

Abstract
Voltage-gated Ca2+ channels couple membrane depolarization to Ca2+-dependent intracellular signaling events. This is achieved by mediating Ca2+ ion influx or by direct conformational coupling to intracellular Ca2+ release channels. The family of Cav1 channels, also termed L-type Ca2+ channels (LTCCs), is uniquely sensitive to organic Ca2+ channel blockers and expressed in many electrically excitable tissues. In this review, we summarize the role of LTCCs for human diseases caused by genetic Ca2+ channel defects (channelopathies). LTCC dysfunction can result from structural aberrations within their pore-forming α1 subunits causing hypokalemic periodic paralysis and malignant hyperthermia sensitivity (Cav1.1 α1), incomplete congenital stationary night blindness (CSNB2; Cav1.4 α1), and Timothy syndrome (Cav1.2 α1; reviewed separately in this issue). Cav1.3 α1 mutations have not been reported yet in humans, but channel loss of function would likely affect sinoatrial node function and hearing. Studies in mice revealed that LTCCs indirectly also contribute to neurological symptoms in Ca2+ channelopathies affecting non-LTCCs, such as Cav2.1 α1 in tottering mice. Ca2+ channelopathies provide exciting disease-related molecular detail that led to important novel insight not only into disease pathophysiology but also to mechanisms of channel function.
Keywords: Channels, Channel gating, Channel activity, Neuronal excitability
Introduction
Voltage-gated Ca2+ channels are Ca2+-selective pores linked to voltage-sensing domains that couple membrane depolarization to intracellular signaling events. Among the three families of voltage-gated Ca2+ channels (VGCCs; Cav1, Cav2, and Cav3, [14]), the family of Cav1 channels, also termed L-type Ca2+ channels (LTCCs), is uniquely sensitive to organic Ca2+ channel blockers and expressed in many electrically excitable tissues. LTCCs were first described in heart and smooth muscle. Today, we know that these cardiovascular channels are almost exclusively of the Cav1.2 subtype and their block by clinically used Ca2+ channel blockers (such as nifedipine, amlodipine, verapamil, and diltiazem) explains most of their therapeutic effects, such as blood pressure lowering and cardiodepression. In addition to Cav1.2, three other isoforms (Cav1.1, Cav1.3, and Cav1.4) exist. Cav1.3 is expressed together with Cav1.2 in many tissues, such as the sinoatrial node and heart atria, neurons, chromaffin cells, and pancreatic islets. Available Ca2+ channel blockers inhibit both of these isoforms with similar affinities, such that their physiological roles could not be separated pharmacologically. This was possible by genetically modified mice revealing distinct functions of these two isoforms based on differences in their biophysical properties [62, 68]. In particular, Cav1.3 can serve pacemaker functions in neurons [57], the sinoatrial node [47], and in chromaffin cells [49, 50]. In the brain, both isoforms couple neuronal activity to transcriptional events: Cav1.2 mediates long-term potentiation and spatial learning and memory in the hippocampus [55]. Cav1.3 mediates long-term potentiation in the amygdala and participates in the consolidation of fear memory [25].
Cav1.1 and Cav1.4 possess a much more restricted expression pattern, with expression almost exclusively in skeletal muscle and the retina, respectively. Cav1.1 channels (which also contain a γ-subunit) carry very slowly activating Ca2+ inward currents, too slow for providing Ca2+ to the contractile machinery in response to millisecond depolarizations eliciting muscle contraction. Although the fast conformational changes of their voltage-sensing domains induce pore opening very slowly, they are quickly transmitted to the sarcoplasmic reticulum (SR) ryanodine receptors (RyR1), thus serving as fast voltage sensors for SR Ca2+ release. This seems to be accomplished through a close physical association of Cav1.1 channels in the T-tubular membrane and RyR1 in the junctional SR of the skeletal muscle triads [45].
Transcripts for all four LTCC α1 subunit isoforms and accessory β3- and β4-subunits are also present in immune cells [2, 36]. Although reduced expression of Cav1.1, β3, or β4 was each associated with reduced Ca2+ influx after T-cell receptor cross-linking in T-cells [52], the exact role of LTCCs for T-cell signaling remains unknown.
Here, we summarize the role of LTCCs for human diseases caused by genetic Ca2+ channel defects (channelopathies) in Ca2+ channel α1 subunits. LTCC dysfunction can result from structural aberrations within their pore-forming α1 subunit (L-type Ca2+ channelopathies), such as in retinal Cav1.4 α1 found in patients with incomplete congenital stationary night blindness (CSNB2), or in skeletal muscle Cav1.1 α1 found in patients with hypokalemic periodic paralysis (HPP) or malignant hyperthermia susceptibility (MHS). However, LTCC dysfunction can also occur in Ca2+ channelopathies with structural aberrations in the α1 subunit of non-LTCCs [13] (non-L-type Ca2+ channelopathies), such as Cav2.1 α1 mutations in tottering mice. Ca2+ channelopathies involving defects of auxiliary subunits (which may not selectively affect only LTCCs) will not be discussed in this review.
Cav1.1 channelopathies (CACNA1S gene)
Hypokalemic periodic paralysis type 1
Familial HPP is an autosomal dominant disorder caused by mutations in the pore-forming Cav1.1 α1-(hypokalemic periodic paralysis type 1,HPP-1) or Na+-channel α-subunit (Nav1.4, SCN4A gene; HPP-2; see chapter on skeletal muscle Na+-channel channelopathies in this issue). CACNA1S mutations are found in about 75% of patients and SCN4A mutations in about 15% [41]. HPP symptoms generally manifest around the second decade of life and are characterized by hypotonia and attacks of local or generalized skeletal muscle weakness or paralysis. The frequency of the attacks is variable. A lower penetrance often occurs in females. Attacks are accompanied by hypokalemia, and therapeutic potassium supplementation relieves symptoms. Precipitating factors are high-carbohydrate meals, insulin intake, acute stress, sudden exposure to heat or cold, and sudden rest after exercise. The long-term prognosis is generally good, and crises may decrease in midlife. However, severely affected families were reported, and involvement of respiratory muscles may lead to death [7]. The discovery of single missense CACNA1S mutations in humans with HPP-1 which still allow expression of a full-length Cav1.1 α1 subunit protein suggested that changes in channel gating or channel expression on the cell surface may account for altered skeletal muscle function. The most frequent mutations affect arginine residues in two of the channel's voltage sensors (R528, R1239; Fig. 1). In contrast to skeletal muscle Na+-channels, Cav1.1 channels are difficult to express in heterologous systems [56]. Results from such studies, and even from recordings of mutant Ca2+ currents from myotubes cultured from affected patient muscle [69], were rather controversial and did not reveal a clear unifying picture of how the reported biophysical changes may explain the episodic failure of muscle excitability in association with a decrease in serum potassium.
Fig. 1.
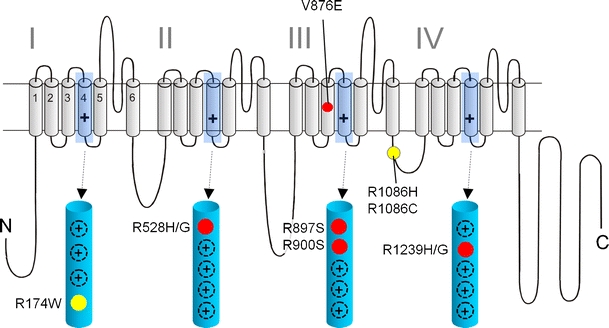
Mutations in Ca2+ channel Cav1.1 α1 subunits identified in patients with HPP-1 and MHS: a folding model of α1-subunits based on hydrophobicity analysis is shown. Plus sign indicates several positive charges in the transmembrane S4 helices within the hydrophobic repeats I–IV. S4 helices and their positively charged residues are shown in the enlarged structures. Together with S1, S2, and S3 helices, they form the four voltage-sensing domains of the channel controlling the opening and closing of a single pore domain formed by S5 and S6 helices together with the connecting linkers. HPP-1 mutations are indicated in red; MHS mutations are shown in yellow. The location of other positive charges in the S4 domains is indicated as black circles (plus sign)
A fresh perspective for a unified hypothesis for HPP pathophysiology came from several independent observations.
