

Abstract
Five hereditary sodium channelopathies of skeletal muscle have been identified. Prominent symptoms are either myotonia or weakness caused by an increase or decrease of muscle fiber excitability. The voltage-gated sodium channel NaV1.4, initiator of the muscle action potential, is mutated in all five disorders. Pathogenetically, both loss and gain of function mutations have been described, the latter being the more frequent mechanism and involving not just the ion-conducting pore, but aberrant pores as well. The type of channel malfunction is decisive for therapy which consists either of exerting a direct effect on the sodium channel, i.e., by blocking the pore, or of restoring skeletal muscle membrane potential to reduce the fraction of inactivated channels.
Keywords: Myotonia, Paramyotonia congenita, Hyperkalemic periodic paralysis, Hypokalemic periodic paralysis, Congenital myasthenic syndrome, Excitability, Muscle, Channels, Sodium channel, Muscle strength
Introduction
Membrane excitability, which is critical for function of skeletal muscle, is regulated by ion channels. It is therefore not surprising that voltage-gated ion channels are involved in the pathogenesis of diseases of these tissues. Early research on muscle tissue of patients with hereditary episodic weakness demonstrated the underlying defect to be a persistent inward sodium current which depolarized the membrane causing inexcitability and weakness [28]. Cloning and analysis of the gene encoding the voltage-gated sodium channel of skeletal muscle confirmed the electrophysiological results and revealed the first mutations associated with impaired ion channel function that were responsible for the skeletal muscle sodium channel disorder hyperkalemic periodic paralysis [11, 40]. In the 1990s, the term ion channelopathies was coined and defined for disorders that are caused by malfunction or altered regulation of ion channel proteins. Therefore, they may be either hereditary (for example by mutations in ion channel genes) or acquired (for example by autoantibodies). Since then, over 50 channelopathies in human beings have been described, 12 of which affect skeletal muscle. Of these, five are caused by mutations in its voltage-gated sodium channel, NaV1.4: potassium-aggravated myotonia (PAM), paramyotonia congenita (PMC), hyperkalemic periodic paralysis (HyperPP), hypokalemic periodic paralysis (HypoPP), and a form of congenital myasthenic syndrome (CMS).
This review focuses on the recurrent mutation patterns, functional consequences, and possible interpretations of the findings of these diseases. Clinical symptoms are briefly described and therapeutic options are discussed since many drugs exist that modulate cell excitability and particularly sodium channel function. A brief overview of muscle physiology is provided to outline the significance of the channels for muscle function.
The resting membrane potential
In skeletal muscle, the resting membrane potential, here denoted potential 1 (P1), is strongly dependent on inwardly rectifying potassium channels (Kir2.1). These channels increase their conductance with hyperpolarization. There are two factors which can decrease Kir2.1 conductance: membrane depolarization and drop of serum potassium, [K+]o. Problems for muscle function may arise especially when both factors change. Even though the Nernst equation predicts hyperpolarization when [K+]o is reduced, the hypokalemia-induced Kir2.1 conductance reduction may lead to depolarization if [K+]o is less than 1.5 mM [54]. This phenomenon is called paradoxical depolarization. The two conditions may form a vicious cycle in which Kir2.1 conductance further decreases with increasing depolarization. Then, the resting potential becomes instable and drops to the next stable state, denoted potential 2 (P2), at which other outward potassium currents activate and counterbalance the depolarization. Therefore, muscle fibers show a bimodal distribution of membrane potentials especially at low [K+]o [44]. While P1 follows the predictions of the Goldman-Hodgkin-Katz equation (−83 mV at 4 mM K+ and −99 mV at 1 mM K+), P2 is about −60 mV and largely independent of extracellular potassium. The fraction of depolarized fibers increases when extracellular potassium is only slightly or moderately lowered [23].
The action potential
The condition for initiating an action potential is that the net membrane current be inward, in the direction that results in sufficient depolarization for activation of sodium channels. The potential at which this condition is reached is termed the “threshold potential” and is, under normal conditions, always exceeded by the endplate potential. The upstroke of the action potential is mediated by opening of voltage-gated sodium channels that conduct a fast sodium inward current along both the electrical and concentration gradient. Due to the resulting high conductance of the membrane for sodium ions, the membrane suddenly depolarizes from the resting value of −84 mV to approximately +25 mV. Immediate repolarization of the membrane to the highly negative resting value is made possible by an intrinsic fast inactivation of sodium channels. Repolarization is driven by the opening of delayed rectifier potassium channels. Specifically in skeletal muscle, repolarization is additionally enforced by a high-chloride conductance that also buffers the resting membrane potential. This allows the muscle fibers to repolarize within about 5 ms despite the large membrane capacitance resulting from the T-tubular system. After an action potential, the membrane is inexcitable for a short period of time, the so-called refractory period, determined by the kinetics of recovery from inactivation of sodium channels.
The sodium channel complex
The α-subunit, NaV1.4 consists of 1,836 amino acid residues [61] and is encoded by the SCN4A gene on chromosome 17q23.1–25.3. It can be functionally expressed whereby co-expression of the β1 subunit will modify kinetics and voltage dependence of channel gating [18]. The tetrameric structure of the α-subunit consists of four domains (DI–DIV) of six transmembrane helical segments (S1–S6; Fig. 1). Four voltage sensors, each made of helices S1–S4, surround the pore domain and control its gates. The exact mechanisms of voltage sensing and the following conformational changes leading to channel opening are still unclear and under intensive investigations [3, 50].
Fig. 1.
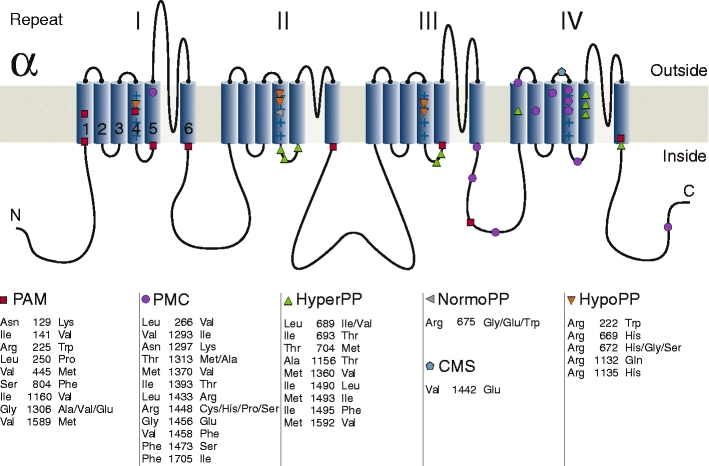
α-Subunit of the voltage-gated sodium channel of skeletal muscle, NaV1.4. The alpha-subunit is composed of four highly homologous domains (DI–DIV) each consisting of six transmembrane segments (S1–S6). When inserted in membrane, the four domains of the protein fold to generate a central pore whereby the S5–S6 loops form the ion-selective pore. The S4 segments contain positively charged residues conferring voltage dependence to the protein. Domains are connected by intracellular loops; one of them, the DIII−DIV linker, contains the inactivation particle of the channel. The sketch gives an overview of locations of known NaV1.4 mutations
Sodium channel activation results from depolarization-induced reorientation of the highly charged S4 segments, which leads to a conformational change of the protein resulting in the opening of the ion-conducting pore. While an immediate hyperpolarization closes the channel by deactivation, an ongoing depolarization will close the channel by inactivation. Inactivation of sodium channels may occur by one of several kinetically distinct processes referred to as fast, intermediate, and slow inactivation, with time constants in the order of milliseconds, tens to hundreds of milliseconds, and seconds to minutes, respectively.
