

Abstract
Introduction
The fimA-encoded fimbriae of the periodontal pathogen Porphyromonas gingivalis display genetic diversity. Type I fimbriated P. gingivalis (Pg-I) has been most widely studied at the molecular level, whereas Pg-II is the most frequent isolate from severe periodontitis.
Methods
To investigate virulence differences between Types I and II fimbriae, we examined strains 33277 (Pg-I) and OMZ314 (Pg-II), reciprocal swap mutants (i.e., expressing the heterologous fimbrial type), and their respective FimA-deficient derivatives. These organisms were tested in a mouse periodontitis model and in interactions with mouse macrophages, a cell type that plays important roles in chronic infections.
Results
Strain 33277 induced significantly more periodontal bone loss than OMZ314, and substitution of Type II fimbriae with Type I in OMZ314 resulted in a more virulent strain than the parent organism. However, the presence of Type II fimbriae was associated with increased proinflammatory and invasive activities in macrophages.
Conclusion
The inverse relationship between proinflammatory potential and ability to cause experimental periodontitis may suggest that an aggressive phenotype could provoke a host response that would compromise the persistence of the pathogen.
Introduction
Porphyromonas gingivalis is a major pathogen in human periodontitis, an inflammatory disease leading to destruction of the tooth-supporting tissues (12, 18). This bacterium is moreover implicated in certain systemic conditions, such as atherosclerosis (9). Its pathogenicity is attributed to several virulence factors, including cysteine proteinases, hemagglutinins, and cell surface fimbriae (24). The fimbriae constitute adhesive filamentous appendages and comprise two types: fimA-encoded “major” fimbriae and mfa1-encoded “minor” fimbriae of considerably shorter length (37). The major fimbriae (henceforth referred to as “fimbriae”) are traditionally recognized as a critical colonization factor (24). It is now appreciated that the fimbriae contribute to P. gingivalis virulence also through immune subversion of innate host responses (17, 34).
On the basis of sequence diversity of the fimA gene, the fimbriae are currently classified into six genotypes (I-V and Ib) (3). P. gingivalis expressing Type II fimbriae (designated Pg-II) is the most common genotype, while the second most prevalent genotype has been variably found to be Type IV, Ib, or I depending on the ethnic population studied (reviewed in refs. (3, 11). Type I fimbriae display 97% amino-acid identity with Type Ib, 76% identity with Type II, but only 46% identity with Type IV (reviewed in ref. (11). In terms of clinical significance, Pg-II is the most frequent isolate from deep periodontal pockets (2). Interestingly, despite lower prevalence, Pg-I is associated with diseased sites that are more refractory to periodontal treatment than Pg-II–infected sites (8).
Most studies examining the virulence properties of P. gingivalis have predominantly used Pg-I strains (e.g., 33277 or 381) or their purified fimbriae, which have been considered as prototypical (4, 9, 13, 36, 37). However, due to diverse amino-acid sequences of the various types of fimbriae and, therefore, the possibility that these may display distinct virulence features, there is a recent interest in comparative studies of P. gingivalis fimbrial genotypes. These studies have shown that Pg-II and, to a lesser extent, Pg-I are the most potent genotypes in terms of adhesive and cell-invasive activities (28, 29). However, Pg-II displays increased adhesion to and invasion of epithelial cells compared to Pg-I (28). Moreover, Pg-II was found to be more proinflammatory than Pg-I (6, 30).
P. gingivalis strains 33277 and OMZ314 have been widely studied as model Pg-I and Pg-II strains, respectively (14, 21, 29, 31). Although the genetic diversity of their fimA genes may contribute to virulence differences, additional factors may play a role. To control for potential differences in non-fimbrial virulence factors and, thereby, validly determine virulence variations attributed to specific fimbrial types, an elegant genetic system was developed (21). Specifically, the Type I and II fimA genes were exchanged between the 33277 and OMZ314 strains, resulting in two swap mutants, i.e., Type II fimA-expressing 33277 and Type I fimA-expressing OMZ314 (21). This study revealed that substitution of Type I FimA with Type II promotes P. gingivalis invasion of epithelial cells and enhances its proinflammatory properties (21). Conversely, the reverse substitution (Type II FimA replaced by Type I) rendered the modified strain less potent in the same assays (21).
In this paper, using 33277 and OMZ314 in parallel with their fimA swap mutants, we determined for the first time the relative contribution of Type I and II fimbriae to P. gingivalis-induced bone loss in the mouse periodontitis model. At the host cellular level, we examined the phagocytic uptake of Type I and II clones and, moreover, investigated their intracellular fate in correlation with the macrophage antimicrobial response. Our findings suggest that an overt aggressive/proinflammatory phenotype could compromise the ability of P. gingivalis to persist and cause disease in the mouse periodontitis model.