First, even normal skeletal muscle cells are known to show a bistable membrane behavior. Initial lowering of extracellular K+ (K ex) hyperpolarizes, but further lowering (usually to below 1 mM in normal muscle) then abruptly (and paradoxically) depolarizes the sarcolemmal membrane to about -50 to -60 mV [79]. This behavior reflects the existence of two stable resting membrane potentials (V R), one near the K+-equilibrium potential (around -80 mV) and one around -50 to -60 mV resulting from two opposing conductances: a Ba2+-sensitive inward rectifier K+-current (which determines the more negative V R) and a linear, non-selective leak inward current. With decreasing K ex, first hyperpolarization occurs as expected from the Nernst equation, but with the inward rectifier conductance declining the hyperpolarizing K+-current will become smaller than the depolarizing leak current with decreasing K ex. V R is then uncoupled from the K+-equilibrium potential and becomes more depolarized. Accordingly, the sensitivity of this paradoxical depolarization to K ex-lowering (i.e., a shift to higher K ex) is increased by either blocking the inward rectifier K+-current (e.g., by Ba2+) or by enhancing the depolarizing leak currents. Indeed, HPP muscle fibers are more susceptible to K+-lowering than normal muscle [41]. Since K+-channels are not mutated in HPP-1 or HPP-2, the only possibility is that mutations observed in the pore-forming subunits of Cav1.1 α1 or Nav1.4 α somehow increase leak current.
Second, a large number of Nav1.4 α-subunit point mutations, also outside of the S4 helices, are known to cause different muscle channelopathies (for review, see [39]) but as in Cav1.1 α1 for HPP-1, only neutralizing mutations in S4 arginines cause HPP-2. This strongly pointed to a specific role of these residues but it was unclear how the voltage-sensing domains of two different ion channels with different ion selectivity could account for the paradoxical depolarization associated with low K ex.
The third and intriguing finding was that mutations of S4 arginines in Shaker K+-channels can create a pore in the voltage-sensing domain independent of the main K+-selective pore. This new pore can selectively conduct protons when mutated to histidine [73] or other cations when mutated to non-charged amino acids [81]. It was termed ω-current or gating pore current. Gating of this pore is voltage-dependent because the position of the S4 arginines strongly depends on the position of the S4 helix which moves during gating (Fig. 2). Mutating the outermost arginine appears to create a pore in the closed state (Fig. 2a, b) that gets plugged by an inner arginine [74], once the S4 moves outward and tilts upon depolarization (Fig. 2c). An opposite voltage dependence would be expected for a mutation of arginines further inside S4, such as arginine in position 3 (Fig. 2d). The finding that a single residue could transform the voltage-sensing domain into a pore was further strengthened by the fact that the voltage-gated proton channel Hv1 contains the typical four transmembrane segments S1–S4 of a voltage-sensing domain but lacks the two transmembrane segments that form the classical pore domain in other voltage-gated channels [82]. Together, these observations paved the way for studies on HPP-2 and HPP-1, demonstrating that these mutations indeed induced a gating pore current which represents the depolarizing conductance predicted from the susceptibility to “paradoxical” depolarization. For Nav1.4 mutations, this could be directly shown from recordings in heterologous expression systems [70]. As mentioned above, heterologous expression is more difficult with Cav1.1. However, in a series of elegant experiments in myofibers from HPP-1 patients with R528H and R1239H Cav1.1 mutations, Jurkat-Rott and colleagues [41] measured a non-selective cation leak of 12–19.5 µS/cm from steady-state current density–voltage relationships, consistent with the assumption that the Cav1.1 α1 mutations also induce gating pore currents. This may also explain the high intracellular Na+ concentrations found in the muscle of these patients in vivo and in vitro [41]. However, these experiments do not allow predictions about the cation selectivity of the Cav1.1 α1 mutations, especially because Cav1.1 α1 mutations to histidines are expected to conduct only protons, as shown for corresponding arginine mutations in Nav1.4 and Shaker K+ channels.
Fig. 2.
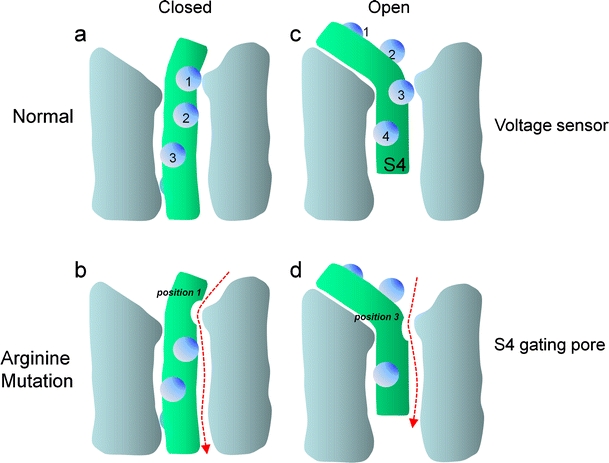
Simplified scheme illustrating the membrane potential-dependent conformations of the voltage sensor: only one of the four voltage-sensing domains is illustrated. S4 helices are shown in green, positively charged residues (mostly arginines) as blue spheres. In the closed state, the positively charged S4 helix is pulled inside by the negative resting potential. The outermost arginine residue (1) interacts with residues of other helices forming the voltage-sensing domain (e.g., a key negative charge in S2; [70]) (a). In Shaker K+, Cav1.1, or Nav1.4 channels, a mutation of arginine in position 1 (1) to an uncharged residue (e.g., serine or glycine) opens a new permeation pathway (arrow) as long as the channel is in the closed state (b). Upon depolarization, the S4 helix is driven outward, rotates, and its extracellular portion tilts (c). This movement shifts the arginine in position (3) outward and would close the gating pore induced by a mutation in position 1. The mechanism can account for the depolarizing current observed in muscle cells from HPP-1 patients carrying the Cav1.1 α1 subunit mutations in S4 helices illustrated in Fig. 1 (HPP-1) or analogous mutations in Nav1.4 (HPP-2, not illustrated, [51]). Conversely, whenever the sensor is in the open state, mutation of an arginine in position 3 (3) would enable a gating pore current (d), which would be closed upon repolarization by inward movement of arginine 1. Such a mechanism can explain the depolarization-activated gating pore current conducted by mutant Nav1.4 channels in potassium-sensitive normokalemic periodic paralysis [70]
The HPP-1 mutations currently known are illustrated in Fig. 1. Two additional mutations affecting the first and second arginine in S4 of domain III (R897S, R900S) were discovered more recently [51] and are in agreement with the gating pore current theory. The first mutation not affecting a S4 arginine, V876E, was reported in a HPP-1 family in South America [42]. V876E is located within the transmembrane helix S3 and replaces a hydrophobic residue by a negative charge. S3 helices are located close to the S4 helix in different models of voltage-gated cation channels [90] and help to stabilize the S4 helix. Upon activation, the S4 helix moves outward, rotates clockwise, and its extracellular end tilts away from the pore axis (Fig. 2). Although the relative movements of the adjacent S1–S3 helices with respect to S4 are a matter of debate [90], the negative charges in these helices (including S3) were shown to form salt bridges with the S4 positive charges, and these interactions change dynamically upon gating-induced S4 movements (as shown, e.g., for a “sliding helix model” [90]). Therefore, a negative charge in the S3 helix is likely to disturb this delicate network of charges. It is possible that this leads to conformational changes that create an ion pore within the voltage sensor. Although this hypothesis needs to be addressed in future studies, the location of this mutation outside S4 is not a priori contradicting the gating pore concept underlying HPP pathophysiology.
