

Abstract
The role of the C-domain sites of cardiac troponin C in the modulation of the calcium signal remains unclear. In this study we investigated the effects of hypertrophic cardiomyopathy linked mutations A8V, E134D and D145E in cardiac troponin C on the properties of the C-domain sites. The A8V mutation had essentially no effect on the calcium or magnesium binding properties of the C-domain sites, while E134D mutation moderately decreased calcium and magnesium binding affinities. On the other hand, D145E mutation affected cooperative interactions between sites III and IV, significantly reducing calcium binding affinity of both sites. Binding of the anchoring region of cardiac troponin I (corresponding to residues 34-71) to cardiac troponin C with D145E mutation was not able to recover normal calcium binding to the C-domain. Experiments utilizing the fluorescent hydrophobic probe bis-ANS suggest that D145E mutation dramatically reduced the extent of calcium-induced hydrophobic exposure by the C-domain. At high non-physiological calcium concentration, A8V, E134D and D145E mutations minimally affected the affinity of cardiac troponin C for the regulatory region of cardiac troponin I (corresponding to residues 128-180). In contrast, at lower physiological calcium concentration, D145E mutation led to ~8-fold decrease in the affinity of cardiac troponin C for the regulatory region of cardiac troponin I. Our results suggest that calcium binding properties of the C-domain sites might be important for the proper regulatory function of cardiac troponin C.
Familial hypertrophic cardiomyopathy (HCM)1 is a genetic disorder of cardiac muscle, characterized by a hypertrophied left ventricle, with a wide range of clinical phenotypes and outcomes (for review, see (1–3)). HCM has been linked to hundreds of mutations of various sarcomeric proteins, including the thin filament proteins actin, tropomyosin, cardiac troponin I (cTnI) and cardiac troponin T (cTnT) (for review, see (4, 5)). Until recently, cardiac troponin C (cTnC), which is highly conserved among vertebrate species, was believed to have fewer HCM linked mutations. However, new evidence indicates that prevalence of HCM linked mutations in cTnC might be similar to that of other thin filament proteins (6, 7).
cTnC, comprised of the N- and C-terminal globular domains connected by a central α-helix (for review, see (8, 9)), is a member of the EF-hand super-family of Ca2+ binding proteins. A canonical helix-loop-helix EF-hand Ca2+ binding motif consists of a 12 residue loop flanked by α-helices. The loop residues at positions 1 (+X), 3 (+Y), 5 (+Z), 7 (−Y), 9 (−X) and 12 (−Z) coordinate the Ca2+ ion through seven oxygen atoms (for review, see (10–12)). Each domain of cTnC contains a pair of EF-hands numbered I-IV, but EF-hand I is not capable of binding Ca2+ due to several loop residue substitutions (13). The α-helices are designated A-H, with an additional 14-residue N-helix at the N-terminus. Because of the defunct first EF-hand, the N-domain of cTnC does not undergo a large conformational “opening” upon Ca2+ binding (14).
Binding of Ca2+ to the second EF-hand of cTnC is believed to play a direct role in regulation of muscle contraction. The C-domain EF-hands, which possess dramatically higher Ca2+ affinity and slower exchange rates compared to the N-domain EF-hand (for review see (15)), are occupied by either Ca2+ or Mg2+ under resting physiological conditions, and thus thought to play a structural role of anchoring cTnC into the thin filament.
Despite the fact that the C-domain sites are considered structural rather than regulatory, a number of mutations associated with either dilated cardiomyopathy (DCM) or HCM are located in the C-domain of cTnC (for review, see (4)). Recently, several novel mutations of cTnC (A8V, E134D and D145E) were linked to HCM (6, 7). Two of the mutations (A8V and D145E) led to higher force recovery and increased Ca2+ sensitivity of force development in skinned fibers, despite being located in separate domains of cTnC (6, 7). Since E134D mutation did not affect either the extent of force recovery or the Ca2+ sensitivity of force generation, it was hypothesized to be a polymorphism (4).
Considering that mutations in the C-domain sites of cTnC have been linked to both HCM and DCM, the importance of the C-domain in the modulation of muscle contraction might have been under-appreciated. The objective of this study was to determine the effect of recently discovered cTnC mutations linked to HCM (A8V, E134D and D145E) on the Ca2+/Mg2+ binding properties of the C-domain sites, and on interactions of cTnC with the regulatory region of cTnI.
EXPERIMENTAL PROCEDURES
Materials
Phenyl-Sepharose CL-4B, CaCl2 and EGTA were purchased from Sigma-Aldrich (St. Louis, MO). Quin-2 was purchased from Calbiochem (La Jolla, CA). IANBD was purchased from Invitrogen (Carlsbad, CA). The human cardiac TnI peptides: residues 34-71, herein designated as TnI34-71, and residues 128-180, herein designated as TnI128-180, were synthesized and purified by GenScript USA, Inc (Piscataway, NJ).
Protein Mutagenesis and Purification
The pET3a plasmid encoding human cTnC was a generous gift from Dr. Lawrence B. Smillie (University of Alberta, Canada). The cTnC construct used in this work contained C35S, T53C and C84S substitutions, to enable fluorescent labeling of cTnC on Cys53. Fluorescently labeled cTnC was used to determine the effect of the mutations on the affinity of cTnC for the regulatory fragment of cTnI (cTnI128-180). The HCM cTnC mutants were generated as previously described, and confirmed by DNA sequencing (16, 17). Expression and purification of cTnC and its mutants was carried out as previously described (16, 17).
Labeling of cTnC and its Mutants
cTnC and its mutants were labeled with the environmentally sensitive thiol-reactive fluorescent probe IANBD on Cys53 as previously described (16, 17).
Determination of Ca2+ Binding Sensitivities
All steady-state fluorescence measurements were performed using a Perkin-Elmer LS55 fluorescence spectrometer at 15°C. Tyr fluorescence was excited at 275 nm and monitored at 303 nm as microliter amounts of CaCl2 were added to 2 ml of each unlabeled TnC protein (0.5 μM) in titration buffer (200 mM MOPS (to prevent pH changes upon addition of Ca2+), 150 mM KCl, 2 mM EGTA, 1 mM DTT, 3 mM MgCl2, pH 7.0) at 15°C with constant stirring. The [Ca2+]free was calculated using the computer program EGCA02 developed by Robertson and Potter (18). The Ca2+ sensitivities of conformational changes were reported as a dissociation constant Kd, representing a mean of at least three titrations ± SE. The data were fit with a logistic sigmoid function (mathematically equivalent to the Hill equation), as previously described (19).