Materials and methods
Bacteria
P. gingivalis was grown anaerobically at 37°C in GAM medium, containing 5 μg/ml hemin and 1 μg/ml menadione (Nissui Pharmaceutical). The Type I and II fimA genes were exchanged between strains 33277 and OMZ314, as previously described (21), resulting in two swap mutants (SM), i.e., Type II fimA-expressing 33277 (strain KDC2; designated SM I→II) and Type I fimA-expressing OMZ314 (strain KDC1; SM II→I). The swapped fimA genes were expressed from their native promoters and the expected fimbrial phenotype was confirmed genetically, biochemically, and morphologically (21). The strains used also included FimA-deficient (“nonfimbriated”) isogenic mutants of 33277 and OMZ314 (Table 1). Type I fimbriae were extracted by a washing method and purified as previously described (38). Type II fimbriae were extracted from bacterial cell lysates using a French Press and ultracentrifugation. The extracts were then subjected to 50% saturated ammonium sulfate precipitation, dialyzed against 20mM Tris-HCl, pH 8.0, and purified by gel filtration (Sephacryl S-400; GE Healthcare) and ion-exchange chromatography (DEAE-Sepharose; GE Healthcare). The fimbrial preparations were free of contaminating substances on silver-stained SDS-PAGE, and tested negative for endotoxin (<6 EU/mg protein) according to the BioWhittaker LAL assay.
Table 1.
P. gingivalis strains used in this study
| Strain | Remarks | FimA phenotype | Designation used in this study | Source/Reference |
|---|---|---|---|---|
| 33277 | Wild-type | Type I | WT I | ATCC |
| KDC2 | Type I fimA substituted by Type II Isogenic with 33277 | Type II | * SM I→II | (21) |
| JI-1 | Nonfimbriated Isogenic with 33277 | Not applicable | FimA(−)I | (34) |
| OMZ314 | Wild-type | Type II | WT II | (29) |
| KDC1 | Type II fimA substituted by Type I Isogenic with OMZ314 | Type I | * SM II→I | (21) |
| KDC3 | Nonfimbriated Isogenic with OMZ314 | Not applicable | FimA(−)II | (21) |
Swap mutants (SM); the arrow indicates the current FimA type after substitution.
Mammalian cell culture
Thioglycollate-elicited macrophages were isolated from the peritoneal cavity of BALB/cByJ mice (Jackson Labs) (15). Mouse macrophages and monocytic THP-1-Blue™ cells (InVivogen) were cultured in RPMI 1640 (InVitrogen) containing 10% heat-inactivated fetal bovine serum, 2mM L-glutamine, 10mM HEPES, 100 U/ml penicillin G, 100 μg/ml streptomycin, and 0.05 mM 2-mercaptoethanol. Cell viability was monitored by the CellTiter-Blue™ assay (Promega). None of the treatments affected cell viability compared to medium-only control.
Phagocytosis
Flow cytometry was performed to assess the uptake of P. gingivalis (34). Briefly, mouse macrophages were incubated at 37°C with FITC-labeled P. gingivalis for 30 min (MOI=10:1; 5×105 macrophages used per assay). Phagocytosis was stopped by cooling the incubation tubes on ice. After washing to remove nonadherent bacteria, in some groups, extracellular fluorescence (representing attached but not internalized bacteria) was quenched with 0.2% trypan blue. The cells were washed again, fixed, and analyzed by flow cytometry (% positive cells for FITC-P. gingivalis and mean fluorescence intensity [MFI]) using the FACSCalibur and the CellQuest software (Becton-Dickinson). Association (i.e., representing both adherence and phagocytosis) or phagocytic indices were calculated using the formula (% positive cells × MFI)/100.
Intracellular survival assay
The capacity of phagocytosed P. gingivalis for intracellular persistence in macrophages was determined by an antibiotic protection-based survival assay, as we previously described (34). Briefly, viable counts (CFU) of internalized P. gingivalis were determined by plating serial dilutions of macrophage lysates on blood agar plates subjected to anaerobic culture. Prior to macrophage lysis, extracellular nonadherent bacteria were removed by washing, while extracellular adherent bacteria were killed by addition of gentamicin and metronidazole.
Cell activation assays
Induction of cytokine release in culture supernatants of activated mouse macrophages was measured by ELISA (eBioscience). Induction of nitric oxide (NO) was assessed by measuring the amount of NO2− (stable metabolite of NO) in stimulated culture supernatants using a Griess reaction-based assay (R&D Systems). Activation of NF-κB was assessed using THP-1-Blue™ cells stably transfected with NF-κB-inducible reporter system, following the manufacturer’s protocol (InVivogen). In all three assays, a MOI of 10:1 was used (2 ×106 bacteria and 2×105 host cells).
Confocal microscopy
Confocal laser scanning microscopy was used to determine colocalization of P. gingivalis with lysosomes, as previously described (33). Briefly, macrophages were exposed for 30 min to FITC-labeled P. gingivalis (MOI=10:1; 5×105 macrophages), washed, and incubation was allowed to proceed for an additional 90 min at 37°C. Subsequently, the cells were stained with LysoTraker Red DND-99, which targets acidified late endosomes and lysosomes. The cells were then fixed and imaged on an Olympus FV500 confocal microscope. Shown in figure 5 are representative single optical sections and 2-color overlay (merge) confocal images, as well as additional overlay with differential interference contrast (DIC) images. The degree of colocalization of P. gingivalis with lysosomes was quantified using the Image J software with the Intensity Correlation Analysis plugin (NIH; http://rsb.info.nih.gov/ij).
