-5. [DOI] [PubMed] [Google Scholar]
- 19.Illum L. Chitosan and its use as a pharmaceutical excipient. Pharm Res. 1998;15:1326–31. doi: 10.1023/a:1011929016601. [DOI] [PubMed] [Google Scholar]
- 20.Soane RJ, Hinchcliffe M, Davis SS, Illum L. Clearance characteristics of chitosan based formulations in the sheep nasal cavity. Int J Pharm. 2001;217:183–91. doi: 10.1016/s0378-5173(01)00602-0. [DOI] [PubMed] [Google Scholar]
- 21.Dodane V, Amin Khan M, Merwin JR. Effect of chitosan on epithelial permeability and structure. Int J Pharm. 1999;182:21–32. doi: 10.1016/s0378-5173(99)00030-7. [DOI] [PubMed] [Google Scholar]
- 22.Davis SS, Illum L. Absorption enhancers for nasal drug delivery. Clin Pharmacokinet. 2003;42:1107–28. doi: 10.2165/00003088-200342130-00003. [DOI] [PubMed] [Google Scholar]
- 23.Illum L, Watts P, Fisher AN, Hinchcliffe M, Norbury H, Jabbal-Gill I, Nankervis R, Davis SS. Intranasal delivery of morphine. J Pharmacol Exp Ther. 2002;301:391–400. doi: 10.1124/jpet.301.1.391. [DOI] [PubMed] [Google Scholar]
- 24.Dussy FE, Hamberg C, Briellmann TA. Quantification of benzodiazepines in whole blood and serum. Int J Legal Med. 2006;120:323–30. doi: 10.1007/s00414-005-0042-1. [DOI] [PubMed] [Google Scholar]
- 25.Schachinger H, Cox D, Linder L, Brody S, Keller U. Cognitive and psychomotor function in hypoglycemia: response error patterns and retest reliability. Pharmacol Biochem Behav. 2003;75:915–20. doi: 10.1016/s0091-3057(03)00167-9. [DOI] [PubMed] [Google Scholar]
- 26.Penkler LJ, Whittaker D, Worthington M, Reyneke-Barnard C, Roelofse J. Intranasal midazolam: pre-formulation, formulation and pilot clinical experience. [Abstract]. Published in the online journal of the American Association of Pharmaceutical Scientists (AAPS) on September 19, 2007. Available at http://www.aapsj.org/abstracts/AM_1999/652.htm. [Google Scholar]
- 27.Persson MP, Nilsson A, Hartvig P. Relation of sedation and amnesia to plasma concentrations of midazolam in surgical patients. Clin Pharmacol Ther. 1988;43:324–31. doi: 10.1038/clpt.1988.39. [DOI] [PubMed] [Google Scholar]