Determination of Mg2+ Binding Sensitivities
Mg2+ sensitivities were calculated from a decrease in the apparent Ca2+ affinities caused by 3 mM Mg2+, assuming competitive binding of Ca2+ and Mg2+, as described (20).
Determination of Ca2+ Dissociation Kinetics
All kinetic measurements were performed utilizing an Applied Photophysics Ltd. (Leatherhead, UK) model SX.18 MV stopped-flow instrument with a dead time of ~1.4 ms at 15°C. The rates of conformational changes induced by EGTA removal of Ca2+ from unlabeled cTnC or its mutants were measured following intrinsic Tyr fluorescence. Tyr was excited at 275 nm. The Tyr emission was monitored through a UG1 interference filter from Oriel (Stratford, CT). Ca2+ dissociation rates were also measured using the fluorescent Ca2+ chelator Quin-2. Quin-2 fluorescence was excited at 330 nm with its emission monitored through a 510 nm BrightLine Basic™ filter from Semrock (Rochester, NY). The changes in Quin-2 fluorescence were converted to moles of Ca2+ dissociating from unlabeled cTnC or its mutants by mixing increasing concentrations of Ca2+ with Quin-2, as previously described (21). The data were fit using a program (by P. J. King, Applied Photophysics Ltd) that utilizes the nonlinear Levenberg-Marquardt algorithm. Each koff represents an average of at least three separate experiments, each averaging at least five traces fit with a single exponential equation.
Determination of cTnI128-180 Peptide Affinities
IANBD fluorescence was monitored with excitation at 480 nm and emission at 525 nm. Microliter amounts of cTnI128-180 were added to 2 ml of each labeled cTnC protein (0.15 μM) in 10 mM MOPS, 150 mM KCl, 3 mM MgCl2, 1 mM CaCl2 or 2 μM CaCl2, 0.02 % Tween-20, 1 mM DTT, pH 7.0 at 15°C. Each peptide affinity represents a mean of at least three titrations ± SE fit to the root of a quadratic equation for binary complex formation as previously described (17, 22).
Statistical Analysis
Statistical significance was determined by an unpaired two-sample t-test using the statistical analysis software Minitab (State College, PA). The two means were considered to be significantly different when the P value was < 0.05. All data is shown as a mean value ± SE.
RESULTS
Location of the HCM Linked Mutations in Different Domains of cTnC
Figure 1 shows that the A8V mutation is located in the N-helix of the N-domain of cTnC, while E134D and D145E mutations are located in the C-domain of cTnC. The E134D mutation is located between Ca2+ binding sites III and IV, while the D145E mutation is located in the +Z chelating loop position of site IV.
Figure 1. Location of HCM linked mutations in the N- and C-domains of cTnC.

The figure shows ribbon representation of the cTnC in the Ca2+ bound state (Protein Data Bank entry 1AJ4 (14)). The A8V mutation (shown in red) is located in the N-helix of the N-domain, the E134D mutation (shown in green) is located between Ca2+ binding sites III and IV, and the D145E mutation (shown in blue) is located in the +Z position of Ca2+ binding site IV. This figure was generated using PyMOL (http://www.pymol.org/).
Effect of HCM Linked cTnC Mutations on the Ca2+and Mg2+ Binding Properties of the C-domain Sites
Tyr fluorescence was utilized to determine the effect of HCM linked cTnC mutations on the Ca2+ sensitivity of the C-domain sites in the absence and presence of 3 mM Mg2+. The Ca2+ dependent changes in intrinsic Tyr fluorescence are due to Ca2+ binding to sites III and IV of cTnC (23, 24). The C35S, T53C and C84S substitutions present in all the proteins had no effect on the Ca2+ affinity (in the absence or presence of Mg2+) or the rate of Ca2+ dissociation from the C-domain sites of cTnC (data not shown). The Ca2+ induced increases in Tyr fluorescence, occurring when Ca2+ binds to the C-domain site of cTnC, cTnCA8V, cTnCE134D and cTnCD145E in the absence or presence of 3 mM Mg2+ are shown in Figure 2. In the absence of Mg2+, cTnC exhibited a half-maximal Ca2+ dependent increase in its Tyr fluorescence at 224 ± 2 nM. In the presence of 3 mM Mg2+, cTnC exhibited a half-maximal Ca2+ dependent increase in its Tyr fluorescence at 534 ± 13 nM. Thus, 3 mM Mg2+ produced ~ 2.4-fold decrease in the Ca2+ sensitivity of the C-domain sites of cTnC. Assuming competitive Mg2+ binding, the Kd(Mg) of the C-domain sites of cTnC was calculated to be 2.17 mM. The Kd(Ca) of the C-domain sites of cTnCA8V was measured at 196 ± 1 nM and 550 ± 5 nM in the absence and presence of 3 mM Mg2+, respectively (Figure 2A). Assuming competitive Mg2+ binding, the Kd(Mg) of the C-domain sites of cTnCA8V was calculated to be 1.66 mM. For cTnCE134D, the half-maximal Ca2+ dependent increase in Tyr fluorescence occurred at 301 ± 2 nM in the absence of Mg2+ and at 605 ± 33 nM in the presence of 3 mM Mg2+ (Figure 2B). Assuming competitive Mg2+ binding, the Kd(Mg) of the C-domain sites of cTnCE134D was calculated to be 2.97 mM. These results indicate that E134D mutation produced ~1.3- and 1.4-fold decreases in the Ca2+ and Mg2+ affinities, respectively, of the C-domain sites. The cTnCD145E underwent a biphasic increase in its Tyr fluorescence in both absence and presence of 3 mM Mg2+ (Figure 2C). The half-maximal increase of the first phase occurred at 314 ± 31 nM (Kd(Ca)1) in the absence of Mg2+ and at 3801 ± 1093 nM in the presence of 3 mM Mg2+. The half-maximal increase of the second phase occurred at 513 ± 36 μM (Kd(Ca)2) in the absence of Mg and at 464 ± 53 μM in the presence of 3 mM Mg2+. The Kd(Ca)2 values were not significantly different from each other in the absence and presence of Mg2+. Since D145E mutation is located in the +Z position of site IV, while site III was unchanged, Kd(Ca)1 was tentatively assigned to site III and Kd(Ca)2 was tentatively assigned to site IV. These results suggest that D145E mutation produced a dramatic 2,290-fold decrease in the Ca2+ affinity of site IV, and abolished Mg2+ binding to that site. Furthermore, D145E mutation produced ~1.4-fold decrease in the Ca2+ affinity of site III. Assuming competitive Mg2+ binding, the Kd(Mg) of site III of cTnCE134D was calculated to be 0.27 mM.