Fig. 5. Colocalization of P. gingivalis strains 3227 and KDC2 with lysosomes.

Primary mouse macrophages were exposed to FITC-labeled P. gingivalis 33277 (Type I FimA strain) or its isogenic KDC2 mutant (Type I fimA substituted by Type II) at a MOI of 10:1 for 30 min. After washing, incubation was allowed to proceed for an additional 90 min at 37°C. Subsequently, lysosomes were stained with LysoTraker Red for 15 min followed by cell fixation and imaging by confocal microscopy. Shown are representative single optical sections; green fluorescent bacteria colocalizing with red fluorescent lysosomes are manifested as yellow spots in merged images.
Mouse periodontitis model
BALB/cByJ mice (10–12 week-old; The Jackson Laboratory) were orally infected with P. gingivalis strains (Table 1) for induction of periodontal bone loss, essentially as originally described by Baker (5) with slight modifications (34). Briefly, upon suppression of the normal oral flora with antibiotics, mice were infected by oral gavage three times at 2-day intervals with 109 CFU P. gingivalis suspended in 2% carboxymethylcellulose/phosphate-buffered saline. Sham-infected controls received 2% carboxymethylcellulose alone. Mice were euthanized six weeks later and assessment of periodontal bone loss in defleshed maxillae was performed under a dissecting microscope (×40) fitted with a video image marker measurement system (VIA-170K; Fryer). Specifically, the distance from the cementoenamel junction (CEJ) to the alveolar bone crest (ABC) was measured on 14 predetermined points on the buccal surfaces of the maxillary molars. To calculate bone loss, the 14-site total CEJ-ABC distance for each mouse was subtracted from the mean CEJ-ABC distance of sham-infected mice. The results were expressed in mm and negative values indicate bone loss relative to sham-infected controls. This morphometric method is well established and validated (5, 10) and correlates very well with histomorphometric and microcomputed tomographic analysis (all three methods were found to accurately quantify periodontal bone loss)(25). All animal procedures were approved by the Institutional Animal Care and Use Committee, in compliance with established Federal and State policies.
Statistical analysis
Data were evaluated by ANOVA and the Tukey-Kramer Multiple Comparisons Test using the GraphPad InStat software. Where appropriate (comparison of two groups only), two-tailed t tests were also performed. P < 0.05 was taken as the level of significance.
Results
Type I vs. Type II fimbriae and P. gingivalis-induced bone loss
We and others have shown that Type I fimbriae play an important virulence role in P. gingivalis-induced periodontal bone loss in mice (9, 34). To determine and compare the relative contributions of Type I and Type II fimbriae to P. gingivalis-induced bone loss, mice were orally infected with Pg-I (33277) or Pg-II (OMZ314) strains, their reciprocal swap mutants (SM I→II and SM II→I), and their respective non-fimbriated mutants (FimA(−)I and FimA(−)II) (Table 1). Periodontal bone loss was induced to varying degrees by all tested strains (Fig. 1). As expected, strain 33277 caused significantly more bone loss than its isogenic FimA(−)I mutant (p < 0.001) but, interestingly, was also more potent in this activity than the OMZ314 strain (p < 0.05). The latter finding indirectly suggested that Type I fimbriae may confer additional virulence potential over Type II in this model. This inference was substantiated by the observation that substitution of Type II fimbriae with Type I resulted in a more virulent strain (SM II→I) than the parent organism, OMZ314 (p < 0.001; Fig. 1). However, the observed reduction in virulence by the reverse substitution (i.e., 33277 vs. SM I→II) did not reach statistical significance (p > 0.05; Fig. 1), suggesting that factors other than FimA also play a role in pathogenesis. Quite unexpectedly, OMZ314 was not significantly more virulent than its isogenic FimA(−)II mutant (p > 0.05); rather, this comparison was suggestive that the presence of Type II fimbriae tended to decrease induction of bone loss (Fig. 1). In summary, at least in the mouse periodontitis model, the Type I fimbriae appear to contribute to P. gingivalis virulence more than the Type II fimbriae.
Fig. 1. Induction of periodontal bone loss by Type I or Type II P. gingivalis strains or mutants.

BALB/cByJ mice were orally infected with P. gingivalis 33277 (WT I) or OMZ314 (WT II), their reciprocal fimbrial swap mutants (SM I→II and SM II→I), and their isogenic FimA-deficient mutants (FimA(−)I and FimA(−)II), or treated with vehicle only (sham). At termination of the experiment, the distancefrom the cementoenamel junction to the alveolar bone crestwas measured at 14 predetermined buccal sites on the maxillary molars and the values were transformed to directly indicate bone loss. Data are means ± SD (n = 5). Asterisks indicate statistically significant differences (p < 0.001) between infected and sham-infected mice. The inverted triangle denotes significant difference (p < 0.05) between WT II and WT I, and the black circle shows significant difference (p < 0.001) between SM II→I and WT II.