Fast inactivation, which is an important factor in shaping action potentials, occurs during the first milliseconds after membrane depolarization. Fast inactivation is believed to function in a so-called hinged-lid mechanism: a hydrophobic particle is occluding the channel’s conducting pore from the intracellular side of the membrane. Fast inactivation depends on a conserved hydrophobic cluster of three amino acids IFM (isoleucine, phenylalanine, methionine) in the DIII–DIV cytoplasmic linker [55, 64]. Residues in the S4–S5 loops of DIII and DIV are thought to influence hydrophobic interactions of the IFM motif with its receptor leading to channel inactivation [38]. Recent studies showed that the C terminus plays an important role, in stabilizing the inactivated state [14, 65]. Inactivated channels do not immediately pass back into to the resting state after hyperpolarization, but require a certain amount of recovery time to do so.
Slow inactivation occurs after depolarization for seconds or minutes. Slow inactivation plays an important role by contributing to the regulation of resting sodium channel availability [43] and by aiding in slow activity-dependent changes in excitability such as spike frequency adaption or burst termination [58]. The molecular mechanism of the slow-inactivation process is still poorly understood. However, slow inactivation is distinct from fast inactivation because mutations that eliminate fast inactivation do not abolish slow inactivation [9, 56]. As large rearrangements are involved in slow inactivation several channel regions determine slow inactivation: pore regions, the voltage sensors, and segments S5 and S6.
Channelopathies
Five sodium channelopathies of skeletal muscle have been identified to date. All of them follow an autosomal dominant mode of transmission. Four of the disorders which are caused by similar NaV1.4 gain-of-function effects have distinct clinical features and therapies which may even be contrary despite common pathogenesis. Although the α subunit’s function is modulated by the β1 subunit, all mutations that cause a muscle disease are situated in NaV1.4. The only known β1 mutation causes generalized epilepsy with febrile seizures plus for which skeletal muscle dysfunction has not been described [60].
The cardinal symptoms of the diseases are myotonia and muscle weakness. Myotonic muscle stiffness is the result of uncontrolled repetitive muscle fiber discharges, which is based on increased membrane excitability most likely originating from the T-tubular system. Muscle weakness or even paralysis is usually caused by fiber inexcitability or, as in congenital myasthenic syndrome, by a reduced safety factor of synaptic transmission at the motor endplate.
Potassium-aggravated myotonia
PAM includes myotonia fluctuans, moderate myotonia, myotonia permanens, acetazolamide-responsive myotonia, and painful myotonia, i.e., a spectrum of diseases with overlapping clinical features which have in common that, in contrast to the allelic disorders paramyotonia congenita, hyperkalemic periodic paralysis and hypokalemic periodic paralysis, no weakness occurs [25, 37, 48]. The prevalence of PAM is estimated at ∼1:400,000 [29].
At first glance, myotonia fluctuans and moderate PAM are clinically very similar to the better known Thomsen myotonia, which is caused by mutations in the main chloride channel of skeletal muscle, ClC-1 [24]. However, in addition to stiffness of Thomsen patients which is most expressed during the first contractions after rest and improves with further contractions (warm-up phenomenon), PAM patients become stiff 10 to 30 min after strenuous work. This delayed and sometimes painful stiffness may hinder the patient’s movements for several hours. It should not be confused with paradoxical myotonia, i.e., myotonia worsening with repeated contractions. Usually, most limb muscles show the warm-up phenomenon. Furthermore, potassium and other depolarizing agents (and sometimes cold) aggravate the myotonia, a reaction that is not observed in chloride channel myotonia. The reaction to potassium was responsible for origination of the term potassium-aggravated myotonia [17, 34].
Most PAM mutations are situated in the N terminus of loops that connect domains (Fig. 1)—particularly in the inactivation gate. Three of the mutations in this gate (G1306A/V/E) affect different amino acid substitutions for one of a pair of glycines proposed to act as the “hinge” for the inactivation gate. The more the substituting amino acid differs from the physiological G1306, the more severe are the clinical symptoms. Alanine, with a short side-chain, results in a benign, often “subclinical” form of myotonia (myotonia fluctuans), valine, having a side-chain of intermediate size, causes moderate myotonia, and glutamic acid, an amino acid with a long side-chain, causes permanent myotonia, the most severe form of the disease [31, 33].
According to disease severity, G1306A shows the mildest, G1306V the moderate (Fig. 2), and G1306E the most severe slowing of fast inactivation and acceleration of recovery from inactivation in the heterologous expression system [31, 33]. In addition to fast channel inactivation, also activation and deactivation are affected whereas slow inactivation is normal. Variation of extracellular potassium has no direct effect on the mutant channels. However, measurements on excised fibers from patients showed that potassium exerts its effect via physiological depolarization, an effect which cannot be observed under experimental voltage-clamp conditions. The increased sodium inward current generates after-depolarizations across the T-tubular membrane which re-approaches activation threshold and, triggers repetitive action potentials [1]. Normally, influences from the activation threshold, resting membrane potential, and the size of after-depolarizations are tuned to avoid re-activations. In myotonia, usually more than one of these factors is changed, i.e., the activation threshold of the action potential is shifted or its amplitude and time-course are changed which might amplify after-depolarizations. An increase of potassium concentration in the T-tubules is pivotal, since this would depolarize the T-tubular membrane and thereby facilitate re-excitation [1]. With every additional action potential, T-tubular potassium concentration rises further. Finally, T-tubular potassium accumulation will terminate the bursts by depolarization and subsequent inactivation of NaV1.4.
Fig. 2.