Malignant hyperthermia susceptibility
Malignant hyperthermia (MH) is a potentially lethal autosomal dominant disorder with susceptibility of otherwise healthy individuals to severe adverse reactions to volatile anesthetics (e.g., halothane) or depolarizing muscle relaxants. Exposure to these drugs can quickly lead to skeletal muscle hypermetabolism resulting from an uncontrolled increase in the concentration of free myoplasmic Ca2+ released from the SR Ca2+ stores [40]. This state results in skeletal muscle contractures with adenosine triphosphate-depletion, excessive activation of glycogenolysis and cell metabolism, hypercapnia, hypoxemia and lactic acid acidosis, and an increase in body temperature. Rhabdomyolysis occurs with subsequent creatine kinase elevation, hyperkalemia, cardiac arrhythmias, myoglobinuria, and the possibility of renal failure. Treatment of a crisis by early administration of dantrolene, an inhibitor of SR Ca2+ release, substantially reduces mortality. Anesthesia-induced MH incidence is estimated to about 1:10,000. However, the true prevalence must be higher because the clinical penetrance is low. The skeletal muscle ryanodine receptor RyR1 gene (RYR1) has been identified as the primary MHS locus and there are about 180 missense mutations described across RYR1 that co-segregate with MHS [12]. Several alternative loci have also been proposed, but so far, only the Cav1.1 α1 subunit gene (CACNA1S) has been identified as an additional causative gene. HPP-1 and MHS can therefore be considered allelic diseases. The Cav1.1 α1 mutations associated with MHS are located in the cytoplasmic linker between repeats III and IV (R1086H, R1086C [54]) or replace the innermost arginine in S4 of repeat I (Fig. 1). Because Cav1.1 mainly serves as the voltage sensor of RyR1 rather than a Ca2+ channel (see above), these mutations may alter the voltage-dependent signaling between these two Ca2+ channels. In a porcine model of MHS (RyR1 point mutation), the typical increased sensitivity to a broad range of pharmacological stimuli was accompanied by a lower threshold for SR Ca2+ release and contraction [24]. The fast depolarization-induced conformational changes of Cav1.1 α1 subunits (also termed dihydropyridine receptors, DHPRs, in muscle) mechanically activate RyR1 and elicit SR Ca2+ release. In addition to this orthograde coupling, there is also a retrograde signaling because the activity of DHPRs is strongly influenced by its RyR1 interaction. Both forms of coupling are mediated through a “critical domain” in the cytoplasmic II–III linker [26]. Obviously, measurements of MHS mutation-induced effects on Cav1.1-mediated ion currents appear of limited value. Instead, the functional coupling needs to be studied, which requires introduction of the mutated channels into a skeletal muscle environment. This can either be achieved by homologous expression of mutant constructs in cultured muscle cells devoid of Cav1.1 α1 subunits or by engineering of MHS mutations into the CACNA1S gene in mice. Muscle cells can then be isolated to monitor changes of Cav1.1-mediated excitation–contraction coupling. Cav1.1-deficient skeletal muscle myotubes were successfully used to demonstrate that the Cav1.1 α1 R1086H mutation lowers the half-maximal voltage required for the induction of SR Ca2+ release by about 5 mV and enhances the sensitivity of SR release to caffeine [24], a drug that is used as a primary diagnostic measure for MHS. This finding is compatible with a mutation-induced facilitation of SR Ca2+ release by both pharmacologic (caffeine) and endogenous (voltage sensor) activators. Notably, a lower activation threshold for Ca2+ release was also found for RyR1 mutations, including a heterozygous RyR1 mutation in a MHS mouse model. Sensitization of Ca2+ release therefore appears as the unifying principle underlying susceptibility to MH. Given the strategically important location of the voltage sensor arginine, it is quite possible that the novel mutation R174W acts through the same pathophysiological mechanism.
Cav1.3 channelopathies (CACNA1D gene)
So far, no human diseases resulting from mutations in the CACNA1D gene encoding the Cav1.3 α1 subunit have been reported. This could be due to the fact that loss-of-function mutations cause no phenotype in the heterozygous state (as in mice) but are lethal in the homozygous state. However, spontaneous gain-of-function mutations may cause a clinical syndrome compatible with life. In the case of Cav1.2 (CACNA1C gene), such a scenario leads to Timothy syndrome (see article in this issue). Homozygous Cav1.2 knockout mice die during development before day 14.5 post-coitum which may be due to their prominent role in the cardiovascular system [65]. Like for Cav1.2, heterozygous Cav1.3 knockout mice were not distinguishable from wild type, suggesting that heterozygous loss-of-function mutations would also be clinically silent in humans. However, based on data from homozygous Cav1.3 knockout mice, it is very likely that complete loss of Cav1.3 function would not be lethal. Homozygous Cav1.3 knockouts are viable and have been successfully used to establish the role of this LTCC isoform for physiology (for review, see [77]). If Cav1.3 serves a similar role in humans, this mouse model predicts no clinical symptoms in heterozygous patients but congenital hearing impairment and sinoatrial node dysfunction in homozygous individuals. Sinoatrial node dysfunction is unlikely to be lethal because the bradycardia and sinoatrial node arrhythmia observed in Cav1.3 knockout mice are pronounced at rest and largely disappear during exercise. Such a syndrome may therefore be rare and present mainly in consanguineous deafness families.
Cav1.4 channelopathies (CACNA1F gene)
Incomplete congenital stationary night blindness type 2
Incomplete congenital stationary night blindness type 2 (CSNB2) is an X-linked form of congenital stationary night blindness which is caused by mutations in the voltage-gated calcium-channel gene CACNA1F encoding Cav1.4 LTCCs (OMIM: 300110). CSNB2 is characterized by variable and usually mild clinical symptoms. The term is, however, misleading because night blindness may not be the major complaint, unlike in the complete form of stationary night blindness (CSNB1) which is caused by different genetic defects either in the nyctalopin (OMIM: 300278) or the metabotropic glutamate receptor-6 (OMIM: 604096). Typical symptoms in CSNB2 are moderately low visual acuity, myopia, nystagmus, and variable levels of night blindness, but one or more of these symptoms may be absent [6]. The eye fundus is normal but electroretinograms (ERGs) are abnormal [83]. CSNB2 patients show a very abnormal dim scotopic ERG and a typical negative bright-flash ERG which has large a-waves, but severely reduced b-waves. Oscillatory potentials are also missing [83]. The ERG data are compatible with a defect in neurotransmission within the retina between photoreceptors and second-order neurons [83]. LTCCs are the predominant channels controlling neurotransmitter secretion at the ribbon synapses of retinal photoreceptors (see references in [44]) and of cochlear inner hair cells [62]. These cell types show “tonic” neurotransmitter release in response to graded changes in the membrane potential, unlike in most other fast, chemical synapses in which non-LTCCs (such as Cav2.1 and Cav2.2) trigger neurotransmitter release during bursts of short action potentials (“phasic release”) [14]. In the dark, photoreceptors depolarize to a resting membrane potential of -36 to -40 mV [17], enhancing tonic release. Light absorption in the photoreceptor outer segments and closure of cyclic guanosine monophosphate (cGMP)-gated cation channels hyperpolarizes the cells to below -55 mV [86]. Release occurs at so-called ribbon-type synapses where Ca2+ channels appear clustered. To support tonic release, retinal Ca2+ channels must activate rapidly at relatively negative voltages (below -40 mV) and inactivate slowly [63]. Identification of the genetic defect responsible for CSNB2 led to the discovery of a novel Ca2+ channel α1 subunit, Cav1.4 (see references in [44]), which carries the disease-related mutations, and is preferentially expressed in retinal synapses [5, 16]. It took several years until cloned Cav1.4 channel complexes could be functionally expressed in mammalian cells [44] to investigate their functional and pharmacological properties [4, 19, 20, 44, 53, 58, 59]. Similar to photoreceptor Ca2+ currents, recombinant Cav1.4 currents in cultured mammalian cells activate rapidly and inactivate very slowly during depolarizing pulses. Interestingly, this was due to a very slow voltage-dependent inactivation accompanied by complete absence of so-called calcium-dependent inactivation (CDI) [44]. CDI is considered an important negative feedback mechanism that protects cells from excess Ca2+ influx [1]. Similar to Cav1.3, Cav1.4 channels open at more negative membrane potentials than Cav1.2 [44], allowing the channel to conduct Ca2+ at potentials negative to -40 mV. Together, inactivation and activation characteristics of Cav1.4 channels reveal a substantial window current, which permits ion influx under constant depolarized conditions. Peloquin and colleagues observed that at near physiological temperatures, inactivation kinetics is accelerated but the window current is still preserved [58]. These biophysical properties make them ideally suited for tonic glutamate release from photoreceptor terminals. Cav1.4 α1 subunits are expressed at release sites of mammalian photoreceptors in the outer plexiform layer [3, 16] and channel loss-of-function would therefore be expected to decrease photoreceptor neurotransmitter release capacity, impair signaling to second-order retinal neurons, and thus explain the ERG abnormalities in CSNB2. Cav1.4 may also contribute to the LTCC currents measured in bipolar cell terminals, explaining punctate Cav1.4 α1 immunostaining in the mouse inner plexiform layer [5].
So far, more than 40 structural aberrations were identified in the Cav1.4 α1 subunit gene of CSNB2 patients (Fig. 3). Most of them are predicted to cause severe structural changes, such as truncated α1 subunits, unlikely to support significant channel activity. Moreover, pre-mature stop codons in regions followed by splice sites at a distance of 50–55 nucleotides downstream-yield mRNAs should be eliminated by nonsense-mediated mRNA decay [48] and thus might not even lead to expression of the truncated Cav1.4 α1 subunit protein. Due to the X-linked condition, CSNB2 results in a complete loss of Cav1.4 channel function only in affected males. However, some missense mutations are unlikely to lead to a complete loss-of-channel function (Fig. 3). Hoda et al. [32] characterized a mutation G369D in the pore-lining region of segment IS6 that caused pronounced changes of the channel's inactivation gating and also shifted the V 0.5,act to more negative voltages compatible with an overall Cav1.4 channel gain-of-function. Furthermore, ion selectivity was affected, suggesting that the negative charge introduced by the G369D mutation at the cytoplasmic side of IS6 not only affects conformational changes associated with channel activation but also interferes with cation permeation through the pore. Interestingly, G369 corresponds to G402 in Cav1.2 α1, which is mutated to serine in some patients with Timothy syndrome [71] and strongly inhibits voltage-dependent inactivation (VDI). In Cav1.2, VDI is also inhibited by mutation of nearby residues such as a serine residue important for slow inactivation in IS6 and G406 in Timothy syndrome (G406R) [72]. Obviously, channelopathies in different LTCC α1 subunits have identified a region forming a critical “hotspot” for channel gating.