Fimbrial genotype-dependent differences in proinflammatory activities
It is conceivable that the reduced P. gingivalis virulence associated with Type II fimbriae in the mouse periodontitis model (Fig. 1) might be due to stronger protective host responses induced by Type II compared to Type I fimbria-expressing strains. We previously showed that P. gingivalis is susceptible to nitric oxide (NO)-mediated macrophage killing, although it is resistant to reactive oxygen species (16). We thus investigated potentially differential induction of NO by mouse macrophages in response to the strains used in the in vivo study. All strains tested were capable of inducing NO production to varying degrees, although the FimA(−)I and FimA(−)II were the least potent (Fig. 2A). However, WT-II (i.e., OMZ314) induced significantly higher NO levels than both WT-I (33277) and SM II→I (p < 0.01), although comparable NO levels to SM I→II (Fig. 2A). Therefore, expression of Type II fimbriae results in stronger induction of NO. This conclusion is further supported by the significant increase of NO production upon substitution of Type I fimbriae with Type II (p < 0.05 for SM I→II vs. 33277; Fig. 2A). Since induction of NO is NF-κB-dependent, we examined whether the NO-inducing capacities of the various strains tested correlates with differential NF-κB activation. Indeed, induction of NF-κB activation followed a similar pattern, i.e., expression of Type II fimbriae was associated with stronger NF-κB activation than expression of Type I fimbriae (Fig. 2B). This notion was confirmed by direct comparison for the first time of purified Type I and II fimbriae. Specifically, Type II fimbriae from OMZ314 were significantly more potent in activating NF-κB than Type I fimbriae from 33277 (Fig. 2C).
Fig. 2. Induction of NO production and activation of NF-κB by Type I or Type II P. gingivalis strains or mutants.
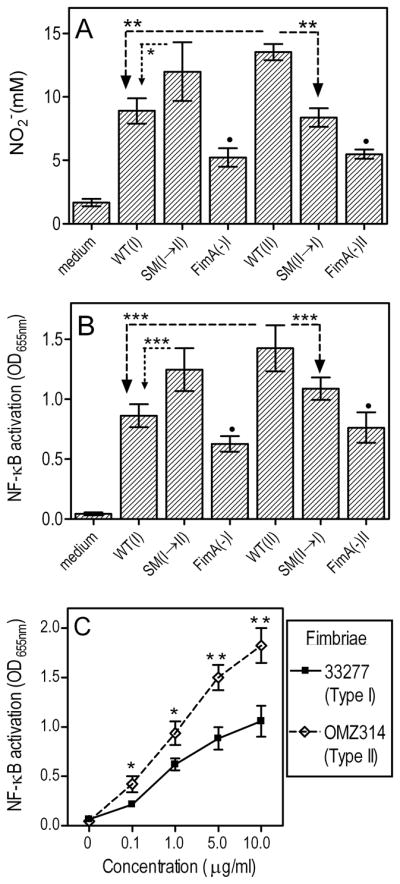
Primary mouse macrophages (A) or the reporter THP-1-Blue™ cell line (B) were incubated with medium only or with the indicated strains of P. gingivalis (MOI = 10:1). After 24h, production of NO2− (stable metabolite of NO) was assayed by the Griess reaction (A) and NF-κB activation was determined colorimetrically by measuring the activity of NF-κB-inducible alkaline phosphatase secreted in culture supernatants (B). In C, a similar NF-κB assay was performed using purified Type I or Type II fimbriae, at the indicated concentrations. Data are means ± SD (n = 3) from one of three sets of independent experiments that yielded consistent results. Asterisks show statistically significant differences (*, p < 0.05; **, p < 0.01; ***, p < 0.001) among WT and SM strains (A-B), or between purified Type I and II fimbriae (C). Black circles indicate significantly reduced activities (p < 0.05) in FimA-deficient mutants compared to their corresponding parent strains (A-B).
Moreover, expression of Type II fimbriae was associated with stronger induction of cytokine responses as revealed by comparison of the OMZ314 and 33277 strains (p < 0.01; Fig. 3). In addition, depending on the type of FimA substitution, cytokine induction tended to be either positively (SM I→II) or negatively (SM II→I) influenced, although some of these differences did not always reach statistical significance (Fig. 3). Taken together, the data from figures 1–3 suggest an inverse relationship between induction of bone loss and activation of NF-κB-dependent activities, such as production of NO and proinflammatory cytokines.
Fig. 3. Induction of proinflammatory cytokine production by Type I or Type II P. gingivalis strains or mutants.

Primary mouse macrophages were incubated with medium only or with the indicated strains of P. gingivalis (MOI = 10:1). After 24h, induction of release of TNF-α (A), IL-6 (B), or IL-12p40 in culture supernatants was determined by ELISA. Data are means ± SD (n = 3) from one of two experiments with consistent results. Asterisks show statistically significant differences (*, p < 0.05; **, p < 0.01; ***, p < 0.001) among WT and SM strains. Black circles indicate significantly reduced activities (p < 0.05) in FimA-deficient mutants compared to their corresponding parent strains.