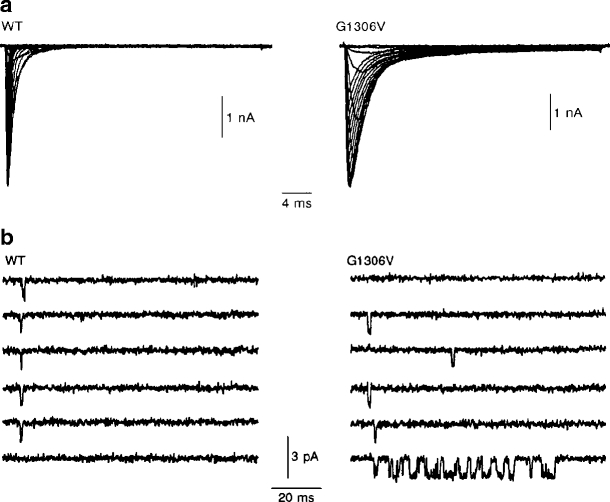
Currents through WT and mutant sodium channels. a Whole-cell sodium current traces of WT and PAM mutant (G1306V) sodium channels. b Single-channel recordings for WT and mutant sodium channels. Sodium currents were elicited by step depolarizations from a holding potential of −120 mV in 10 mV steps to +30 mV. Re-openings were more frequent for mutant channels, thereby leading to a small persistent current. Modified after Mitrovic et al. [34]
Paramyotonia congenita—stiffness followed by flaccid weakness
The cardinal symptom is cold-induced muscle stiffness that increases with continued activity (paradoxical myotonia). In the cold (or even in a cool wind), the face may appear mask-like, and the eyes cannot be opened for several seconds or minutes. On intensive cooling, the stiffness gives way to flaccid weakness or even to paralysis. Families with R1448 substitutions also have episodes of generalized periodic paralysis [29]. Such attacks occur spontaneously and can be triggered by rest or potassium. They are of short duration (an hour or less) in comparison to the cold-induced weakness which usually lasts for several hours even when the muscles are immediately re-warmed after a short bout of exposure to cold. Under warm conditions, most patients have no complaints because impaired muscle relaxation improves at higher temperatures. The prevalence is about 1:200,000 [29].
Most of the 16 PMC mutations are situated in protein parts relevant for channel inactivation: in the inactivation particle IFM itself like F1311V (unpublished observation) and nearby like T1313M/A [4, 66], in the outermost arginine of the voltage sensor of DIV (R1448H/C/S/P/L), in intracellular S4–S5 loops of DIII or DIV (e.g., F1473S) [10], or in the C terminus [65]. Functional expression revealed slowed fast inactivation and accelerated recovery from the inactivated state [6, 15]. These gain of function changes lead to action potential facilitation as already explained. A recent study pointed out, that resurgent currents, i.e., currents conducted by sodium channels which re-open during the repolarization, may contribute to the pathogenesis of PMC [19]. In neurons, these unusual sodium currents are proposed to result from a putative intracellular blocking factor that binds to open channels preventing classical inactivation, but unbinds during repolarization at potentials at which channels normally remain inactivated. The blocking factor has been hypothesized to be a beta subunit, for example beta-4, which is present in skeletal muscle and could contribute to the hyperexcitability of PMC.
Except for one controversially discussed study [36], all studies carried out so far showed minor differences in temperature sensitivity between WT and PC mutations [7, 10, 32]. This leads to the conclusion that the effect of temperature on excitability in PC is simply a result of the normal slowing of channel kinetics with cooling and is not the consequence of any alteration in the temperature sensitivity of sodium channel properties. Therefore, temperature sensitivity in PC might be explained as a threshold phenomenon [10, 32]. A recent study of Webb and Cannon [62] suggests that temperature dependency of slow inactivation may contribute to the phenotype.
Hyperkalemic periodic paralysis
HyperPP patients present with increased serum potassium during episodes of weakness. They may not have any interictal symptoms and are therefore often thought to exhibit a conversion reaction, and the subsequent omission of adequate therapy may cause them to suffer needlessly. Weakness is triggered by a variety of circumstances including rest after exercise, potassium-rich food, cold environment, emotional stress, fasting, and pregnancy. Between episodes, the disease is often associated with myotonia, which is mild and does not impede voluntary movements but may exacerbate at the beginning of the weakness spell.
Mutations causing HyperPP, slow fast inactivation to an extent, that channels fail to inactivate completely even after long-lasting depolarizations. As a result, there is persistent sodium inward current as shown in excised fibers obtained from patients [28] and as intracellular sodium accumulation in vivo [63]. The persistent sodium conductance leads to membrane depolarization and drives an outward flux of potassium into the extracellular space. Fiber depolarization, persistent sodium current, and hyperkalemia form a vicious cycle which spreads out and affects the surrounding muscle tissue. The resulting hyperkalemia can be so severe that cardiac complications arise.
Most of the HyperPP Nav1.4 mutations are situated at inner parts of the transmembrane segments and in the intracellular loop connecting transmembrane segments S5 and S6 of DII. They affect structures that form the three-dimensional docking site for the fast inactivation particle. Any malformation may reduce the affinity between the “latch bar and the catch”. The mutant channels avoid the inactivated state and, in contrast to normal sodium channels, re-open or flicker between the inactivated and the open state [16, 59]. In HyperPP, slow inactivation of the mutant channels is incomplete, especially for mutations situated at the S5/S6 loop.
During a paralytic episode, death may occur due to respiratory insufficiency. Independently of the severity and frequency of the paralytic episodes, many patients develop a chronic progressive myopathy in the forties, an age at which the attacks of weakness decrease. Patients without interictal myotonia are much more prone to develop the progressive myopathy and permanent weakness than individuals with myotonia. This becomes especially obvious in individuals with the most common T704M mutation which is not associated with EMG myotonia in half of the patients, and about half of the T704M patients develop permanent myopathy. The second most frequent mutation, M1592V, always is associated with EMG myotonia and permanent myopathy has never been reported.
Hypokalemic periodic paralysis
HypoPP differs from the hyperkalemic form in the sense that a spontaneous attack is associated with hypokalemia, potassium is a remedy, whereas carbohydrate- and sodium-rich food triggers an attack. In general, the attacks last longer and are more severe. Usually, the patients are weakest during the second half of the night and in the morning, and become stronger as the day goes by.