Fig. 3.
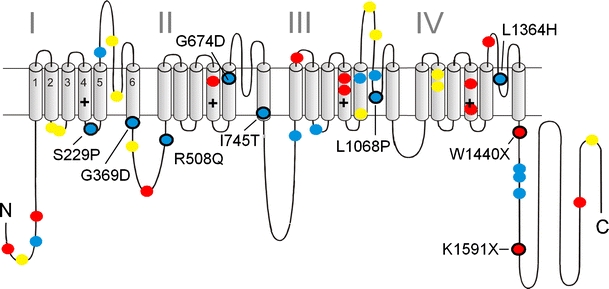
Mutations in Ca2+ channel Cav1.4 α1 subunits identified in patients with CSNB2: a folding model of α1 subunits based on hydrophobicity analysis is shown. Plus sign indicates several positive charges within the transmembrane S4 helices within the hydrophobic repeats I–IV. Position of CSNB2 mutations is indicated. Colors indicate the predicted structural changes: blue, single missense mutations; yellow, in-frame amino acid deletions or insertions; red, truncated protein due to single mutations that introduce stop codons. Black circles refer to mutations that are functionally characterized [31–33, 53, 59, 67]
Another gain-of-function mutation was discovered in a New Zealand family showing a similar but more severe clinical phenotype than in CSNB2. The missense mutation I745T in the pore helix IIS6 produced a remarkable -30-mV shift in the voltage dependence of Cav1.4 channel activation as well as significantly slower inactivation kinetics when expressed in tsA-201 cells [31]. This observation triggered a detailed analysis of the role of the equivalent residue in Cav1.2 for channel gating [34], indicating that substitution of this residue destabilizes the closed and favor the open conformation of the pore. Molecular dynamics simulations suggest that this may also involve mutation-induced conformational alterations of other interacting transmembrane segments [75, 76].
In contrast, no channel activity could be measured for mutants S229P and W1440X after expression in Xenopus oocytes, and mutant L1068P yielded currents only in the presence of the channel activator BayK8644 [32]. Mutations S229P, G369D, and L1068P α1 subunits were expressed at levels indistinguishable from wild-type channels, but no protein was detected for the truncation mutation W1440X after expression in tsA-201 cells [32]. Two other missense mutations, R508Q and L1364H, reduced protein expression in transfected tsA-201 cells, which may, although not yet proven, also decrease retinal Ca2+ current density [33]. However, McRory et al. found that two missense mutations, G674D and A928D, and the W1459X truncation mutation in the C-terminus exerted no detectable changes in the activation, inactivation, or conductance properties of expressed Cav1.4 channels. For the mutation G369D, they only found a slight, but statistically significant increase in the slope factor of the activation curve and a less pronounced shift of the half-activation potential with Ca2+ as compared to Ba2+ as charge carrier. This discrepant finding might be explained by the fact that their Cav1.4 α1 subunit [44] differed in four amino acid positions from the human Cav1.4 α1 subunits used by Hoda et al. [53]. This also includes neutralization of a negative charge in the IS6 helix which may by required to “sense” the additional negative charge introduced by the G369D mutation. The possibility that the mutations affect Cav1.4 α1 protein expression has not been tested in their study.
Clinical CSNB2 symptoms might therefore result not only from complete loss of function and/or decreased expression of mutant channels with unchanged gating behavior but also from gating changes including a channel gain-of-function. The gain-of-function mutations should promote Ca2+ entry through the channel raising the important question about how increased channel function could impair light-induced signaling between photoreceptors and second-order neurons. One possible interpretation is as follows. Because the half-maximal voltage of activation for retinal LTCCs (and Cav1.4) [44, 53] is clearly above -40 mV [17, 85], the retinal operating range of membrane potential changes is at the “foot” of the LTCC activation curve and thus Ca2+-influx becomes very small or not measurable [86] at hyperpolarized voltages (e.g., -55 mV, Fig. 4) during illumination. From the activation curve, an about 50-fold increase of Cav1.4 inward current can be predicted upon depolarization to -35 mV. A pronounced negative shift of the activation curve by a CSNB2 mutation would result in a significant increase of Ca2+ influx during illumination at negative voltages, but at the same time, would reduce the increase upon depolarization, leading to a reduced dynamic range (Fig. 4). The corresponding change in the dynamic range of tonic glutamate release could then explain how the synaptic gain between first- and second-order neurons is reduced in CSNB2 retinas.
Fig. 4.
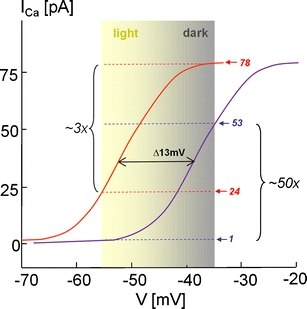
Functional CSNB2 mutations in Cav1.4 α1 cause a decreased dynamic range of photoreceptor signaling: the operation range of photoreceptors (between -35 mV (dark) and approximately -55 mV (light) is near the foot of the I Ca activation curve at physiological Ca2+ concentrations to ensure Ca2+ influx necessary for tonic glutamate release (see also text). A hyperpolarizing shift of the current–voltage relationship (I–V) is predicted to result in higher glutamate release at a given illumination level, causing a decreased dynamic range of photoreceptor signaling (here shown for mutation K1591X). According to the L-type current I–V relationship measured in photoreceptors (black curve [80]), a 13-mV hyperpolarizing shift of the I Ca I–V relationship as observed for K1591X [67] would predict a smaller increase of I Ca and exocytosis (predicted: normal ∼50-fold, K1591X ∼3-fold) when moving from the light (-55 mV) to the dark membrane potential (-35 mV)
In addition to CACANA1F, mutations in other genes can also cause incomplete forms of CSNB. Ca2+-binding protein 4 (CaBP4) belongs to a protein family structurally similar to calmodulin (CaM). It is specifically found in photoreceptor synaptic terminals [29], modulates Cav1.4 Ca2+ channels by binding to the C-terminus [29], and the phenotype of CaBP-/- mice shares similarities with that of CSNB2 patients [29]. It therefore appeared as a disease candidate in CSNB2 patients without CACNA1F mutations. Zeitz and colleagues indeed found mutations in CaBP4 that account for an autosomal recessive form of CSNB2.
A homozygous nonsense mutation in the human gene for the accessory Ca2+ channel α2-δ4-subunit (CACNA2D4) was also found in patients with an electronegative electroretinogram and an initial diagnosis of night blindness [88]. Detailed clinical examination finally revealed a mild form of cone dystrophy. In mice, a protein-truncating frameshift of this subunit leads to abnormal electroretinograms, a reduction in the photoreceptor synaptic layer and a profound loss of synaptic ribbons between rods and rod bipolar cells [64, 87]. This emphasizes a key role of this accessory subunit for normal retinal function in humans and mice.