Figure 2. Effect of HCM linked cTnC mutations on the Ca2+ and Mg2+ binding properties of the C-domain sites.
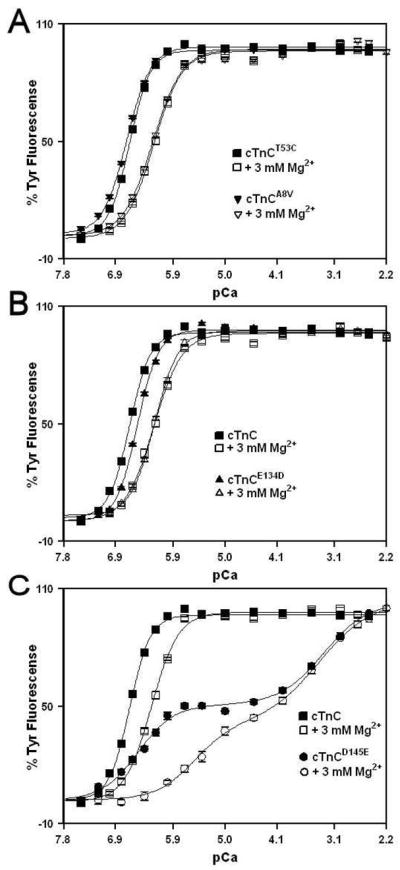
Panel A shows the Ca2+ dependent increases in Tyr fluorescence for cTnC (■) and cTnCA8V (▼) in the absence of Mg2+; and for cTnC (□) and cTnCA8V (▽) in the presence of 3 mM Mg2+. Panel B shows the Ca2+ dependent increases in Tyr fluorescence for cTnC (■) and cTnCE134D (▲) in the absence of Mg2+; and for cTnC (□) and cTnCE134D (△) in the presence of 3 mM Mg2+.. Panel C shows the Ca2+ dependent increases in Tyr fluorescence for cTnC (■) and cTnCD145E (●) in the absence of Mg2+; and for cTnC (□) and cTnCD145E (○) in the presence of 3 mM Mg2+. Microliter amounts of Ca2+ were added to 2 ml of each protein (0.5 μM) in 200 mM MOPS, 150 mM KCl, 2 mM EGTA, 1 mM DTT with or without 3 mM MgCl2, pH 7.0 at 15°C. Tyr fluorescence was excited at 275 nm and monitored at 303 nm at 15 °C. The data sets were normalized individually for each mutant. Each data point represents the mean ± SE of at least three titrations fit with a single logistic sigmoid function for cTnC, cTnCA8V and cTnCE134D, and with a double logistic sigmoid function for cTnCD145E.
Effect of HCM Linked TnC Mutations on the Rates of Ca2+ Dissociation from the C-domain Sites
Fluorescence stopped-flow measurements, utilizing intrinsic Tyr fluorescence, were conducted to determine the effect of HCM linked cTnC mutations on the kinetics of Ca2+ dissociation from the C-domain sites. Figure 3A shows that excess EGTA removed Ca2+ from the C-domain sites of cTnC, cTnCA8V, cTnCE134D and cTnCD145E at 1.02 ± 0.01, 0.91 ± 0.01, 1.64 ± 0.02 and 3.09 ± 0.05/s, respectively. To verify that Tyr signal changes were accurately reporting the true Ca2+ dissociation rates and not slower or faster structural change, Ca2+ dissociation rates were also measured using the fluorescent Ca2+ chelator Quin-2. Figure 3B shows the time course of the increases in Quin-2 fluorescence as Ca2+ dissociated from the C-domain sites of cTnC and HCM linked cTnC mutants. Similar Ca2+ dissociation rates were measured using Quin-2 fluorescence for cTnC, cTnCA8V, cTnCE134D and cTnCD145E at 1.25 ± 0.02, 1.16 ± 0.01, 1.79 ± 0.01, and 3.03 ± 0.08/s, respectively, as were measured by EGTA induced Tyr changes. Therefore, E134D and D145E mutations led to ~1.6- and 3.0-fold faster rate of Ca2+ dissociation from the C-domain sites, respectively. The stoichiometry of Ca2+ dissociation from the C-domain sites of cTnC, cTnCA8V and cTnCE134D was 1.91 ± 0.07, 1.83 ± 0.07, and 1.94 ± 0.08 mol of Ca2+/mol of protein, respectively. In contrast, the stoichiometry of Ca2+ dissociation from the C-domain sites of cTnCD145E was 0.87 ± 0.03 mol of Ca2+/mol of protein. Thus, at 15 μM Ca2+, the C-domain of cTnCD145E bound ~ one-half of mol Ca2+/mol of protein bound by the C-domains of cTnC, cTnCA8V and cTnCE134D.
Figure 3. Effect of HCM linked TnC mutations on the Rates of Ca2+ dissociation from the C-domain sites.