HypoPP is caused by mutations in two voltage-gated cation channels in skeletal muscle Cav1.1 (HypoPP-1) and Nav1.4 (HypoPP-2) [12, 21, 22]. Almost all mutations neutralize a positively charged amino acid in one of the outermost arginines or lysines of a voltage sensor. The Nav1.4 mutations are situated in the voltage sensors of DI, DII, and DIII. The electrophysiological characterization of the gating defects induced by these mutations revealed a loss of channel function, which does not explain the phenotype [27]. By expressing HypoPP mutations in Xenopus oocytes, Sokolov et al. [45] and Cannon et al. [47] discovered a cation leak with the typical characteristics found for the ω-current in Shaker K+-channels [49]. The ω-current, so called to differentiate it from the (α-)current through the ion-conducting pore, is a hyperpolarization-activated current of monovalent cations that is thought to flow through the S4 gating pore (Fig. 3). The ω-current counteracts the Kir2.1 current and therefore depolarizes and destabilizes the resting membrane potential so that the fraction of fibers in P2 is increased [20]. This has been observed in preparations obtained from patients [23]. In vivo, the muscles from these patients exhibited an intracellular sodium accumulation and edema.
Fig. 3.
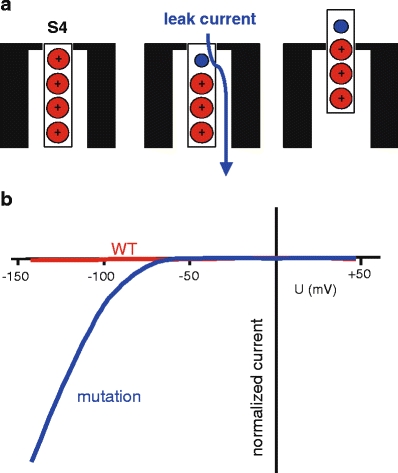
Leak currents. a A replacement of the outermost arginine (red) by a smaller amino acid (blue), e.g., glycine, opens a conductive pathway at hyperpolarized potentials, resulting in an inward cation current (blue). At depolarized potentials at which the S4 segment moves outward, the conductive pathway is closed and the cation current ceases. b Schematic of cation currents through sodium channels carrying charge-neutralizing substitutions in S4 voltage sensors. Note the large inward current in the hyperpolarized potential range corresponding to the resting state of the leaky S4 voltage sensor
As muscle fibers with a severe voltage sensor mutation are depolarized not only during hypokalemia but also at potassium levels in the normal range, this membrane leak might not only explain episodes of weakness, but interictal (permanent) weakness as well. The permanent weakness associated with a fatty replacement myopathy is very frequently found in patients harboring DIV mutations in the calcium channel, i.e., Cav1.1 R1239H [23].
Normokalemic periodic paralysis
The term normokalemic periodic paralysis (NormoPP) was originally given to a variant described in the 1960s. The disorder resembled hyperkalemic PP in many aspects; the only real differences were the lack of increase in the concentration of serum potassium even during serious attacks, and the lack of a beneficial effect of glucose administration [29]. The existence of NormoPP as a nosologic entity was questioned because of the potassium sensitivity of the patients and the identification of the most frequent hyperkalemic PP mutations T704M or M1592V in such families including the original family.
Recently, a potassium-sensitive type of periodic paralysis with normokalemia and episodes of weakness reminiscent of those in both hyperkalemic (initiation of an attack by potassium) and hypokalamic forms (duration of attacks) was reported [57]. This phenotype, also named NormoPP, is caused by SCN4A mutations at deeper locations of the voltage sensor of domain II at codon 675 (Fig. 1). Functionally, R675 mutations generate an ω-current with a reversed voltage dependence compared to mutations causing HypoPP-2 [46], since this site is exposed to the extracellular space at stronger depolarizations. Future studies will show whether NormoPP is a separate clinical entity. The diagnostics are as described for the two more common forms of the disease. The prophylaxis consists of avoidance of hyperkalemia and the administration of acetazolamide.
Pathophysiological basics of weakness
Primary myogenic paralysis is based on muscle inexcitability which is caused by depolarization beyond −60 mV. This may be due to the aforementioned paradoxical depolarization described for [K+]o reduction. The paralytic state corresponds to P2. At this potential, Nav1.4 is subject to closed-state inactivation. Transition jumps between the two states (P1 and P2) occur when any trigger drives the system closer to or shifts a limit point, so that the current state becomes instable (Fig. 4a). The limit points of both states (LP1 and LP2) have different locations in the state space. Therefore, hysteresis is a typical feature of such a system. If [K+]o is considered as a control parameter as in Fig. 4a, it reveals its important role in driving the system to its limits.
Fig. 4.
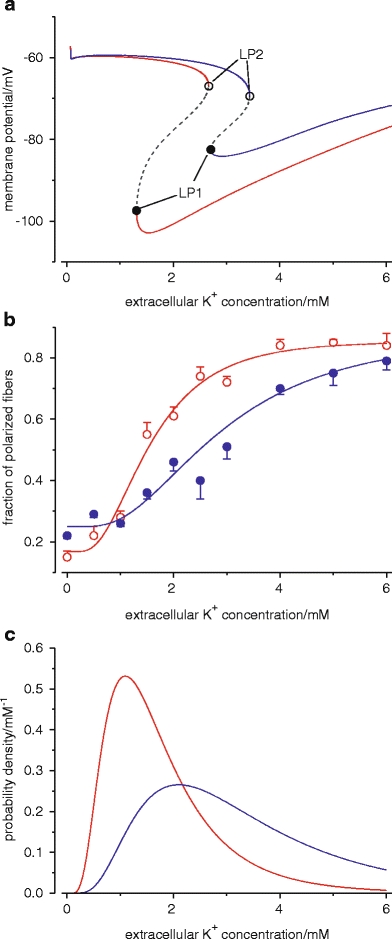
Bistable membrane potentials and pathogenesis of hypokalemic periodic paralysis. a Bifurcation diagram of a mathematical model of skeletal muscle with the extracellular K+ concentration as the control parameter. Beginning at high K+ concentrations, a reduction reveals an instability of the resting potential at the limit point, LP1 (closed circles), from which a sudden transition to about −60 mV occurs. Increasing the K+ concentration from low values takes the muscle fiber to another limit point, LP2 (open circles), where the system jumps back into a state with normal membrane potentials. Control (red line), additional small (5 µS/cm2) depolarizing current and insulin-induced reduction of Kir conductance as in HypoPP (blue line), regions of instability (dashed line). b Fraction of rat diaphragm muscle fibers with normal membrane potentials after the reduction of [K+]o to different values, without (red) and with (blue) amphotericin B, a Na+ and K+ ionophore. Curves represent fits of a log-normal cumulative distribution function (modified from [23]). c Probability density functions computed with fit parameters from (b). These functions give the probability of a transition from the normal to the depolarized state for responding fibers at different [K+]o. LP1 may be defined as the mode or the median of the curves. Amphotericin B causes an increment of mode, median, and SD by 1 mM, 1.5 mM, and 1.2 mM, respectively. Therefore, in the presence of a small depolarizing leak current, LP1 is shifted to the right and a paradoxical depolarization is more likely to occur even at normal [K+]o
Normal muscle does not become paralytic because the limit points are located at very low potassium concentrations that do not occur physiologically. The location of LP1 has been measured in diaphragm of rat muscle (Fig. 4b, c) [23]. Results from a single fiber of mouse lumbricalis muscle have been published by Geukes-Foppen et al. [13] and suggest a hysteresis of about 1 mM. If the limit points were shifted to higher potassium concentrations, transitions between the states would be possible under physiological conditions and result in periodic paralysis. Hypokalemic periodic paralysis is caused by such a shift. The shift is induced by a small depolarizing leak current (ω-current) and by a reduction of resting potassium conductance [42, 52]. As an in vitro model, the pharmacological induction of a small depolarizing leak current by amphotericin B has been successfully applied (Fig. 4b, c).