A truncating CSNB2 mutation reveals an intrinsic gating modulator in Cav1.4
Upon functional characterization of the CSNB2 C-terminal truncation mutant K1591X, Singh et al. [67] recently discovered that the absence of CDI in Cav1.4 channels is due to its active suppression by a C-terminal inhibitory domain. Like other VGCCs (such as Cav1.2 and Cav1.3) Cav1.4 channels are capable of undergoing robust CDI in a CaM-dependent manner [67] when this inhibitory domain is removed. In wild-type Cav1.4, this intrinsic gating modulator resides within the C-terminal tail downstream of an IQ domain which is required for CaM binding (Figs. 5 and 6). K1591X channels lack this modulator and therefore exhibit fast CaM-dependent CDI and a more negative activation voltage range than the wild type. These findings [67, 27, 84] revealed inhibition of CDI as a novel modulatory concept that contributes to the fine-tuning of Cav1.4 gating to prevent inactivation and thus support tonic neurotransmitter release in sensory cells and normal visual function in humans. The molecular basis of this modulatory mechanism itself is discussed controversially. Wahl-Schott and colleagues postulated binding of the distal C-terminus (termed ICDI, inhibitor of CDI, in their publication) to the EF hand motif in the proximal C-terminus, thereby, uncoupling the EF hand from the Ca2+ sensing apparatus. Based on their co-immunoprecipitation studies, loss of CaM-interaction with the C-terminus as underlying mechanism was excluded [84]. Instead, Singh and colleagues [67] postulated that the distal C-terminus (ICDI) binds to a segment comprising the EF hand, the pre-IQ and the IQ regions (Fig. 5a). In addition, their functional experiments also suggested a role for the post-IQ domain. Notably, they found that deletion of the C-terminal domain not only restored robust CDI but also induced a strong hyperpolarizing shift of the voltage dependence of Cav1.4 activation [67]. Therefore, they termed this domain “C-terminal modulator” (CTM) instead of ICDI, emphasizing this additional regulatory effect. Protein–protein interactions of C-terminal channel fragments and CaM expressed in HEK-293 cells measured using fluorescence resonance energy transfer (FRET), revealed that at resting calcium concentrations, apo-CaM binds to a C-terminal fragment containing the known CaM binding domains identified previously in other L-type Ca2+ channels (pre-IQ, IQ domains; [23, 60, 94]). Calcification of CaM at higher Ca2+ concentrations further stimulated CaM binding. In contrast, when the complete C-terminus was expressed (also containing the CTM) no apo-CaM binding occurred at resting Ca2+ concentrations (Fig. 5b) but was restored at higher Ca2+ concentrations, suggesting that the CTM modulates pre-association of CaM with the C-terminus. This could explain the lack of CDI in the wild-type Cav1.4 channel. By generation of different Cav1.4 truncation mutants, the critical residues comprising the CTM (and ICDI) were restricted to a stretch of about 25 amino acid residues within the distal C-terminus, which is highly conserved between Cav1.4, Cav1.3, and Cav1.2 (Fig. 6). Further FRET data were recently reported by the Biel group [27], which support the hypothesis that motifs further downstream of the EF hand are important for the intramolecular interaction in the Cav1.4 α1 C-terminus. More recently, David Yue's group confirmed the interference of the CTM with apoCaM binding. They provided evidence for a competitive mechanism in which CTM reduces the apparent affinity for apoCaM for the channel [46]. As the concentration of the CTM remains constant, the channel occupancy by apoCaM (and therefore CDI) becomes a function of the intracellular concentration of CaM.
Fig. 5.
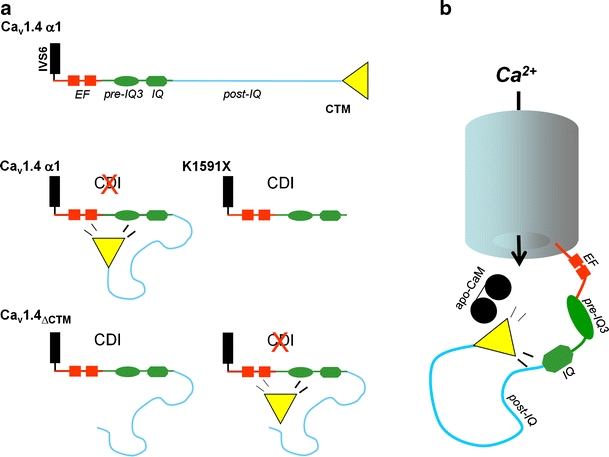
Hypothetical model of Cav1.4 C-terminal modulation. a Motifs previously demonstrated to be important for CaM modulation of other Ca2+ isoforms (red: EF hand; green: pre-IQ regions, IQ domain) are illustrated. In wild-type Cav1.4 channels, the CTM predominantly interacts with a region comprising the EF hand, pre-IQ, and IQ domains and thereby inhibits CDI [67]. The CTM and the post-IQ motif (light blue) are missing in truncation mutant K1591X and therefore intrinsic CDI of Cav1.4 becomes apparent. CDI is present after deletion of the last 122 residues which comprises the CTM. When co-expressed with the truncated channel  , the CTM-peptide inhibits CDI and restores wild-type gating properties. This modulation requires the presence of the post-IQ region. In addition, Singh et al. imply a role of the post-IQ motif for voltage-dependent inactivation [67]. b As shown in FRET experiments [70], the Cav1.4 CTM interferes with CaM binding to one or more sites responsible for CaM pre-association (apo-CaM) in intact cells. Therefore, interference with CaM coordination is suggested, the likely mechanism explaining the inhibition of CDI
, the CTM-peptide inhibits CDI and restores wild-type gating properties. This modulation requires the presence of the post-IQ region. In addition, Singh et al. imply a role of the post-IQ motif for voltage-dependent inactivation [67]. b As shown in FRET experiments [70], the Cav1.4 CTM interferes with CaM binding to one or more sites responsible for CaM pre-association (apo-CaM) in intact cells. Therefore, interference with CaM coordination is suggested, the likely mechanism explaining the inhibition of CDI
Fig. 6.

Sequence alignment of C-terminal tails of human Cav1.3 and Cav1.4 L-type channels: a sequence alignment of human Cav1.3 (Genbank accession number EU363339) and Cav1.4 (Genbank accession number AJ224874) α1 subunits is shown. Sequence identity (blue) and gaps (-) are indicated. Regions previously shown to be important for channel modulation by CaM in other voltage-gated Ca2+ channel isoforms are depicted (EF hand, pre-IQ, and IQ domain). The position of long and short Cav1.3 channels is indicated by black arrows (Cav1.3L and Cav1.3S, respectively). Position of the Cav1.4 CTM is given in yellow; - indicates residues absent in this sequence
CSNB2 mutations reveal an intrinsic gating modulator in Cav1.3
Cav1.4 α1 subunit mutations have provided valuable insight into the molecular mechanisms underlying the regulation not only of Cav1.4 but also Cav1.3 LTCCs. Given the high sequence homology in the C-terminus of LTCCs (Fig. 6), channel modulation by an intramolecular C-terminal protein–protein interaction may represent a general regulatory concept of LTCCs not limited to Cav1.4. Notably, alternative splicing in exon 42 in the C-terminus of Cav1.3 channels gives rise to naturally occurring channels with different lengths [35, 66]. Singh and colleagues [66] exploited the presence of a Cav1.3 CTM by functionally investigating the two human Cav1.3 α1 subunit splice variants. Similar to the Cav1.4 truncation mutant K1591X, the short splice form terminates shortly after the IQ motif, and therefore, also lacks the conserved region forming the CTM (Fig. 6). Indeed, the existence of a C-terminal modulation in human Cav1.3 is manifested by the pronounced gating differences between the long and short splice variant. This revealed an exciting novel mechanism by which Cav1.3 channel activity can be adjusted by splicing. Like Cav1.4 K1491X, the absence of the CTM in the short splice form led to Cav1.3 channels that activate and inactivate at lower voltages, resulting in a hyperpolarizing shift in the window current. Its stronger CDI also caused more pronounced inactivation of I Ca without affecting the voltage-dependent inactivation (VDI) time course. Interestingly, this regulation has not been reported for rat Cav1.3 analogs [89]. Many unique physiological functions of Cav1.3, including sensory and neuroendocrine cell signaling [49, 50, 62], pacemaking in neurons [57] and sinoatrial node cells [47], as well as its proposed role in the pathology of Parkinson's disease [15, 28] depend on the negative activation range and the amount of Ca2+ ions entering during plateau [57] or single action potentials [30]. Accordingly, the Cav1.3-CTM and factors that modify its activity (such as alternative splicing or interaction with other proteins [8, 43, 93]) appear as crucial determinants of electrical excitability. It can be predicted that the expression of short Cav1.3 channels would allow a cell to promote Ca2+ entry through Cav1.3 channels at sub-threshold voltages due to the more negative window current. Stronger activation at more negative voltages may also facilitate the onset of upstate potentials in neurons. Whereas negative activation of an even small Cav1.3 current could trigger pacemaking, faster CDI would limit Ca2+ entry during ensuing action potentials. This effect may be important in neurons which are susceptible to Ca2+ toxicity and neurodegeneration in Parkinson's disease [15]. In contrast, the CTM in the long Cav1.3 channels may be required for longer lasting Ca2+ signals triggered by stronger depolarization inducing cyclic adenosin monophosphate response element binding protein (CREB) phosphorylation and synaptic plasticity [92], or in sensory cells with tonic neurotransmitter release, such as cochlear inner hair cells or photoreceptors [62, 91] .