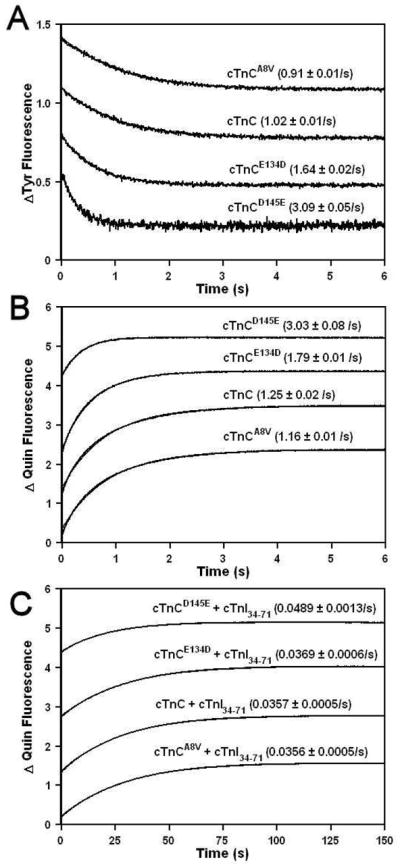
Panel A shows the time course of the decrease in Tyr fluorescence as Ca2+ was removed by EGTA from the C-domain sites of cTnC, cTnCA8V, cTnCE134D and cTnCD145E at 15 °C. Each protein (5 μM) with 500 μM Ca2+ in the stopped-flow buffer (10 mM MOPS, 150 mM KCl, 3 mM MgCl2 and 1 mM DTT, pH 7.0) was rapidly mixed with an equal volume of EGTA (10 mM) in the stopped-flow buffer. The traces have been normalized and displaced vertically for clarity. Panel B shows the time course of the increase in Quin-2 fluorescence as Ca2+ was removed by Quin-2 from the C-domain sites of cTnC, cTnCA8V, cTnCE134D and cTnCD145E at 15 °C. Each protein (6 μM) with 15 μM Ca2+ in the stopped-flow buffer was rapidly mixed with an equal volume of Quin-2 (150 μM) in the stopped-flow buffer. The traces are not normalized but have been displaced vertically for clarity. Panel C shows the time course of the increase in Quin-2 fluorescence as Ca2+ was removed by Quin-2 from the C-domain sites of cTnC, cTnCA8V, cTnCE134D and cTnCD145E at 15°C in the presence of cTnI34-71. Each protein (6 μM) in the presence of cTnI34-71 (18 μM) with 15 μM Ca2+ in the stopped-flow buffer was rapidly mixed with an equal volume of Quin-2 (150 μM) in the stopped-flow buffer. The traces are not normalized but have been displaced vertically for clarity.
The rates of Ca2+ dissociation from the C-domain sites of cTnC and HCM linked cTnC mutants were also measured in the presence of anchoring fragment of cTnI that binds to the C-domain of cTnC (residues 34-71). Figure 3C shows the time course of the increases in Quin-2 fluorescence as Ca2+ dissociated from the C-domain sites of cTnC, cTnCA8V, cTnCE134D and cTnCD145E in the presence of cTnI34-71 peptide. The rates of Ca2+ dissociation from the C-domain sites of cTnC, cTnCA8V, cTnCE134D and cTnCD145E in the presence of cTnI34-71 were measured at 0.0357 ± 0.0005, 0.0356 ± 0.0005, 0.0369 ± 0.0006, and 0.0489 ± 0.0013/s, respectively. The stoichiometry of Ca2+ dissociation from the C-domain sites of cTnC, cTnCA8V and cTnCE134D in the presence of cTnI34-71 was 1.37 ± 0.07, 1.25 ± 0.07 and 1.20 ± 0.07 mol of Ca2+/mol of protein. The stoichiometry for the C-domain sites of cTnC, cTnCA8V and cTnCE134D in the presence of cTnI34-71 was less than expected 2 mol of Ca2+/mol of protein, likely due to the fact that Quin-2 was unable to remove all the Ca2+ from the C-domain sites of cTnC proteins in the presence of cTnI34-71. Similarly, Quin-2 was unable to remove all the Ca2+ from the C-domain of cTnC in the cTn complex (21), likely due to the fact that Ca2+ affinity of the C-domain sites of cTnC in the cTn complex is ~ 24-fold higher that that of isolated cTnC (25). The stoichiometry of Ca2+ dissociation from the C-domain sites of cTnCD145E in the presence of cTnI34-71 was 0.69 ± 0.03 mol of Ca2+/mol of protein, or ~ one-half of that for cTnC. Thus, cTnI34-71 was not able to recover normal Ca2+ binding to the C-domain of cTnCD145E.
Effect of HCM Linked Mutations on the Interactions of cTnC with bis-ANS
Non-covalent binding of bis-ANS to the hydrophobic segments of proteins is accompanied by an increase in its fluorescence, and has been widely used to follow conformational changes. Addition of Ca2+ to a bis-ANS solution in the presence of cTnC (in the absence or presence of Mg2+) causes a biphasic increase in fluorescence (26). The first phase of the increase in bis-ANS fluorescence is related to the Ca2+ induced binding of bis-ANS to the C-domain sites of cTnC (26). The second phase is likely associated with non-specific Ca2+ binding to the additional weak binding sites for Ca2+/Mg2+ present in the C-domain (27). Figure 4 shows that Ca2+ binding to unlabeled cTnC, cTnCA8V and cTnCE134D induced a biphasic increase in the fluorescence of bis-ANS in the absence (Figure 4A) or the presence of 3 mM Mg2+ (Figure 4B). In the absence of Mg2+, the half-maximal increase of the first phase occurred at 277 ± 3, 308 ± 10, and 408 ± 3 nM for cTnC, cTnCA8V and cTnCE134D, respectively. In the presence of 3 mM Mg2+, the half-maximal increase of the first phase occurred at 666 ± 12, 760 ± 14, and 1087 ± 50 nM for cTnC, cTnCA8V and cTnCE134D, respectively. The Kd(Ca) values determined from the first phase of the bis-ANS titration are in reasonable agreement with those measured using intrinsic Tyr fluorescence. These results indicate that the C-domains of cTnC, cTnCA8V and cTnCE134D gradually expose hydrophobic surface to the solvent upon binding Ca2+, leading to the first phase of the increase in bis-ANS fluorescence. It is worth noting that both A8V and E134D mutations reduced the magnitude of the first phase of bis-ANS fluorescence, suggesting a reduction in hydrophobic exposure. The Ca2+ binding to the C-domain of cTnCD145E did not lead to an increase in bis-ANS fluorescence at the pCa range where site III was binding Ca2+ in either absence or presence of 3 mM Mg2+. These results suggest that the C-domain of cTnCD145E either does not undergo a conformational “opening” upon the Ca2+ binding to site III, or the exposed hydrophobic pocket is substantially altered to prevent bis-ANS binding.
Figure 4. Effect of HCM linked mutations on the interactions of cTnC with bis-ANS.