Paralytic attacks are triggered by certain endocrine or physical challenges. The triggers seem to have in common that they affect [K+]o by either changing internal potassium balance or shifting the limit points. Typical triggers that reduce [K+]o and therefore provoke attacks in hypokalemic periodic paralysis are: insulin, catecholamines (β2-agonists), glucocorticoids, and rest after exercise. Triggers for hyperkalemic periodic paralysis, where an instability of P1 emerges at increasing [K+]o values [5] are: potassium-rich meals and exercise. The fact that a limit point or threshold has to be reached to provoke a change in stability might explain the periodicity of the paralytic attacks. Hysteresis is reflected in the clinical fact, that, for recovery from paralysis, [K+]o has to change more than it did when it provoked weakness.
Muscle membrane potentials provide an explanation for the weakness observed directly following cold-induced paramyotonia as well. During cooling to 27°C in vitro, excised PMC muscle fibers slowly depolarized from −85 mV to about −45 mV whereas normal muscle fibers depolarized by not more than 5 mV. The depolarization was associated with a long-lasting burst of action potentials which stop as soon as the membrane potential approximates values of −45 to −50 mV [30, 32]. At this voltage, also the mutant sodium channels are inactivated and therefore the muscle fibers become inexcitable and paralyzed.
Congenital myasthenic syndrome
CMS is a heterogeneous group of inherited disorders with defective transmission of neuromuscular excitation resulting in muscle fatigue [8]. They result from defects in presynaptic, synaptic, and postsynaptic proteins. Of the postsynaptic CMS that are usually caused by mutations in the nicotinic acetylcholine receptor, one form has been identified as sodium channelopathy. Two alterations in Nav1.4 have been identified, S246L in the S4–S5 cytoplasmic linker in DI, and V1442E in the S3–S4 extracellular linker in DIV [53]. While S246L is likely a benign polymorphism, V1442E revealed a marked enhancement of fast inactivation even at hyperpolarized potentials, and enhanced use-dependent inactivation on high-frequency stimulation. The reduced availability of sodium channels decreases the safety factor of synaptic transmission.
Medication of skeletal muscle sodium channelopathies
The aim of drug therapy in myotonia is to reduce the involuntary action potential bursts without blocking voluntary high-frequency muscle stimulation. This is a delicate balance between drug-induced reduction of voluntary tetanic muscle strength and too little antimyotonic effects. Fortunately, local anesthetics and anti-arrhythmic drugs of class IB effectively relieve stiffness in PAM and prevent muscle stiffness and weakness occurring in PMC with cooling [2, 41]. These agents stabilize the inactivated channel by a hyperpolarizing shift in steady-state inactivation and by slowing the recovery from inactivation. Agents such as mexiletine, flecainide, and other lidocaine analogs, prevent repetitive firing of action potentials due to their "use dependence", a dependence of the depth of block on the frequency of action potentials. The degree of use dependence varies with the structure (charge and hydrophobicity) of the drug. Beyond this “unspecific” antimyotonic effect, the agents seem to be more effective on certain mutant sodium channels than on wild-type channels [35]. Unfortunately, the spontaneous and potassium-induced attacks of weakness typical for hyperkalemic PP and also occurring in some PMC patients are not improved by mexiletine [39]. However, diuretics such as hydrochlorothiazide and acetazolamide can decrease frequency and severity of paralytic episodes, probably by lowering serum potassium and perhaps by shifting the pH to lower values.
In HypoPP, acute weakness spells can be treated by potassium and be prevented by certain carbonic anhydrase inhibitors, aldosterone antagonists, and potassium-sparing diuretics. Serum potassium levels in the high normal range help reduce the paradoxical membrane depolarization and therefore shift the resting potential from P2 to P1 values. Acetazolamide also lowers intracellular sodium accumulation in these patients [23] addressing both pathogenetic factors in HypoPP: depolarization and sodium accumulation. The repolarizing effect of acetazolamide may be explained at least partially by opening of big conductance potassium channels [51].
Perspectives
As ion channels constitute one of the only protein families that allow functional examination on the molecular level, expression studies of putative mutations have become standard in supporting the disease-causing nature of mutations. While this is quite helpful, one must not over-interpret functional changes that a mutation produces because these changes may not necessarily indicate a disease-causing mutation but a functional polymorphism instead [26]. Therefore, while functional studies are essential, they do not alleviate from the need for careful observation of clinical phenotype, response to therapy, and genetic screening of large and adequately matched control populations, for interpretation of pathogenesis of ion channel disorders.