Effects of FimA type on P. gingivalis intracellular entry into macrophages
Although macrophages are professional phagocytes which actively internalize bacteria, the status of Type I fimbriae (presence or absence, or genetically altered) dictates the degree of P. gingivalis uptake (34). To further characterize potential functional differences of Type I and II fimbriae, we examined macrophage uptake of 33277 and OMZ314, their reciprocal swap mutants (SM I→II and SM II→I), and their respective FimA-deficient mutants (Table 1). As expected, the FimA-deficient mutants were significantly less active than their corresponding parent strains in both macrophage association and uptake assays (p < 0.001; Fig. 4A). However, no significant differences were noted between the wild-type strains in the way they associate or are taken up by macrophages, although substitution of Type I FimA with Type II (clone SM I→II) resulted in enhanced P. gingivalis association with and phagocytosis by macrophages (p < 0.001; Fig. 4A). Conversely, the reverse substitution (clone SM II→I) resulted in decreased association or uptake activities (p < 0.001; Fig. 4A). Therefore, all other factors being equal, Type II fimbriae endow P. gingivalis with increased adhesive/invasive activities, although, alternatively, Type I fimbriae may impede phagocytosis.
Fig. 4. Role of FimA type on the uptake and intracellular fate of P. gingivalis in mouse macrophages.

(A) Primary mouse macrophages were incubated for 30 min with the indicated FITC-labeled P. gingivalis strains (MOI = 10:1). Association (i.e., representing both adherence and phagocytosis) or phagocytic indices were determined by flow cytometry, using the formula % positive cells × MFI/100. (B-C) Primary mouse macrophages were incubated at a MOI of 10:1 with Type I or II P. gingivalis or mutants, for 1.5 (B) or 15h (C). The persistence of viable internalized bacteria was determined using an antibiotic protection-based survival assay. Data are means ± SD (n = 3), from one of three independent sets of experiments that yielded consistent results. Double asterisks indicate statistically significant differences (p < 0.01) between FimA-deficient or swap mutants compared to their corresponding wild-type strains. In C, a single asterisk denotes significant (p < 0.05) difference between the indicated wild-type groups.
We then followed the intracellular fate of these P. gingivalis strains, to determine whether the type of fimbriae impacts on their survival capacity within macrophages. Viable CFU counts for the two FimA-deficient mutants were recovered at significantly lower levels than their corresponding parent strains at the 1.5-h time point (Fig. 4B) and were hardly recoverable following overnight incubation for 15h (Fig. 4C). There were no significant differences between 33277 and OMZ314 in terms of recovered CFU counts after 1.5-h incubation (Fig. 4B), although modest but statistically significant differences were noted at the 15-h time point (p < 0.05; Fig. 4C).
Whether Type I fimbriae confer a survival advantage to P. gingivalis can be more validly determined by comparing the wild-type strain with its isogenic swap mutant. However, 33277 did not display increased intracellular survival compared to SM I→II (Fig. 4B,C). In fact, SM I→II CFU counts were about three-fold higher than the counts corresponding to 33277 (Fig. 4B,C), which may reflect the higher uptake efficiency of SM I→II compared to 33277 (Fig. 4). Indeed, no significant differences in intracellular survival were noted between SM I→II and 33277 when the MOI values were adjusted to allow equilibration of the initial intracellular bacterial load (not shown). Moreover, although confocal microscopy confirmed the higher uptake efficiency of SM I→II (i.e., strain KDC2), it did not support possible enhanced survival of this strain (Fig. 5). Specifically, the proportions of 33277 and SM I→II bacteria trafficking to lysosomes (where they are presumably degraded) were comparable (Fig. 5). In support of this visual observation, when the confocal colocalization of LysoTraker-labeled lysosomes with FITC-labeled 33277 or SM I→II was quantified by ImageJ analysis, no significant differences were found (% lysosome colocalization of 33277 and SM I→II were 56.1±11.3 and 64.7±10.7, respectively, upon examining 15 macrophages from each group). Thus, although the Type of FimA has a major impact on the internalization of P. gingivalis by macrophages, it does not significantly influence the subsequent intracellular fate of the pathogen.
Discussion
It is becoming increasingly evident from this and other work that P. gingivalis represents a heterogeneous group of strains with distinct properties, in terms of proinflammatory potential, immune evasion capacity, cell invasive activity, and ability to cause disease (7, 20, 21, 30, 37). The genetic diversity of the fimA gene may, at least in part, mediate some of the virulence differences among distinct strains.
The findings that substitution of Type I FimA with Type II (clone SM I→II) results in enhanced P. gingivalis phagocytosis by macrophages, whereas the reverse substitution (clone SM II→I) decreases these activities, suggest that Type II fimbriae interact more efficiently with macrophage phagocytic receptors than Type I fimbriae. However, there were no substantial differences between the original strains (33277 and OMZ314) in terms of phagocytosis, despite expressing different fimbrial types. This may be related to the fact that OMZ314, but not 33277, is encapsulated (19). Indeed, the presence of a hydrophilic capsule is negatively correlated with phagocytosis, whereas surface hydrophobicity, which is strongly associated with the presence of fimbriae, promotes P. gingivalis interactions with phagocytes (32, 35). In the OMZ314 strain, therefore, the increased interactive potential of its Type II fimbriae with phagocytes may be offset by the presence of its capsule, which is also a potential virulence factor. In this regard, although the potential contribution of the capsule of P. gingivalis in the induction of periodontal bone loss has not been addressed yet, it is known that this factor contributes to disseminating infections in the mouse subcutaneous abscess model (23, 32).