Panel A shows the Ca2+ dependent increases in bis-ANS fluorescence in the presence of cTnC (■), cTnCA8V (▼), cTnCE134D (▲) and cTnCD145E (●) in the absence of Mg2+. Microliter amounts of Ca2+ were added to 2 ml of each protein (2 μM) with bis-ANS (2 μM) in 200 mM MOPS, 150 mM KCl, 2 mM EGTA, 1 mM DTT, pH 7.0 at 15°C. Panel B shows the Ca2+ dependent increases in bis-ANS fluorescence for cTnC (□), cTnCA8V (▽), cTnCE134D (△) and cTnCD145E (○) in the presence of 3 mM Mg2+. The experimental conditions were identical to those in Panel A, except the buffer contained 3 mM MgCl2. Bis-ANS fluorescence was excited at 400 nm and monitored at 495 nm at 15°C. F0 is the fluorescence value (F) of bis-ANS before addition of Ca2+. Each data point represents the mean ± SE of at least three titrations fit with a double logistic sigmoid function for cTnC, cTnCA8V and cTnCE134D, and with a single logistic sigmoid for cTnCD145E.
Effect of HCM Linked cTnC Mutations on the cTnI128-180 Binding Properties of Ca2+ Saturated cTnC
The HCM linked TnC mutations could have affected the strength of regulatory cTnC-cTnI interactions, considering that the N-domain of cTnC interacts with the switch region of cTnI (corresponding to residues 150-159 (28)) in a Ca2+ dependent manner, while the C-domain of cTnC interacts with the inhibitory region of cTnI (corresponding to residues 128-147) (29). A change in fluorescence of Ca2+ saturated cTnC labeled with environmentally sensitive probe IANBD on Cys53 was utilized to determine whether the HCM linked mutations affected the affinity of cTnC for cTnI peptide corresponding to residues 128-180 of human cTnI (cTnI128-180). The cTnI128-180 peptide is homologous to the skeletal TnI96-148 peptide, shown to be a good model system to study the Ca2+-dependent interactions between skeletal TnI (sTnI) and skeletal TnC (sTnC) (22, 30), and includes both the inhibitory and the switch region. Figure 5A demonstrates that at 1 mM Ca2+, the affinity of cTnC for cTnI128-180 was at ~ 73 ± 3 nM. At 1 mM Ca2+, the affinities of cTnCA8V, cTnCE134D and cTnCD145E for cTnI128-180 were ~ 56 ± 2 nM, 62± 2 nM and 75 ± 2 nM, respectively. Thus, at 1 mM Ca2+, HCM linked mutations A8V, E134D and D145E had minimal effect on the affinity of cTnC for cTnI128-180.
Figure 5. Effect of HCM linked mutations on cTnI128-180 binding affinity of cTnC.

Panel A shows the effect of the HCM linked mutations on the cTnI128-180 binding properties of cTnC in the presence of 1 mM Ca2+. The cTnI128-180 dependent changes in IANBD fluorescence are shown as a function of [cTnI128-180] for the cTnC (□), cTnCA8V (▽), cTnCE134D (△) and cTnCD145E (○). Panel B shows the effect of the HCM linked mutations on the cTnI128-180 binding properties of the Ca2+ saturated cTnC in the presence of 2 μM Ca2+. The cTnI128-180 dependent changes in IANBD fluorescence are shown as a function of [cTnI128-180] for the cTnC (■), cTnCA8V (▼), cTnCE134D (▲) and cTnCD145E (●). 100% IANBD fluorescence corresponds to the Ca2+ -bound state, whereas 0% corresponds to the Ca2+-cTnI128-180 bound state for each individual cTnC protein. In the case of cTnCD145E in the presence of 2 μM Ca2+, the IANBD fluorescence increased upon addition of cTnI128-180, so the plot of the data was inverted for the sake of comparison.
During cardiac muscle contraction, intracellular Ca2+ concentration elevates to a level of 1–10 μM (for review, see (31)). Thus, the effect of HCM linked mutations on the affinity of cTnC for cTnI128-180 peptide was also determined at 2 μM Ca2+. Figure 5B demonstrates that at 2μM Ca2+, the affinity of cTnC for cTnI128-180 was at 447 ± 10 nM. At 2 μM Ca2+, the affinities of cTnCA8V, cTnCE134D and cTnCD145E for cTnI128-180 were at 286 ± 5 nM, 336 ± 6 nM and 3561 ±157 nM, respectively. Thus, at 2 μM Ca2+, HCM linked mutations A8V and E134D led to modest ~1.6 and ~1.3-fold, respectively, increases in the affinity of cTnC for cTnI128-180, while D145E mutation led to a dramatic ~8.0-fold decrease in the affinity of cTnC for cTnI128-180.
DISCUSSION
While the N-domain site of cTnC responds to Ca2+ to regulate muscle contraction, the C-domain sites are thought to be permanently occupied by Ca2+ and/or Mg2+, thus playing a structural role of anchoring cTnC into the thin filament. However, a number of recently discovered cTnC mutations linked to DCM and HCM are located in the C-domain (for review, see (4)), indicating that properties of this domain might play an important role in the modulation of contraction. The main objective of this study was to examine the effect of HCM linked mutations A8V, E134D and D145E of cTnC on the Ca2+/Mg2+ binding properties of the C-domain sites. We also wanted to examine whether HCM linked mutations affected affinity of cTnC for the regulatory region of cTnI. First, we investigated whether A8V, E134D and D145E mutations affected the Ca2+ binding affinity and rate of Ca2+ dissociation from the C-domain of isolated cTnC by following the intrinsic Tyr fluorescence. The A8V mutation had very little effect on the Ca2+/Mg2+ binding affinities and the rate of Ca2+ dissociation from the C-terminal of cTnC. Likely, the main mechanism by which the A8V mutation leads to HCM is by altering the Ca2+ binding properties of the N-domain (6). In contrast, the E134D and D145E mutations significantly affected the Ca2+ and Mg2+ binding properties of the C-terminal sites. The E134D mutation moderately decreased both Ca2+ and Mg2+ binding affinities. The decrease in Ca2+ affinity was caused by the faster rate of Ca2+ dissociation from the C-domain sites of cTnCE134D. Since the effects of E134D mutation on the properties of the C-domain were rather modest, further studies are needed to examine if this mutation causes altered regulation under certain physiological conditions, or whether it is simply a rare polymorphism.