Acknowledgments
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Adrian RH, Marshall MW. Action potentials reconstructed in normal and myotonic muscle fibres. J Physiol. 1976;258:125–143. doi: 10.1113/jphysiol.1976.sp011410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alfonsi E, Merlo IM, Tonini M, Ravaglia S, Brugnoni R, Gozzini A, Moglia A. Efficacy of propafenone in paramyotonia congenita. Neurology. 2007;68:1080–1081. doi: 10.1212/01.wnl.0000257825.29703.e8. [DOI] [PubMed] [Google Scholar]
- 3.Bezanilla F. How membrane proteins sense voltage. Nat Rev Mol Cell Biol. 2008;9:323–332. doi: 10.1038/nrm2376. [DOI] [PubMed] [Google Scholar]
- 4.Bouhours M, Sternberg D, Davoine CS, Ferrer X, Willer JC, Fontaine B, Tabti N. Functional characterization and cold sensitivity of T1313A, a new mutation of the skeletal muscle sodium channel causing paramyotonia congenita in humans. J Physiol. 2004;554:635–647. doi: 10.1113/jphysiol.2003.053082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cannon SC, Brown RH, Jr, Corey DP. Theoretical reconstruction of myotonia and paralysis caused by incomplete inactivation of sodium channels. Biophys J. 1993;65:270–288. doi: 10.1016/S0006-3495(93)81045-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chahine M, George AL, Jr, Zhou M, Ji S, Sun W, Barchi RL, Horn R. Sodium channel mutations in paramyotonia congenita uncouple inactivation from activation. Neuron. 1994;12:281–294. doi: 10.1016/0896-6273(94)90271-2. [DOI] [PubMed] [Google Scholar]
- 7.Dice MS, Abbruzzese JL, Wheeler JT, Groome JR, Fujimoto E, Ruben PC. Temperature-sensitive defects in paramyotonia congenita mutants R1448C and T1313M. Muscle Nerve. 2004;30:277–288. doi: 10.1002/mus.20080. [DOI] [PubMed] [Google Scholar]
- 8.Engel AG, Ohno K, Shen XM, Sine SM. Congenital myasthenic syndromes: multiple molecular targets at the neuromuscular junction. Ann N Y Acad Sci. 2003;998:138–160. doi: 10.1196/annals.1254.016. [DOI] [PubMed] [Google Scholar]
- 9.Featherstone DE, Richmond JE, Ruben PC. Interaction between fast and slow inactivation in Skm1 sodium channels. Biophys J. 1996;71:3098–3109. doi: 10.1016/S0006-3495(96)79504-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fleischhauer R, Mitrovic N, Deymeer F, Lehmann-Horn F, Lerche H. Effects of temperature and mexiletine on the F1473S Na+ channel mutation causing paramyotonia congenita. Pflugers Arch. 1998;436:757–765. doi: 10.1007/s004240050699. [DOI] [PubMed] [Google Scholar]
- 11.Fontaine B, Khurana TS, Hoffman EP, Bruns GA, Haines JL, Trofatter JA, Hanson MP, Rich J, McFarlane H, Yasek DM, et al. Hyperkalemic periodic paralysis and the adult muscle sodium channel alpha-subunit gene. Science. 1990;250:1000–1002. doi: 10.1126/science.2173143. [DOI] [PubMed] [Google Scholar]
- 12.Fontaine B, Vale-Santos J, Jurkat-Rott K, Reboul J, Plassart E, Rime CS, Elbaz A, Heine R, Guimaraes J, Weissenbach J, et al. Mapping of the hypokalaemic periodic paralysis (HypoPP) locus to chromosome 1q31-32 in three European families. Nat Genet. 1994;6:267–272. doi: 10.1038/ng0394-267. [DOI] [PubMed] [Google Scholar]
- 13.Geukes Foppen RJ, van Mil HG, Siegenbeek van Heukelom J. Osmolality influences bistability of membrane potential under hypokalemic conditions in mouse skeletal muscle: an experimental and theoretical study. Comp Biochem Physiol A Mol Integr Physiol. 2001;130:533–538. doi: 10.1016/S1095-6433(01)00430-5. [DOI] [PubMed] [Google Scholar]
- 14.Glaaser IW, Bankston JR, Liu H, Tateyama M, Kass RS. A carboxyl-terminal hydrophobic interface is critical to sodium channel function. Relevance to inherited disorders. J Biol Chem. 2006;281:24015–24023. doi: 10.1074/jbc.M605473200. [DOI] [PubMed] [Google Scholar]
- 15.Goldman L. On mutations that uncouple sodium channel activation from inactivation. Biophys J. 1999;76:2553–2559. doi: 10.1016/S0006-3495(99)77408-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hayward LJ, Brown RH, Jr, Cannon SC. Inactivation defects caused by myotonia-associated mutations in the sodium channel III-IV linker. J Gen Physiol. 1996;107:559–576. doi: 10.1085/jgp.107.5.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heine R, Pika U, Lehmann-Horn F. A novel SCN4A mutation causing myotonia aggravated by cold and potassium. Hum Mol Genet. 1993;2:1349–1353. doi: 10.1093/hmg/2.9.1349. [DOI] [PubMed] [Google Scholar]
- 18.Isom LL. Sodium channel beta subunits: anything but auxiliary. Neuroscientist. 2001;7:42–54. doi: 10.1177/107385840100700108. [DOI] [PubMed] [Google Scholar]
- 19.Jarecki BW, Piekarz AD, Jackson JO, 2nd, Cummins TR. Human voltage-gated sodium channel mutations that cause inherited neuronal and muscle channelopathies increase resurgent sodium currents. J Clin Invest. 2010;120:369–378. doi: 10.1172/JCI40801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jurkat-Rott K, Lehmann-Horn F. Do hyperpolarization-induced proton currents contribute to the pathogenesis of hypokalemic periodic paralysis, a voltage sensor channelopathy? J Gen Physiol. 2007;130:1–5. doi: 10.1085/jgp.200709834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jurkat-Rott K, Lehmann-Horn F, Elbaz A, Heine R, Gregg RG, Hogan K, Powers PA, Lapie P, Vale-Santos JE, Weissenbach J, et al. A calcium channel mutation causing hypokalemic periodic paralysis. Hum Mol Genet. 1994;3:1415–1419. doi: 10.1093/hmg/3.8.1415. [DOI] [PubMed] [Google Scholar]