In assays of macrophage activation, the immunostimulatory potential of P. gingivalis 33277 was significantly lower than that of OMZ314, in agreement with differential in vivo cytokine responses upon systemic P. gingivalis infection with these strains (21). The findings that both strains displayed altered proinflammatory activities upon mutual exchange of fimbrial type indicate that their fimbriae are largely responsible for their cell activation capacity. The fimbriae could influence these activities either indirectly, i.e., by facilitating P. gingivalis-macrophage interactions, and/or directly by activating fimbria-specific signaling. The latter possibility is supported by our findings that purified Type II fimbriae were more potent than Type I in cytokine induction, thus reflecting the activities of their respective strains. These differences in proinflammatory cytokine release were modest despite being statistically significant and reproducible. However, it is likely that initial small changes in cytokine production (upon initial encounter of the pathogen with phagocytes) are amplified in vivo through mobilization and activation of additional cells or cell types.
Activated macrophages may help control bacterial infection, but can also contribute to periodontal tissue destruction, depending on the quality, magnitude, and duration of activation (22). At least in certain in vivo models, induction of macrophage nitric oxide responses correlates with increased clearance of P. gingivalis (16, 17). Studies with additional pathogens also support the notion that suppressed activation of innate host responses leads to enhanced bacterial survival (27). It could be speculated, therefore, that reduced activation of the immune system by 33277 relative to OMZ314 (as seen here in vitro or in vivo in a previous report (30)) may allow 33277 to persist at higher levels than OMZ314 in the mouse host and eventually cause greater bone loss. Consistent with this notion, when the immunostimulatory potential of P. gingivalis OMZ314 was suppressed upon substitution of its Type II FimA with Type I (clone SM II→I), this resulted in enhanced induction of periodontal bone loss. At least in part, the decreased Type II FimA-associated virulence could be due to reduced colonization by the OMZ314 strain, although Type II FimA is more adhesive than Type I FimA (1). It is conceivable, however, that possible reduced colonization by OMZ314 could be the result of enhanced innate host response, as suggested above.
Despite their aggressive phenotype which could provoke a clearance response in mice, Pg-II may have more opportunities to establish chronic infection in humans, perhaps through supportive interactions with other members of the subgingival biofilm that may be absent from the murine oral cavity. Indeed, according to epidemiological studies, Pg-II strains represent the most frequently isolated strains from periodontal lesions in diverse populations (2, 26), although more studies are warranted to fully validate the generality of these observations. Periodontitis can be modeled in certain animal species, although no model can faithfully reproduce all aspects of periodontal pathogenesis. However, each of the currently validated models can be very informative in terms of their capacity to test specific hypotheses, and different models can measure distinct aspects of bacterial virulence (10). For example, the subcutaneous abscess model can measure the potential of a periodontal pathogen for systemic dissemination, whereas the oral gavage model can assess the ability of a pathogen to persist in the oral cavity and cause bone loss (10).
This study has provided novel insights into the apparent heterogeneity in P. gingivalis virulence properties. Our findings in macrophages indicate that Type II fimbriae endow P. gingivalis with increased invasive and proinflammatory capacity compared to Type I fimbriae. However, there is an inverse relationship between proinflammatory activity and ability to induce periodontal bone loss. It is possible that different P. gingivalis strains may have developed distinct virulence strategies to cope with the mammalian host (12). Certain strains may be overtly aggressive aiming to overwhelm host defense mechanisms, whereas other may have evolved immunosuppressive strategies to evade immune elimination. For example, strain 33277 utilizes its Type I fimbriae to suppress TLR2-dependent killing by instigating an inhibitory cross-talk with CXCR4 (17). More studies are warranted to further elucidate intricate virulence variations among distinct P. gingivalis fimbrial genotypes.
Acknowledgments
This study was supported by the JSPS and the AUG High-Tech Research Center Project (F.Y.), Grants-in-aid for Scientific Research, Ministry of Education, Japan (A.A.), and U.S. PHS Grants DE015254 and DE018292 from the NIH (G.H.).