Our results suggest that the D145E mutation, located in the +Z position of the fourth Ca2+ EF-hand, dramatically decreased Ca2+ affinity of site IV, and abolished Mg2+ binding to that site. Substitution of Asp residue with a larger Glu residue in the + Z position of site IV might lead to a smaller Ca2+ binding cavity resulting in lower affinity. The effect of D145E mutation is consistent with the results obtained for the synthetic helix-loop-helix peptides, where substitution of Asp residue with Glu in the +Z position caused the peptide to lose the Ca2+ and Mg2+ binding capacities (32).
Binding of the anchoring region of cTnI, cTnI34-71 was not able to restore normal Ca2+ binding to the C-domain of cTnCD145E, as evidenced by the experiments utilizing Quin-2. In addition to drastically affecting Ca2+ binding to site IV, the D145E mutation significantly decreased Ca2+ affinity of site III, due to a faster rate of Ca2+ dissociation from that site. The lower Ca2+ affinity of site III is likely due to the loss of cooperativity between the C-domain sites. Our results are consistent with those observed with closely related EF-hand Ca2+ binding protein calmodulin, where D133E mutation in the +Z position of the fourth Ca2+ binding loop drastically reduced Ca2+ affinity of site IV and significantly reduced that of site III (33). A previous study demonstrated that disruption of Ca2+ binding to site III or IV of sTnC (by the substitution of Asp in the +X position of the Ca2+ binding loop with Ala) results in the increased Ca2+ sensitivity of force development (34). Perhaps cTnCD145E increases the Ca2+ sensitivity of force development via a similar mechanism. However, at this time we do not have clear-cut data indicating that D145E mutation specifically inactivated site IV. Structural studies are needed to unequivocally determine whether D145E mutation specifically inactivates site IV.
The dramatic effect of D145E mutation on the properties of the C-domain sites was further revealed by the characterization of the interactions between the cTnC and fluorescent hydrophobic probe bis-ANS. Binding of Ca2+ to sites III and IV of cTnC, cTnCA8V and cTnCE134D (in the absence or presence of Mg2+) leads to bis-ANS binding to the exposed hydrophobic pocket. It is worth noting that both A8V and E134D mutations reduced the magnitude of the increase in bis-ANS fluorescence associated with Ca2+ binding to sites III and IV. These results suggest that A8V and E134D mutations led to the reduction of hydrophobic surface exposed by the binding of Ca2+ to the C-domain of cTnC. The most dramatic result was observed for cTnCD145E, where the D145E mutation prevented bis-ANS binding to the C-domain sites of cTnC at the pCa range where site III was binding Ca2+. A possible interpretation is that binding of Ca2+ to the C-domain of cTnCD145E either does not lead to the “opening” of the C-terminal hydrophobic pocket, or the exposed hydrophobic pocket differs substantially from that of cTnC. Presence of both Ca2+ and cTnI (residues 147-163) are needed to induce the “opening” of the N-domain of cTnC (35). On the other hand, binding of Ca2+ to the single functional site IV of F1 TnC (TnC isoform responsible for stretch activation in insect muscles) was sufficient to induce modest but clear “opening” of the C-domain (36). Structural studies are needed to unequivocally determine whether the C-domain of cTnCD145E “opens” upon binding of Ca2+ and/or the anchoring region of cTnI.
The interaction of cTnC with the regulatory region of cTnI plays a crucial role in the regulation of muscle contraction (for review, see (37, 38)). Thus, perturbations in affinity of cTnC for cTnI can potentially lead to adverse physiological consequences, such as development of cardiomyopathies. For instance, HCM linked cTnI mutation, R144G, led to ~ 6-fold reduction in the affinity of the Ca2+-satuarated C-domain of cTnC for the inhibitory region of cTnI (residues 128-147)(39). Our results show that A8V, E134D or D145E mutations did not led to substantial alterations in affinity of cTnC for the regulatory region of cTnI (which includes inhibitory and switch regions), cTnI128-180 at 1 mM Ca2+. At this high non-physiological Ca2+ concentration, the C-domain of cTnCD145E should be almost completely saturated with Ca2+, apparently enabling cTnCD145E to bind cTnI128-180 with an affinity similar to that of cTnC. Consistent with these results, co-sedimentation analysis did not detect any changes in the ability of the cTn complex reconstituted with cTnCA8V, cTnCE134D or cTnCD145E to bind to the thin filament at 0.5 mM Ca2+ (6). In contrast, at lower physiological 2 μM Ca2+, D145E mutation led to ~8-fold decrease in the affinity of cTnC for cTnI128-180. At this low Ca2+concentration, the C-domain of cTnCD145E should be only partially saturated with Ca2+, while the C-domain of cTnC should be almost completely saturated with Ca2+. Apparently, reduction in the level of Ca2+ saturation of the C-domain at low Ca2+ resulted in the decreased affinity of cTnCD145E for cTnI128-180, compared to that of cTnC. These results suggest that Ca2+ binding properties of the C-domain sites of cTnC play an important role in the interactions of cTnC with the regulatory region of cTnI.
In conclusion, our data demonstrate that the A8V mutation had minimal effect on the Ca2+/Mg2+ affinities of the C-domain sites. On the other hand, both E134D and D145E mutations altered the Ca2+ and Mg2+ binding affinities of the C-domain sites. While E134D substitution moderately decreased the Ca2+ and Mg2+ affinities, the D145E substitution drastically altered Ca2+ binding by the C-domain of cTnC. The cTnI34-71 peptide was not able to recover normal Ca2+ binding to the C-domain of cTnCD145E. Experiments utilizing the hydrophobic fluorescent probe bis-ANS suggest that D145E mutation led to a dramatic reduction in the Ca2+-induced hydrophobic surface exposure by the C-domain. At high non-physiological Ca2+ concentration, A8V, E134D and D145E mutations had minimal effect on the affinity of cTnC for the regulatory region of cTnI. In contrast, at low physiological Ca2+ concentration, D145E mutation led to ~8-fold decrease in the affinity of cTnC for the regulatory region of cTnI. While more studies are needed to fully understand the role of the C-domain of cTnC in the modulation of the Ca2+ signal, our results suggest that Ca2+ binding properties of the C-domain sites are important for the proper regulatory function of cTnC.
Acknowledgments
We thank Dr. Lawrence Smillie for the generous gift of the human cTnC plasmid. We also thank Kristin Tang and Miranda Willacey for technical assistance.