Non L-type Ca2+ channelopathies leading to altered LTCC function
Brain LTCCs are mainly located at somatodendritic locations. Rather than contributing to fast neurotransmitter release at nerve terminals, their somatodendritic Ca2+ signals play a major role in coupling synaptic activity to gene-transcription through different intracellular signaling pathways (for review, see [18]). These properties allow them to contribute to synaptic plasticity and control neuronal functions of pharmacotherapeutic relevance, including drug taking behavior, mood behavior, and fear memory (for reviews, see [18, 78]). Due to this special role, the question arises whether pathological changes in other (i.e., non-L-type) Ca2+ channel isoforms [14] can lead to secondary changes in LTCC expression and thereby allow them to contribute to disease-related processes. This question has already been addressed in tottering mice, a natural mouse mutant. The tottering phenotype, an autosomal recessive mouse disease, is associated with mild ataxia, spontaneous behavioral arrest associated with synchronous, bilateral cortical polyspike discharges (resembling human absence epilepsy), and attacks of paroxysmal dystonia [10, 61]. A missense mutation (P601L, IIS5-S6 pore-loop) in the Cav2.1 α1 subunit (forming P/Q-type Ca2+ channels, [14]) was found to underlie this phenotype (for review, see [61]). Interestingly, the paroxysmal dystonic symptoms, which can be reproducibly triggered, e.g., by immobilization stress, are prevented by subcutaneous or intracerebroventricular injection of different chemical classes of LTCC blockers, whereas ataxia is not ameliorated [10]. In accordance with these findings, dystonic episodes in tottering are also triggered by the LTCC activator Bay K8644 at doses not affecting wild-type mice [10]. Biochemical studies revealed significant upregulation of Cav1.2 α1 subunits in tottering brains. Enhanced expression is mainly restricted to cerebellar Purkinje cells, suggesting that LTCCs in these cells can mediate episodic dystonia. This finding is surprising because LTCC expression in these neurons is very low, thus mediating only about 7% of the total Ca2+ channel current [21, 38]. L-type currents increased by 2.2-fold were recorded from tottering Purkinje cells already at early postnatal stages (P15), indicating developmental changes preceding the appearance of behavioral deficits [22]. Interestingly, Cav2.1-deficient mice, which also develop severe dystonia, show an increased contribution of L-type currents in Purkinje but not in cerebellar granule cells [38]. Somehow, altered Cav2.1 channel signaling appears to activate pathways that enhance Cav1.2 (but not Cav1.3; [38]) LTCC expression. The finding that enhanced LTCC expression and most likely activity in Purkinje cells contributes to the paroxysmal dystonia of the tottering phenotype is in good agreement with the observation that dystonic episodes lead to neuronal activation in the cerebellum and its relay nuclei in these mice [9], and that the dystonic phenotype is absent in tottering mice lacking Purkinje cells [11].
Further support for an isoform-specific role of Cav1.2 LTCCs in the induction of dystonic behavior comes from a mouse mutant in that a single targeted mutation within the dihydropyridine binding pocket eliminates BayK 8644 sensitivity but causes no detectable changes in Cav1.2 function and expression [68]. These mice are completely resistant to the typical BayK 8644-induced dystonic behavior observed in wild-type mice [37], indicating that this drug effect cannot be mediated by Cav1.3 activation alone but requires Cav1.2 [68].
Taken together, these findings are an important first step to address the general question about the role of LTCCs for the pathophysiology of paroxysmal dyskinesias. As demonstrated here, dysregulation of these channels, even in neurons where they only contribute marginally to total Ca2+ channel currents, can be relevant for disease.
Conclusions
So far, Ca2+ channelopathies have been described for Cav1.1, Cav1.2, and Cav1.4, but not yet for Cav1.3 LTCCs. Cav1.1 mutations associated with HPP-1 have provided valuable insight into the function of the voltage-sensing domains of voltage-gated Ca2+ channels and their dual role as voltage sensors and ion pores. Although the molecular details of how α1 mutations sensitize excitation–contraction coupling between plasmalemmal Cav1.1 and SR RyR1 in skeletal muscle, and thereby cause susceptibility to MH, are not yet fully understood, they point to functionally critical regions in α1 which were not detected in previous mutational studies investigating the orthograde coupling between these two ion channels. Finally, Cav1.4 mutations led to the discovery of a novel intramolecular protein interaction by which LTCCs modulate their gating behavior. This opened a new field of research also on Cav1.3 channels, which use this mechanism to adjust their activity by intracellular Ca2+ activity and alternative splicing. Given their delicate role in the pathophysiology of Parkinson's disease, this mechanism may also become a target for the development of novel therapies.
Acknowledgements
The work of the authors is supported by the Austrian Science Fund (P20670), the European Community (MRTN-CT-2006-035367), and the University of Innsbruck.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Alseikhan BA, DeMaria CD, Colecraft HM, Yue DT. Engineered calmodulins reveal the unexpected eminence of Ca2+ channel inactivation in controlling heart excitation. Proc Natl Acad Sci U S A. 2002;99:17185–17190. doi: 10.1073/pnas.262372999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Badou A, Jha MK, Matza D, Mehal WZ, Freichel M, Flockerzi V, Flavell RA. Critical role for the beta regulatory subunits of Cav channels in T lymphocyte function. Proc Natl Acad Sci U S A. 2006;103:15529–15534. doi: 10.1073/pnas.0607262103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ball SL, Gregg RG. Using mutant mice to study the role of voltage-gated calcium channels in the retina. Adv Exp Med Biol. 2002;514:439–450. doi: 10.1007/978-1-4615-0121-3_26. [DOI] [PubMed] [Google Scholar]
- 4.Baumann L, Gerstner A, Zong X, Biel M, Wahl-Schott C. Functional characterization of the L-type Ca2+ channel Cav1.4α1 from mouse retina. Invest Ophthalmol Vis Sci. 2004;45:708–713. doi: 10.1167/iovs.03-0937. [DOI] [PubMed] [Google Scholar]
- 5.Berntson A, Taylor WR, Morgans CW. Molecular identity, synaptic localization, and physiology of calcium channels in retinal bipolar cells. J Neurosci Res. 2003;71:146–151. doi: 10.1002/jnr.10459. [DOI] [PubMed] [Google Scholar]
- 6.Boycott KM, Pearce WG, Bech-Hansen NT. Clinical variability among patients with incomplete X-linked congenital stationary night blindness and a founder mutation in CACNA1F. Can J Ophthalmol. 2000;35:204–213. doi: 10.1016/s0008-4182(00)80031-9. [DOI] [PubMed] [Google Scholar]
- 7.Caciotti A, Morrone A, Domenici R, Donati MA, Zammarchi E. Severe prognosis in a large family with hypokalemic periodic paralysis. Muscle Nerve. 2003;27:165–169. doi: 10.1002/mus.10298. [DOI] [PubMed] [Google Scholar]
- 8.Calin-Jageman I, Yu K, Hall RA, Mei L, Lee A. Erbin enhances voltage-dependent facilitation of Cav1.3 Ca2+ channels through relief of an autoinhibitory domain in the Cav1.3 α1 subunit. J Neurosci. 2007;27:1374–1385. doi: 10.1523/JNEUROSCI.5191-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Campbell DB, Hess EJ. Cerebellar circuitry is activated during convulsive episodes in the tottering (tg/tg) mutant mouse. Neuroscience. 1998;85:773–783. doi: 10.1016/S0306-4522(97)00672-6. [DOI] [PubMed] [Google Scholar]
- 10.Campbell DB, Hess EJ. L-type calcium channels contribute to the tottering mouse dystonic episodes. Mol Pharmacol. 1999;55:23–31. doi: 10.1124/mol.55.1.23. [DOI] [PubMed] [Google Scholar]