- 22.Jurkat-Rott K, Mitrovic N, Hang C, Kouzmekine A, Iaizzo P, Herzog J, Lerche H, Nicole S, Vale-Santos J, Chauveau D, Fontaine B, Lehmann-Horn F. Voltage-sensor sodium channel mutations cause hypokalemic periodic paralysis type 2 by enhanced inactivation and reduced current. Proc Natl Acad Sci USA. 2000;97:9549–9554. doi: 10.1073/pnas.97.17.9549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jurkat-Rott K, Weber MA, Fauler M, Guo XH, Holzherr BD, Paczulla A, Nordsborg N, Joechle W, Lehmann-Horn F. K+-dependent paradoxical membrane depolarization and Na+ overload, major and reversible contributors to weakness by ion channel leaks. Proc Natl Acad Sci USA. 2009;106(10):4036–4041. doi: 10.1073/pnas.0811277106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koch MC, Steinmeyer K, Lorenz C, Ricker K, Wolf F, Otto M, Zoll B, Lehmann-Horn F, Grzeschik KH, Jentsch TJ. The skeletal muscle chloride channel in dominant and recessive human myotonia. Science. 1992;257:797–800. doi: 10.1126/science.1379744. [DOI] [PubMed] [Google Scholar]
- 25.Kubota T, Kinoshita M, Sasaki R, Aoike F, Takahashi MP, Sakoda S, Hirose K. New mutation of the Na channel in the severe form of potassium-aggravated myotonia. Muscle Nerve. 2009;39:666–673. doi: 10.1002/mus.21155. [DOI] [PubMed] [Google Scholar]
- 26.Kuzmenkin A, Jurkat-Rott K, Lehmann-Horn F, Mitrovic N. Impaired slow inactivation due to a polymorphism and substitutions of Ser-906 in the II-III loop of the human Nav1.4 channel. Pflugers Arch. 2003;447:71–77. doi: 10.1007/s00424-003-1137-5. [DOI] [PubMed] [Google Scholar]
- 27.Lehmann-Horn F, Jurkat-Rott K. Voltage-gated ion channels and hereditary disease. Physiol Rev. 1999;79:1317–1372. doi: 10.1152/physrev.1999.79.4.1317. [DOI] [PubMed] [Google Scholar]
- 28.Lehmann-Horn F, Kuther G, Ricker K, Grafe P, Ballanyi K, Rudel R. Adynamia episodica hereditaria with myotonia: a non-inactivating sodium current and the effect of extracellular pH. Muscle Nerve. 1987;10:363–374. doi: 10.1002/mus.880100414. [DOI] [PubMed] [Google Scholar]
- 29.Lehmann-Horn F, Rüdel R, Jurkat-Rott K. Chapter 46: nondystrophic myotonias and periodic paralyses. In: Engel AG, Franzini-Armstrong C, editors. Myology. New York: McGraw-Hill; 2004. pp. 1257–1300. [Google Scholar]
- 30.Lehmann-Horn F, Rudel R, Ricker K. Membrane defects in paramyotonia congenita (Eulenburg) Muscle Nerve. 1987;10:633–641. doi: 10.1002/mus.880100709. [DOI] [PubMed] [Google Scholar]
- 31.Lerche H, Heine R, Pika U, George AL, Jr, Mitrovic N, Browatzki M, Weiss T, Rivet-Bastide M, Franke C, Lomonaco M, et al. Human sodium channel myotonia: slowed channel inactivation due to substitutions for a glycine within the III-IV linker. J Physiol. 1993;470:13–22. doi: 10.1113/jphysiol.1993.sp019843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lerche H, Mitrovic N, Dubowitz V, Lehmann-Horn F. Paramyotonia congenita: the R1448P Na+ channel mutation in adult human skeletal muscle. Ann Neurol. 1996;39:599–608. doi: 10.1002/ana.410390509. [DOI] [PubMed] [Google Scholar]
- 33.Mitrovic N, George AL, Jr, Heine R, Wagner S, Pika U, Hartlaub U, Zhou M, Lerche H, Fahlke C, Lehmann-Horn F. K(+)-aggravated myotonia: destabilization of the inactivated state of the human muscle Na+ channel by the V1589M mutation. J Physiol. 1994;478(Pt 3):395–402. doi: 10.1113/jphysiol.1994.sp020260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mitrovic N, George AL, Jr, Lerche H, Wagner S, Fahlke C, Lehmann-Horn F. Different effects on gating of three myotonia-causing mutations in the inactivation gate of the human muscle sodium channel. J Physiol. 1995;487(Pt 1):107–114. doi: 10.1113/jphysiol.1995.sp020864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mohammadi B, Jurkat-Rott K, Alekov A, Dengler R, Bufler J, Lehmann-Horn F. Preferred mexiletine block of human sodium channels with IVS4 mutations and its pH-dependence. Pharmacogenet Genomics. 2005;15:235–244. doi: 10.1097/01213011-200504000-00007. [DOI] [PubMed] [Google Scholar]
- 36.Mohammadi B, Mitrovic N, Lehmann-Horn F, Dengler R, Bufler J. Mechanisms of cold sensitivity of paramyotonia congenita mutation R1448H and overlap syndrome mutation M1360V. J Physiol. 2003;547:691–698. doi: 10.1113/jphysiol.2002.033928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Petitprez S, Tiab L, Chen L, Kappeler L, Rosler KM, Schorderet D, Abriel H, Burgunder JM. A novel dominant mutation of the Nav1.4 {alpha}-subunit domain I leading to sodium channel myotonia. Neurology. 2008;71:1669–1675. doi: 10.1212/01.wnl.0000335168.86248.55. [DOI] [PubMed] [Google Scholar]
- 38.Popa MO, Alekov AK, Bail S, Lehmann-Horn F, Lerche H. Cooperative effect of S4-S5 loops in domains D3 and D4 on fast inactivation of the Na+ channel. J Physiol. 2004;561:39–51. doi: 10.1113/jphysiol.2004.065912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ricker K, Bohlen R, Rohkamm R. Different effectiveness of tocainide and hydrochlorothiazide in paramyotonia congenita with hyperkalemic episodic paralysis. Neurology. 1983;33:1615–1618. doi: 10.1212/wnl.33.12.1615. [DOI] [PubMed] [Google Scholar]
- 40.Rojas CV, Wang JZ, Schwartz LS, Hoffman EP, Powell BR, Brown RH., Jr A Met-to-Val mutation in the skeletal muscle Na+ channel alpha-subunit in hyperkalaemic periodic paralysis. Nature. 1991;354:387–389. doi: 10.1038/354387a0. [DOI] [PubMed] [Google Scholar]
- 41.Rudel R, Dengler R, Ricker K, Haass A, Emser W. Improved therapy of myotonia with the lidocaine derivative tocainide. J Neurol. 1980;222:275–278. doi: 10.1007/BF00313157. [DOI] [PubMed] [Google Scholar]
- 42.Ruff RL. Insulin acts in hypokalemic periodic paralysis by reducing inward rectifier K+ current. Neurology. 1999;53:1556–1563. doi: 10.1212/wnl.53.7.1556. [DOI] [PubMed] [Google Scholar]
- 43.Ruff RL, Simoncini L, Stuhmer W. Slow sodium channel inactivation in mammalian muscle: a possible role in regulating excitability. Muscle Nerve. 1988;11:502–510. doi: 10.1002/mus.880110514. [DOI] [PubMed] [Google Scholar]