References
- 1.Amano A. Molecular interaction of Porphyromonas gingivalis with host cells: implication for the microbial pathogenesis of periodontal disease. J Periodontol. 2003;74:90–96. doi: 10.1902/jop.2003.74.1.90. [DOI] [PubMed] [Google Scholar]
- 2.Amano A, Nakagawa I, Kataoka K, Morisaki I, Hamada S. Distribution of Porphyromonas gingivalis strains with fimA genotypes in periodontitis patients. J Clin Microbiol. 1999;37:1426–1430. doi: 10.1128/jcm.37.5.1426-1430.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amano A, Nakagawa I, Okahashi N, Hamada N. Variations of Porphyromonas gingivalis fimbriae in relation to microbial pathogenesis. J Periodontal Res. 2004;39:136–142. doi: 10.1111/j.1600-0765.2004.00719.x. [DOI] [PubMed] [Google Scholar]
- 4.Amano A, Sojar HT, Lee JY, Sharma A, Levine MJ, Genco RJ. Salivary receptors for recombinant fimbrillin of Porphyromonas gingivalis. Infect Immun. 1994;62:3372–3380. doi: 10.1128/iai.62.8.3372-3380.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baker PJ, Dixon M, Roopenian DC. Genetic control of susceptibility to Porphyromonas gingivalis-induced alveolar bone loss in mice. Infect Immun. 2000;68:5864–5868. doi: 10.1128/iai.68.10.5864-5868.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bodet C, Chandad F, Grenier D. Porphyromonas gingivalis-induced inflammatory mediator profile in an ex vivo human whole blood model. Clin Exp Immunol. 2006;143:50–57. doi: 10.1111/j.1365-2249.2005.02956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Enersen M, Olsen I, Kvalheim O, Caugant DA. FimA genotypes and multilocus sequence types of Porphyromonas gingivalis from patients with periodontitis. J Clin Microbiol. 2008;46:31–42. doi: 10.1128/JCM.00986-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fujise O, Miura M, Hamachi T, Maeda K. Involvement of Porphyromonas gingivalis fimA genotype in treatment outcome following non-surgical periodontal therapy. J Periodontol. 2005;76:1661–1666. doi: 10.1902/jop.2005.76.10.1661. [DOI] [PubMed] [Google Scholar]
- 9.Gibson FC, 3rd, Hong C, Chou HH, et al. Innate immune recognition of invasive bacteria accelerates atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2004;109:2801–2806. doi: 10.1161/01.CIR.0000129769.17895.F0. [DOI] [PubMed] [Google Scholar]
- 10.Graves DT, Fine D, Teng Y-TA, Van Dyke TE, Hajishengallis G. The use of rodent models to investigate host-bacteria interactions related to periodontal diseases. J Clin Periodontol. 2008;35:89–105. doi: 10.1111/j.1600-051X.2007.01172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hajishengallis G. Peptide mapping of a functionally versatile fimbrial adhesin from Porphyromonas gingivalis. Int J Pept Res Ther. 2007;13:533–546. [Google Scholar]
- 12.Hajishengallis G. Porphyromonas gingivalis-host interactions: open war or intelligent guerilla tactics? Microbes Infect. 2009;11:637–645. doi: 10.1016/j.micinf.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hajishengallis G, Ratti P, Harokopakis E. Peptide mapping of bacterial fimbrial epitopes interacting with pattern recognition receptors. J Biol Chem. 2005;280:38902–38913. doi: 10.1074/jbc.M507326200. [DOI] [PubMed] [Google Scholar]
- 14.Hajishengallis G, Shakhatreh M-AK, Wang M, Liang S. Complement receptor 3 blockade promotes IL-12-mediated clearance of Porphyromonas gingivalis and negates its virulence in vivo. J Immunol. 2007;179:2359–2367. doi: 10.4049/jimmunol.179.4.2359. [DOI] [PubMed] [Google Scholar]
- 15.Hajishengallis G, Tapping RI, Martin MH, et al. Toll-like receptor 2 mediates cellular activation by the B subunits of type II heat-labile enterotoxins. Infect Immun. 2005;73:1343–1349. doi: 10.1128/IAI.73.3.1343-1349.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hajishengallis G, Wang M, Bagby GJ, Nelson S. Importance of TLR2 in early innate immune response to acute pulmonary infection with Porphyromonas gingivalis in mice. J Immunol. 2008;181:4141–4149. doi: 10.4049/jimmunol.181.6.4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hajishengallis G, Wang M, Liang S, Triantafilou M, Triantafilou K. Pathogen induction of CXCR4/TLR2 cross-talk impairs host defense function. Proc Natl Acad Sci U S A. 2008;105:13532–13537. doi: 10.1073/pnas.0803852105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holt SC, Ebersole JL. Porphyromonas gingivalis, Treponema denticola, and Tannerella forsythia: the “red complex”, a prototype polybacterial pathogenic consortium in periodontitis. Periodontol 2000. 2005;38:72–122. doi: 10.1111/j.1600-0757.2005.00113.x. [DOI] [PubMed] [Google Scholar]
- 19.Inaba H, Nakano K, Kato T, et al. Heterogenic virulence and related factors among clinical isolates of Porphyromonas gingivalis with type II fimbriae. Oral Microbiol Immunol. 2008;23:29–35. doi: 10.1111/j.1399-302X.2007.00386.x. [DOI] [PubMed] [Google Scholar]