- 11.Campbell DB, North JB, Hess EJ. Tottering mouse motor dysfunction is abolished on the Purkinje cell degeneration (pcd) mutant background. Exp Neurol. 1999;160:268–278. doi: 10.1006/exnr.1999.7171. [DOI] [PubMed] [Google Scholar]
- 12.Carpenter D, Ringrose C, Leo V, Morris A, Robinson RL, Halsall PJ, Hopkins PM, Shaw MA. The role of CACNA1S in predisposition to malignant hyperthermia. BMC Med Genet. 2009;10:104. doi: 10.1186/1471-2350-10-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Catterall WA, Goldin AL, Waxman SG. International union of pharmacology. XXXIX. Compendium of voltage-gated ion channels: sodium channels. Pharmacol Rev. 2003;55:575–578. doi: 10.1124/pr.55.4.7. [DOI] [PubMed] [Google Scholar]
- 14.Catterall WA, Perez-Reyes E, Snutch TP, Striessnig J. International union of pharmacology. XLVIII. Nomenclature and structure–function relationships of voltage-gated calcium channels. Pharmacol Rev. 2005;57:411–425. doi: 10.1124/pr.57.4.5. [DOI] [PubMed] [Google Scholar]
- 15.Chan CS, Guzman JN, Ilijic E, Mercer JN, Rick C, Tkatch T, Meredith GE, Surmeier DJ. ‘Rejuvenation’ protects neurons in mouse models of Parkinson's disease. Nature. 2007;447:1081–1086. doi: 10.1038/nature05865. [DOI] [PubMed] [Google Scholar]
- 16.Chang B, Heckenlively JR, Bayley PR, Brecha NC, Davisson MT, Hawes NL, Hirano AA, Hurd RE, Ikeda A, Johnson BA, McCall MA, Morgans CW, Nusinowitz S, Peachey NS, Rice DS, Vessey KA, Gregg RG. The nob2 mouse, a null mutation in Cacna1f: anatomical and functional abnormalities in the outer retina and their consequences on ganglion cell visual responses. Vis Neurosci. 2006;23:11–24. doi: 10.1017/S095252380623102X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Corey DP, Dubinsky JM, Schwartz EA. The calcium current in inner segments of rods from the salamander (Ambystoma tigrinum) retina. J Physiol. 1984;354:557–575. doi: 10.1113/jphysiol.1984.sp015393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deisseroth K, Mermelstein PG, Xia H, Tsien RW. Signaling from synapse to nucleus: the logic behind the mechanisms. Curr Opin Neurobiol. 2003;13:354–365. doi: 10.1016/S0959-4388(03)00076-X. [DOI] [PubMed] [Google Scholar]
- 19.Doering CJ, Hamid J, Simms B, McRory JE, Zamponi GW. Cav1.4 encodes a calcium channel with low open probability and unitary conductance. Biophys J. 2005;89:3042–3048. doi: 10.1529/biophysj.105.067124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doering CJ, Peloquin JB, McRory JE. The Cav1.4 calcium channel: more than meets the eye. Channels (Austin) 2007;1:3–10. [PubMed] [Google Scholar]
- 21.Dove LS, Abbott LC, Griffith WH. Whole-cell and single-channel analysis of P-type calcium currents in cerebellar Purkinje cells of leaner mutant mice. J Neurosci. 1998;18:7687–7699. doi: 10.1523/JNEUROSCI.18-19-07687.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Erickson MA, Haburcak M, Smukler L, Dunlap K. Altered functional expression of Purkinje cell calcium channels precedes motor dysfunction in tottering mice. Neuroscience. 2007;150:547–555. doi: 10.1016/j.neuroscience.2007.09.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Erickson MG, Liang H, Mori MX, Yue DT. FRET two-hybrid mapping reveals function and location of L-type Ca2+ channel CaM preassociation. Neuron. 2003;39:97–107. doi: 10.1016/S0896-6273(03)00395-7. [DOI] [PubMed] [Google Scholar]
- 24.Gallant EM, Lentz LR. Excitation–contraction coupling in pigs heterozygous for malignant hyperthermia. Am J Physiol. 1992;262:C422–C426. doi: 10.1152/ajpcell.1992.262.2.C422. [DOI] [PubMed] [Google Scholar]
- 25.Gamelli AE, McKinney BC, White JA, Murphy GG (2009) Deletion of the L-type calcium channel Cav1.3 but not Cv1.2 results in a diminished sAHP in mouse CA1 pyramidal neurons. Hippocampus 2009, Dec 15 (published online) [DOI] [PMC free article] [PubMed]
- 26.Grabner M, Dirksen RT, Suda N, Beam KG. The II–III loop of the skeletal muscle dihydropyridine receptor is responsible for the bi-directional coupling with the ryanodine receptor. J Biol Chem. 1999;274:21913–21919. doi: 10.1074/jbc.274.31.21913. [DOI] [PubMed] [Google Scholar]
- 27.Griessmeier K, Cuny H, Rotzer K, Griesbeck O, Harz H, Biel M, Wahl-Schott C. Calmodulin is a functional regulator of Cav1.4 L-type Ca2+ channels. J Biol Chem. 2009;284:29809–29816. doi: 10.1074/jbc.M109.048082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guzman JN, Sanchez-Padilla J, Chan CS, Surmeier DJ. Robust pacemaking in substantia nigra dopaminergic neurons. J Neurosci. 2009;29:11011–11019. doi: 10.1523/JNEUROSCI.2519-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haeseleer F, Imanishi Y, Maeda T, Possin DE, Maeda A, Lee A, Rieke F, Palczewski K. Essential role of Ca2+-binding protein 4, a Cav1.4 channel regulator, in photoreceptor synaptic function. Nat Neurosci. 2004;7:1079–1087. doi: 10.1038/nn1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Helton TD, Xu W, Lipscombe D. Neuronal L-type calcium channels open quickly and are inhibited slowly. J Neurosci. 2005;25:10247–10251. doi: 10.1523/JNEUROSCI.1089-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hemara-Wahanui A, Berjukow S, Hope CI, Dearden PK, Wu SB, Wilson-Wheeler J, Sharp DM, Lundon-Treweek P, Clover GM, Hoda JC, Striessnig J, Marksteiner R, Hering S, Maw MA. A CACNA1F mutation identified in an X-linked retinal disorder shifts the voltage dependence of Cav1.4 channel activation. Proc Natl Acad Sci U S A. 2005;102:7553–7558. doi: 10.1073/pnas.0501907102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoda JC, Zaghetto F, Koschak A, Striessnig J. Congenital stationary night blindness type 2 mutations S229P, G369D, L1068P, and W1440X alter channel gating or functional expression of Cav1.4 L-type Ca2+ channels. J Neurosci. 2005;25:252–259. doi: 10.1523/JNEUROSCI.3054-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoda JC, Zaghetto F, Singh A, Koschak A, Striessnig J. Effects of congenital stationary night blindness type 2 mutations R508Q and L1364H on Cav1.4 L-type Ca2+ channel function and expression. J Neurochem. 2006;96:1648–1658. doi: 10.1111/j.1471-4159.2006.03678.x. [DOI] [PubMed] [Google Scholar]
- 34.Hohaus A, Beyl S, Kudrnac M, Berjukow S, Timin EN, Marksteiner R, Maw MA, Hering S. Structural determinants of L-type channel activation in segment IIS6 revealed by a retinal disorder. J Biol Chem. 2005;280:38471–38477. doi: 10.1074/jbc.M507013200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hui A, Ellinor PT, Krizanova O, Wang JJ, Diebold RJ, Schwartz A. Molecular cloning of multiple subtypes of a novel rat brain isoform of the a1 subunit of the voltage-dependent calcium channel. Neuron. 1991;7:35–44. doi: 10.1016/0896-6273(91)90072-8. [DOI] [PubMed] [Google Scholar]
- 36.Jha MK, Badou A, Meissner M, McRory JE, Freichel M, Flockerzi V, Flavell RA. Defective survival of naive CD8+ T lymphocytes in the absence of the beta3 regulatory subunit of voltage-gated calcium channels. Nat Immunol. 2009;10:1275–1282. doi: 10.1038/ni.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jinnah HA, Sepkuty JP, Ho T, Yitta S, Drew T, Rothstein JD, Hess EJ. Calcium channel agonists and dystonia in the mouse. Mov Disord. 2000;15:542–551. doi: 10.1002/1531-8257(200005)15:3<542::AID-MDS1019>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 38.Jun K, Piedras-Renteria ES, Smith SM, Wheeler DB, Lee SB, Lee TG, Chin H, Adams ME, Scheller RH, Tsien RW, Shin HS. Ablation of P/Q-type Ca2+ channel currents, altered synaptic transmission, and progressive ataxia in mice lacking the alpha(1A)-subunit. Proc Natl Acad Sci U S A. 1999;96:15245–15250. doi: 10.1073/pnas.96.26.15245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jurkat-Rott K, Lehmann-Horn F. Muscle channelopathies and critical points in functional and genetic studies. J Clin Invest. 2005;115:2000–2009. doi: 10.1172/JCI25525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jurkat-Rott K, Mitrovic N, Hang C, Kouzmekine A, Iaizzo P, Herzog J, Lerche H, Nicole S, Vale-Santos J, Chauveau D, Fontaine B, Lehmann-Horn F. Voltage-sensor sodium channel mutations cause hypokalemic periodic paralysis type 2 by enhanced inactivation and reduced current. Proc Natl Acad Sci U S A. 2000;97:9549–9554. doi: 10.1073/pnas.97.17.9549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jurkat-Rott K, Weber MA, Fauler M, Guo XH, Holzherr BD, Paczulla A, Nordsborg N, Joechle W, Lehmann-Horn F. K+-dependent paradoxical membrane depolarization and Na+ overload, major and reversible contributors to weakness by ion channel leaks. Proc Natl Acad Sci U S A. 2009;106:4036–4041. doi: 10.1073/pnas.0811277106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ke T, Gomez CR, Mateus HE, Castano JA, Wang QK. Novel CACNA1S mutation causes autosomal dominant hypokalemic periodic paralysis in a South American family. J Hum Genet. 2009;54:660–664. doi: 10.1038/jhg.2009.92. [DOI] [PubMed] [Google Scholar]