- 44.Siegenbeek van Heukelom J. Role of the anomalous rectifier in determining membrane potentials of mouse muscle fibres at low extracellular K+ J Physiol. 1991;434:549–560. doi: 10.1113/jphysiol.1991.sp018485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sokolov S, Scheuer T, Catterall WA. Gating pore current in an inherited ion channelopathy. Nature. 2007;446:76–78. doi: 10.1038/nature05598. [DOI] [PubMed] [Google Scholar]
- 46.Sokolov S, Scheuer T, Catterall WA (2008) Depolarization-activated gating pore current conducted by mutant sodium channels in potassium-sensitive normokalemic periodic paralysis. Proc Natl Acad Sci USA 105:19980–19985 [DOI] [PMC free article] [PubMed]
- 47.Struyk AF, Cannon SC. A Na+ channel mutation linked to hypokalemic periodic paralysis exposes a proton-selective gating pore. J Gen Physiol. 2007;130:11–20. doi: 10.1085/jgp.200709755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stunnenberg BC, Ginjaar HB, Trip J, Faber CG, van Engelen BG, Drost G (2009) Isolated eyelid closure myotonia in two families with sodium channel myotonia. Neurogenetics. doi:10.1007/s10048-009-0225-x [DOI] [PMC free article] [PubMed]
- 49.Tombola F, Pathak MM, Isacoff EY. Voltage-sensing arginines in a potassium channel permeate and occlude cation-selective pores. Neuron. 2005;45:379–388. doi: 10.1016/j.neuron.2004.12.047. [DOI] [PubMed] [Google Scholar]
- 50.Tombola F, Pathak MM, Isacoff EY. How does voltage open an ion channel? Annu Rev Cell Dev Biol. 2006;22:23–52. doi: 10.1146/annurev.cellbio.21.020404.145837. [DOI] [PubMed] [Google Scholar]
- 51.Tricarico D, Barbieri M, Camerino DC. Acetazolamide opens the muscular KCa2+ channel: a novel mechanism of action that may explain the therapeutic effect of the drug in hypokalemic periodic paralysis. Ann Neurol. 2000;48:304–312. doi: 10.1002/1531-8249(200009)48:3<304::AID-ANA4>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 52.Tricarico D, Servidei S, Tonali P, Jurkat-Rott K, Camerino DC. Impairment of skeletal muscle adenosine triphosphate-sensitive K+ channels in patients with hypokalemic periodic paralysis. J Clin Invest. 1999;103:675–682. doi: 10.1172/JCI4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tsujino A, Maertens C, Ohno K, Shen XM, Fukuda T, Harper CM, Cannon SC, Engel AG. Myasthenic syndrome caused by mutation of the SCN4A sodium channel. Proc Natl Acad Sci USA. 2003;100:7377–7382. doi: 10.1073/pnas.1230273100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Mil H, Siegenbeek van Heukelom J, Bier M. A bistable membrane potential at low extracellular potassium concentration. Biophys Chem. 2003;106:15–21. doi: 10.1016/S0301-4622(03)00135-2. [DOI] [PubMed] [Google Scholar]
- 55.Vassilev PM, Scheuer T, Catterall WA. Identification of an intracellular peptide segment involved in sodium channel inactivation. Science. 1988;241:1658–1661. doi: 10.1126/science.2458625. [DOI] [PubMed] [Google Scholar]
- 56.Vedantham V, Cannon SC. Slow inactivation does not affect movement of the fast inactivation gate in voltage-gated Na+ channels. J Gen Physiol. 1998;111:83–93. doi: 10.1085/jgp.111.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vicart S, Sternberg D, Fournier E, Ochsner F, Laforet P, Kuntzer T, Eymard B, Hainque B, Fontaine B. New mutations of SCN4A cause a potassium-sensitive normokalemic periodic paralysis. Neurology. 2004;63:2120–2127. doi: 10.1212/01.wnl.0000145768.09934.ec. [DOI] [PubMed] [Google Scholar]
- 58.Vilin YY, Ruben PC. Slow inactivation in voltage-gated sodium channels: molecular substrates and contributions to channelopathies. Cell Biochem Biophys. 2001;35:171–190. doi: 10.1385/CBB:35:2:171. [DOI] [PubMed] [Google Scholar]
- 59.Wagner S, Lerche H, Mitrovic N, Heine R, George AL, Lehmann-Horn F. A novel sodium channel mutation causing a hyperkalemic paralytic and paramyotonic syndrome with variable clinical expressivity. Neurology. 1997;49:1018–1025. doi: 10.1212/wnl.49.4.1018. [DOI] [PubMed] [Google Scholar]
- 60.Wallace RH, Wang DW, Singh R, Scheffer IE, George AL, Jr, Phillips HA, Saar K, Reis A, Johnson EW, Sutherland GR, Berkovic SF, Mulley JC. Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel beta1 subunit gene SCN1B. Nat Genet. 1998;19:366–370. doi: 10.1038/448. [DOI] [PubMed] [Google Scholar]
- 61.Wang JZ, Rojas CV, Zhou JH, Schwartz LS, Nicholas H, Hoffman EP. Sequence and genomic structure of the human adult skeletal muscle sodium channel alpha subunit gene on 17q. Biochem Biophys Res Commun. 1992;182:794–801. doi: 10.1016/0006-291X(92)91802-W. [DOI] [PubMed] [Google Scholar]
- 62.Webb J, Cannon SC. Cold-induced defects of sodium channel gating in atypical periodic paralysis plus myotonia. Neurology. 2008;70:746–747. doi: 10.1212/01.wnl.0000265397.70057.d8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Weber MA, Nielles-Vallespin S, Essig M, Jurkat-Rott K, Kauczor HU, Lehmann-Horn F. Muscle Na+ channelopathies: MRI detects intracellular 23Na accumulation during episodic weakness. Neurology. 2006;67:1151–1158. doi: 10.1212/01.wnl.0000233841.75824.0f. [DOI] [PubMed] [Google Scholar]
- 64.West JW, Patton DE, Scheuer T, Wang Y, Goldin AL, Catterall WA. A cluster of hydrophobic amino acid residues required for fast Na(+)-channel inactivation. Proc Natl Acad Sci USA. 1992;89:10910–10914. doi: 10.1073/pnas.89.22.10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu FF, Gordon E, Hoffman EP, Cannon SC. A C-terminal skeletal muscle sodium channel mutation associated with myotonia disrupts fast inactivation. J Physiol. 2005;565:371–380. doi: 10.1113/jphysiol.2005.082909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang N, Ji S, Zhou M, Ptacek LJ, Barchi RL, Horn R, George AL., Jr Sodium channel mutations in paramyotonia congenita exhibit similar biophysical phenotypes in vitro. Proc Natl Acad Sci USA. 1994;91:12785–12789. doi: 10.1073/pnas.91.26.12785. [DOI] [PMC free article] [PubMed] [Google Scholar]