- 20.Jandik KA, Belanger M, Low SL, Dorn BR, Yang MC, Progulske-Fox A. Invasive differences among Porphyromonas gingivalis strains from healthy and diseased periodontal sites. J Periodontal Res. 2008;43:524–530. doi: 10.1111/j.1600-0765.2007.01064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kato T, Kawai S, Nakano K, et al. Virulence of Porphyromonas gingivalis is altered by substitution of fimbria gene with different genotype. Cell Microbiol. 2007;9:753–765. doi: 10.1111/j.1462-5822.2006.00825.x. [DOI] [PubMed] [Google Scholar]
- 22.Kornman KS, Page RC, Tonetti MS. The host response to the microbial challenge in periodontitis: assembling the players. Periodontol 2000. 1997;14:33–53. doi: 10.1111/j.1600-0757.1997.tb00191.x. [DOI] [PubMed] [Google Scholar]
- 23.Laine ML, van Winkelhoff AJ. Virulence of six capsular serotypes of Porphyromonas gingivalis in a mouse model. Oral Microbiol Immunol. 1998;13:322–325. doi: 10.1111/j.1399-302x.1998.tb00714.x. [DOI] [PubMed] [Google Scholar]
- 24.Lamont RJ, Jenkinson HF. Life below the gum line: Pathogenic mechanisms of Porphyromonas gingivalis. Microbiol Mol Biol Rev. 1998;62:1244–1263. doi: 10.1128/mmbr.62.4.1244-1263.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li CH, Amar S. Morphometric, histomorphometric, and microcomputed tomographic analysis of periodontal inflammatory lesions in a murine model. J Periodontol. 2007;78:1120–1128. doi: 10.1902/jop.2007.060320. [DOI] [PubMed] [Google Scholar]
- 26.Missailidis CG, Umeda JE, Ota-Tsuzuki C, Anzai D, Mayer MP. Distribution of fimA genotypes of Porphyromonas gingivalis in subjects with various periodontal conditions. Oral Microbiol Immunol. 2004;19:224–229. doi: 10.1111/j.1399-302X.2004.00140.x. [DOI] [PubMed] [Google Scholar]
- 27.Montminy SW, Khan N, McGrath S, et al. Virulence factors of Yersinia pestis are overcome by a strong lipopolysaccharide response. Nat Immunol. 2006;7:1066–1073. doi: 10.1038/ni1386. [DOI] [PubMed] [Google Scholar]
- 28.Nakagawa I, Amano A, Kuboniwa M, Nakamura T, Kawabata S, Hamada S. Functional differences among FimA variants of Porphyromonas gingivalis and their effects on adhesion to and invasion of human epithelial cells. Infect Immun. 2002;70:277–285. doi: 10.1128/IAI.70.1.277-285.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakagawa I, Inaba H, Yamamura T, et al. Invasion of epithelial cells and proteolysis of cellular focal adhesion components by distinct types of Porphyromonas gingivalis fimbriae. Infect Immun. 2006;74:3773–3782. doi: 10.1128/IAI.01902-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakano K, Kuboniwa M, Nakagawa I, et al. Comparison of inflammatory changes caused by Porphyromonas gingivalis with distinct fimA genotypes in a mouse abscess model. Oral Microbiol Immunol. 2004;19:205–209. doi: 10.1111/j.0902-0055.2004.00133.x. [DOI] [PubMed] [Google Scholar]
- 31.Nishikawa K, Yoshimura F, Duncan MJ. A regulation cascade controls expression of Porphyromonas gingivalis fimbriae via the FimR response regulator. Mol Microbiol. 2004;54:546–560. doi: 10.1111/j.1365-2958.2004.04291.x. [DOI] [PubMed] [Google Scholar]
- 32.Sundqvist G, Figdor D, Hanstrom L, Sorlin S, Sandstrom G. Phagocytosis and virulence of different strains of Porphyromonas gingivalis. Scand J Dent Res. 1991;99:117–129. doi: 10.1111/j.1600-0722.1991.tb01874.x. [DOI] [PubMed] [Google Scholar]
- 33.Wang M, Hajishengallis G. Lipid raft-dependent uptake, signalling and intracellular fate of Porphyromonas gingivalis in mouse macrophages. Cell Microbiol. 2008;10:2029–2042. doi: 10.1111/j.1462-5822.2008.01185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang M, Shakhatreh M-AK, James D, et al. Fimbrial proteins of Porphyromonas gingivalis mediate in vivo virulence and exploit TLR2 and complement receptor 3 to persist in macrophages. J Immunol. 2007;179:2349–2358. doi: 10.4049/jimmunol.179.4.2349. [DOI] [PubMed] [Google Scholar]
- 35.Watanabe K, Yamaji Y, Umemoto T. Correlation between cell-adherent activity and surface structure in Porphyromonas gingivalis. Oral Microbiol Immunol. 1992;7:357–363. doi: 10.1111/j.1399-302x.1992.tb00636.x. [DOI] [PubMed] [Google Scholar]
- 36.Yilmaz O, Watanabe K, Lamont RJ. Involvement of integrins in fimbriae-mediated binding and invasion by Porphyromonas gingivalis. Cell Microbiol. 2002;4:305–314. doi: 10.1046/j.1462-5822.2002.00192.x. [DOI] [PubMed] [Google Scholar]
- 37.Yoshimura F, Murakami Y, Nishikawa K, Hasegawa Y, Kawaminami S. Surface components of Porphyromonas gingivalis. J Periodontal Res. 2008;44:1–12. doi: 10.1111/j.1600-0765.2008.01135.x. [DOI] [PubMed] [Google Scholar]
- 38.Yoshimura F, Takahashi K, Nodasaka Y, Suzuki T. Purification and characterization of a novel type of fimbriae from the oral anaerobe Bacteroides gingivalis. J Bacteriol. 1984;160:949–957. doi: 10.1128/jb.160.3.949-957.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]