Footnotes
Abbreviations: Hypertrophic cardiomyopathy, HCM; dilated cardiomyopathy, DCM; cTn, cardiac troponin; cTnC, cardiac troponin C; cTnI, cardiac troponin I; cTnT, cardiac troponin T; sTnC, skeletal troponin C; sTnI, skeletal troponin I; IANBD, N-((2-(iodoacetoxy)ethyl)-N-methyl)amino-7-nitrobenz-2-oxa-1,3-diazole; cTnI34-71, peptide corresponding to residues from 34 to 71 of human cTnI, cTnI128-180, peptide corresponding to residues from 128 to 180 of human cTnI; EGTA, ethylene glycol-bis(2-aminoethyl)-N,N,N′,N′-tetraacetic acid; Quin-2, 2-{[2-bis(carboxymethyl)amino-5-methylphenoxy]methyl}-6-methoxy-8-bis(carboxymethyl)aminoquinoline; bis-ANS, 4,4′-Dianilino-1,1′-binaphthyl-5,5′-disulfonic acid dipotassium salt; DTT, dithiothreitol; MOPS, 3-(N-morpholino)propanesulfonic acid; Tween-20, polysorbate 20; Kd, dissociation constant.
This research was funded by NIH grant 5R00HL087462 (to S.B.T)
References
- 1.Chung MW, Tsoutsman T, Semsarian C. Hypertrophic cardiomyopathy: from gene defect to clinical disease. Cell Res. 2003;13:9–20. doi: 10.1038/sj.cr.7290146. [DOI] [PubMed] [Google Scholar]
- 2.Bashyam MD, Savithri GR, Kumar MS, Narasimhan C, Nallari P. Molecular genetics of familial hypertrophic cardiomyopathy (FHC) J Hum Genet. 2003;48:55–64. doi: 10.1007/s100380300007. [DOI] [PubMed] [Google Scholar]
- 3.Arad M, Seidman JG, Seidman CE. Phenotypic diversity in hypertrophic cardiomyopathy. Hum Mol Genet. 2002;11:2499–2506. doi: 10.1093/hmg/11.20.2499. [DOI] [PubMed] [Google Scholar]
- 4.Willott RH, Gomes AV, Chang AN, Parvatiyar MS, Pinto JR, Potter JD. Mutations in Troponin that cause HCM, DCM AND RCM: What can we learn about thin filament function? J Mol Cell Cardiol. 2009 doi: 10.1016/j.yjmcc.2009.10.031. [DOI] [PubMed] [Google Scholar]
- 5.Rodriguez JE, McCudden CR, Willis MS. Familial hypertrophic cardiomyopathy: basic concepts and future molecular diagnostics. Clin Biochem. 2009;42:755–765. doi: 10.1016/j.clinbiochem.2009.01.020. [DOI] [PubMed] [Google Scholar]
- 6.Pinto JR, Parvatiyar MS, Jones MA, Liang J, Ackerman MJ, Potter JD. A functional and structural study of troponin C mutations related to hypertrophic cardiomyopathy. J Biol Chem. 2009;284:19090–19100. doi: 10.1074/jbc.M109.007021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Landstrom AP, Parvatiyar MS, Pinto JR, Marquardt ML, Bos JM, Tester DJ, Ommen SR, Potter JD, Ackerman MJ. Molecular and functional characterization of novel hypertrophic cardiomyopathy susceptibility mutations in TNNC1-encoded troponin C. J Mol Cell Cardiol. 2008;45:281–288. doi: 10.1016/j.yjmcc.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Farah CS, Reinach FC. The troponin complex and regulation of muscle contraction. Faseb J. 1995;9:755–767. doi: 10.1096/fasebj.9.9.7601340. [DOI] [PubMed] [Google Scholar]
- 9.Filatov VL, Katrukha AG, Bulargina TV, Gusev NB. Troponin: structure, properties, and mechanism of functioning. Biochemistry (Mosc) 1999;64:969–985. [PubMed] [Google Scholar]
- 10.Gifford JL, Walsh MP, Vogel HJ. Structures and metal-ion-binding properties of the Ca2+-binding helix-loop-helix EF-hand motifs. Biochem J. 2007;405:199–221. doi: 10.1042/BJ20070255. [DOI] [PubMed] [Google Scholar]
- 11.Nelson MR, Chazin WJ. Structures of EF-hand Ca(2+)-binding proteins: diversity in the organization, packing and response to Ca2+ binding. Biometals. 1998;11:297–318. doi: 10.1023/a:1009253808876. [DOI] [PubMed] [Google Scholar]
- 12.Yap KL, Ames JB, Swindells MB, Ikura M. Diversity of conformational states and changes within the EF-hand protein superfamily. Proteins. 1999;37:499–507. doi: 10.1002/(sici)1097-0134(19991115)37:3<499::aid-prot17>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 13.van Eerd JP, Takahashi K. The amino acid sequence of bovine cardiac tamponin-C. Comparison with rabbit skeletal troponin-C. Biochem Biophys Res Commun. 1975;64:122–127. doi: 10.1016/0006-291x(75)90227-2. [DOI] [PubMed] [Google Scholar]
- 14.Sia SK, Li MX, Spyracopoulos L, Gagne SM, Liu W, Putkey JA, Sykes BD. Structure of cardiac muscle troponin C unexpectedly reveals a closed regulatory domain. J Biol Chem. 1997;272:18216–18221. doi: 10.1074/jbc.272.29.18216. [DOI] [PubMed] [Google Scholar]
- 15.Davis JP, Tikunova SB. Ca(2+) exchange with troponin C and cardiac muscle dynamics. Cardiovasc Res. 2008;77:619–626. doi: 10.1093/cvr/cvm098. [DOI] [PubMed] [Google Scholar]
- 16.Davis JP, Norman C, Kobayashi T, Solaro RJ, Swartz DR, Tikunova SB. Effects of thin and thick filament proteins on calcium binding and exchange with cardiac troponin C. Biophys J. 2007;92:3195–3206. doi: 10.1529/biophysj.106.095406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tikunova SB, Liu B, Swindle N, Little SC, Gomes AV, Swartz DR, Davis JP. Effect of calcium-sensitizing mutations on calcium binding and exchange with troponin C in increasingly complex biochemical systems. Biochemistry. 2010;49:1975–1984. doi: 10.1021/bi901867s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Robertson S, Potter JD. The regulation of free Ca2+ ion concentration by metal chelators. Methods in Pharmacology. 1984;5:63–75. [Google Scholar]
- 19.Tikunova SB, Rall JA, Davis JP. Effect of hydrophobic residue substitutions with glutamine on Ca(2+) binding and exchange with the N-domain of troponin C. Biochemistry. 2002;41:6697–6705. doi: 10.1021/bi011763h. [DOI] [PubMed] [Google Scholar]
- 20.Tikunova SB, Davis JP. Designing calcium-sensitizing mutations in the regulatory domain of cardiac troponin C. J Biol Chem. 2004;279:35341–35352. doi: 10.1074/jbc.M405413200. [DOI] [PubMed] [Google Scholar]
- 21.Gomes AV, Venkatraman G, Davis JP, Tikunova SB, Engel P, Solaro RJ, Potter JD. Cardiac troponin T isoforms affect the Ca(2+) sensitivity of force development in the presence of slow skeletal troponin I: insights into the role of troponin T isoforms in the fetal heart. J Biol Chem. 2004;279:49579–49587. doi: 10.1074/jbc.M407340200. [DOI] [PubMed] [Google Scholar]
- 22.Davis JP, Rall JA, Alionte C, Tikunova SB. Mutations of hydrophobic residues in the N-terminal domain of troponin C affect calcium binding and exchange with the troponin C-troponin I96-148 complex and muscle force production. J Biol Chem. 2004;279:17348–17360. doi: 10.1074/jbc.M314095200. [DOI] [PubMed] [Google Scholar]
- 23.Dotson DG, Putkey JA. Differential recovery of Ca2+ binding activity in mutated EF-hands of cardiac troponin C. J Biol Chem. 1993;268:24067–24073. [PubMed] [Google Scholar]
- 24.Negele JC, Dotson DG, Liu W, Sweeney HL, Putkey JA. Mutation of the high affinity calcium binding sites in cardiac troponin C. J Biol Chem. 1992;267:825–831. [PubMed] [Google Scholar]
- 25.Holroyde MJ, Robertson SP, Johnson JD, Solaro RJ, Potter JD. The calcium and magnesium binding sites on cardiac troponin and their role in the regulation of myofibrillar adenosine triphosphatase. J Biol Chem. 1980;255:11688–11693. [PubMed] [Google Scholar]
- 26.Pan BS, Johnson RG., Jr Interaction of cardiotonic thiadiazinone derivatives with cardiac troponin C. J Biol Chem. 1996;271:817–823. [PubMed] [Google Scholar]
- 27.Braga CA, Pinto JR, Valente AP, Silva JL, Sorenson MM, Foguel D, Suarez MC. Ca(2+) and Mg(2+) binding to weak sites of TnC C-domain induces exposure of a large hydrophobic surface that leads to loss of TnC from the thin filament. Int J Biochem Cell Biol. 2006;38:110–122. doi: 10.1016/j.biocel.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 28.Takeda S, Yamashita A, Maeda K, Maeda Y. Structure of the core domain of human cardiac troponin in the Ca(2+)-saturated form. Nature. 2003;424:35–41. doi: 10.1038/nature01780. [DOI] [PubMed] [Google Scholar]
- 29.Li MX, Spyracopoulos L, Beier N, Putkey JA, Sykes BD. Interaction of cardiac troponin C with Ca(2+) sensitizer EMD 57033 and cardiac troponin I inhibitory peptide. Biochemistry. 2000;39:8782–8790. doi: 10.1021/bi000473i. [DOI] [PubMed] [Google Scholar]
- 30.Davis JP, Rall JA, Reiser PJ, Smillie LB, Tikunova SB. Engineering competitive magnesium binding into the first EF-hand of skeletal troponin C. J Biol Chem. 2002;277:49716–49726. doi: 10.1074/jbc.M208488200. [DOI] [PubMed] [Google Scholar]
- 31.Endoh M. Signal transduction and Ca2+ signaling in intact myocardium. J Pharmacol Sci. 2006;100:525–537. doi: 10.1254/jphs.cpj06009x. [DOI] [PubMed] [Google Scholar]
- 32.Reid RE, Procyshyn RM. Engineering magnesium selectivity in the helix-loop-helix calcium-binding motif. Arch Biochem Biophys. 1995;323:115–119. doi: 10.1006/abbi.1995.0016. [DOI] [PubMed] [Google Scholar]
- 33.Wu X, Reid RE. Conservative D133E mutation of calmodulin site IV drastically alters calcium binding and phosphodiesterase regulation. Biochemistry. 1997;36:3608–3616. doi: 10.1021/bi962149m. [DOI] [PubMed] [Google Scholar]
- 34.Szczesna D, Guzman G, Miller T, Zhao J, Farokhi K, Ellemberger H, Potter JD. The role of the four Ca2+ binding sites of troponin C in the regulation of skeletal muscle contraction. J Biol Chem. 1996;271:8381–8386. doi: 10.1074/jbc.271.14.8381. [DOI] [PubMed] [Google Scholar]
- 35.Li MX, Spyracopoulos L, Sykes BD. Binding of cardiac troponin-I147-163 induces a structural opening in human cardiac troponin-C. Biochemistry. 1999;38:8289–8298. doi: 10.1021/bi9901679. [DOI] [PubMed] [Google Scholar]
- 36.De Nicola GF, Martin S, Bullard B, Pastore A. Solution structure of the Apo C-terminal domain of the Lethocerus F1 troponin C isoform. Biochemistry. 2010;49:1719–1726. doi: 10.1021/bi902094w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li MX, Wang X, Sykes BD. Structural based insights into the role of troponin in cardiac muscle pathophysiology. J Muscle Res Cell Motil. 2004;25:559–579. doi: 10.1007/s10974-004-5879-2. [DOI] [PubMed] [Google Scholar]
- 38.Kobayashi T, Solaro RJ. Calcium, thin filaments, and the integrative biology of cardiac contractility. Annu Rev Physiol. 2005;67:39–67. doi: 10.1146/annurev.physiol.67.040403.114025. [DOI] [PubMed] [Google Scholar]
- 39.Li MX, Wang X, Lindhout DA, Buscemi N, Van Eyk JE, Sykes BD. Phosphorylation and mutation of human cardiac troponin I deferentially destabilize the interaction of the functional regions of troponin I with troponin C. Biochemistry. 2003;42:14460–14468. doi: 10.1021/bi035408y. [DOI] [PubMed] [Google Scholar]