- 43.Kersten F, van Wijk E, van Reeuwijk J, van der Zwaag B, Maerker T, Peters T, Katsanis N, Wolfrum U, Keunen J, Roepman R, Kremer H (2009) Whirlin associates with the Cav1.3 (α1D) channels in photoreceptors, defining a novel member of the Usher protein network. Invest Ophthalmol Vis Sci 2009 Dec 3 (published online) [DOI] [PubMed]
- 44.Koschak A, Reimer D, Walter D, Hoda JC, Heinzle T, Grabner M, Striessnig J. Cav1.4α1 subunits can form slowly inactivating dihydropyridine-sensitive L-type Ca2+ channels lacking Ca2+-dependent inactivation. J Neurosci. 2003;23:6041–6049. doi: 10.1523/JNEUROSCI.23-14-06041.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kugler G, Weiss RG, Flucher BE, Grabner M. Structural requirements of the dihydropyridine receptor alpha1S II–III loop for skeletal-type excitation–contraction coupling. J Biol Chem. 2004;279:4721–4728. doi: 10.1074/jbc.M307538200. [DOI] [PubMed] [Google Scholar]
- 46.Liu X, Yang PS, Yang W, Yue DT. Enzyme-inhibitor-like tuning of calcium channel connectivity with calmodulin. Nature. 2010;463:968–972. doi: 10.1038/nature08766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mangoni ME, Couette B, Bourinet E, Platzer J, Reimer D, Striessnig J, Nargeot J. Functional role of L-type Cav1.3 Ca2+ channels in cardiac pacemaker activity. Proc Natl Acad Sci U S A. 2003;100:5543–5548. doi: 10.1073/pnas.0935295100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maquat LE. Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat Rev Mol Cell Biol. 2004;5:89–99. doi: 10.1038/nrm1310. [DOI] [PubMed] [Google Scholar]
- 49.Marcantoni A, Baldelli P, Hernandez-Guijo JM, Comunanza V, Carabelli V, Carbone E. L-type calcium channels in adrenal chromaffin cells: role in pace-making and secretion. Cell Calcium. 2007;42:397–408. doi: 10.1016/j.ceca.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 50.Marcantoni A, Vandael DH, Mahapatra S, Carabelli V, Sinnegger-Brauns MJ, Striessnig J, Carbone E. Loss of Cav1.3 channels reveals the critical role of L-type and BK channel coupling in pacemaking mouse adrenal chromaffin cells. J Neurosci. 2010;30:491–504. doi: 10.1523/JNEUROSCI.4961-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matthews E, Labrum R, Sweeney MG, Sud R, Haworth A, Chinnery PF, Meola G, Schorge S, Kullmann DM, Davis MB, Hanna MG. Voltage sensor charge loss accounts for most cases of hypokalemic periodic paralysis. Neurology. 2009;72:1544–1547. doi: 10.1212/01.wnl.0000342387.65477.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Matza D, Flavell RA. Roles of Cav channels and AHNAK1 in T cells: the beauty and the beast. Immunol Rev. 2009;231:257–264. doi: 10.1111/j.1600-065X.2009.00805.x. [DOI] [PubMed] [Google Scholar]
- 53.McRory JE, Hamid J, Doering CJ, Garcia E, Parker R, Hamming K, Chen L, Hildebrand M, Beedle AM, Feldcamp L, Zamponi GW, Snutch TP. The CACNA1F gene encodes an L-type calcium channel with unique biophysical properties and tissue distribution. J Neurosci. 2004;24:1707–1718. doi: 10.1523/JNEUROSCI.4846-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Monnier N, Procaccio V, Stieglitz P, Lunardi J. Malignant-hyperthermia susceptibility is associated with a mutation of the alpha 1-subunit of the human dihydropyridine-sensitive L-type voltage-dependent calcium-channel receptor in skeletal muscle. Am J Hum Genet. 1997;60:1316–1325. doi: 10.1086/515454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moosmang S, Haider N, Klugbauer N, Adelsberger H, Langwieser N, Muller J, Stiess M, Marais E, Schulla V, Lacinova L, Goebbels S, Nave KA, Storm DR, Hofmann F, Kleppisch T. Role of hippocampal Cav1.2 Ca2+ channels in NMDA receptor-independent synaptic plasticity and spatial memory. J Neurosci. 2005;25:9883–9892. doi: 10.1523/JNEUROSCI.1531-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morrill JA, Cannon SC. Effects of mutations causing hypokalaemic periodic paralysis on the skeletal muscle L-type Ca2+ channel expressed in Xenopus laevis oocytes. J Physiol. 1999;520(Pt 2):321–336. doi: 10.1111/j.1469-7793.1999.00321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Olson PA, Tkatch T, Hernandez-Lopez S, Ulrich S, Ilijic E, Mugnaini E, Zhang H, Bezprozvanny I, Surmeier DJ. G-protein-coupled receptor modulation of striatal Cav1.3 L-type Ca2+ channels is dependent on a Shank-binding domain. J Neurosci. 2005;25:1050–1062. doi: 10.1523/JNEUROSCI.3327-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Peloquin JB, Doering CJ, Rehak R, McRory JE. Temperature dependence of Cav1.4 calcium channel gating. Neuroscience. 2008;151:1066–1083. doi: 10.1016/j.neuroscience.2007.11.053. [DOI] [PubMed] [Google Scholar]
- 59.Peloquin JB, Rehak R, Doering CJ, McRory JE. Functional analysis of congenital stationary night blindness type-2 CACNA1F mutations F742C, G1007R, and R1049W. Neuroscience. 2007;150:335–345. doi: 10.1016/j.neuroscience.2007.09.021. [DOI] [PubMed] [Google Scholar]
- 60.Peterson BZ, DeMaria CD, Yue DT. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron. 1999;22:549–558. doi: 10.1016/S0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- 61.Pietrobon D. Calcium channels and channelopathies of the central nervous system. Mol Neurobiol. 2002;25:31–50. doi: 10.1385/MN:25:1:031. [DOI] [PubMed] [Google Scholar]
- 62.Platzer J, Engel J, Schrott-Fischer A, Stephan K, Bova S, Chen H, Zheng H, Striessnig J. Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. Cell. 2000;102:89–97. doi: 10.1016/S0092-8674(00)00013-1. [DOI] [PubMed] [Google Scholar]
- 63.Rabl K, Thoreson WB. Calcium-dependent inactivation and depletion of synaptic cleft calcium ions combine to regulate rod calcium currents under physiological conditions. Eur J NeuroSci. 2002;16:2070–2077. doi: 10.1046/j.1460-9568.2002.02277.x. [DOI] [PubMed] [Google Scholar]
- 64.Ruether K, Grosse J, Matthiessen E, Hoffmann K, Hartmann C. Abnormalities of the photoreceptor-bipolar cell synapse in a substrain of C57BL/10 mice. Invest Ophthalmol Vis Sci. 2000;41:4039–4047. [PubMed] [Google Scholar]
- 65.Seisenberger C, Specht V, Welling A, Platzer J, Pfeifer A, Kuhbandner S, Striessnig J, Klugbauer N, Feil R, Hofmann F. Functional embryonic cardiomyocytes after disruption of the L-type alpha1C (Cav1.2) calcium channel gene in the mouse. J Biol Chem. 2000;275:39193–39199. doi: 10.1074/jbc.M006467200. [DOI] [PubMed] [Google Scholar]
- 66.Singh A, Gebhart M, Fritsch R, Sinnegger-Brauns MJ, Poggiani C, Hoda JC, Engel J, Romanin C, Striessnig J, Koschak A (2008) Modulation of voltage- and Ca2+-dependent gating of Cav1.3 L-type calcium channels by alternative splicing of a C-terminal regulatory domain. J Biol Chem 283:20733–20744 [DOI] [PMC free article] [PubMed]
- 67.Singh A, Hamedinger D, Hoda JC, Gebhart M, Koschak A, Romanin C, Striessnig J. C-terminal modulator controls Ca2+-dependent gating of Cav1.4 L-type Ca2+ channels. Nat Neurosci. 2006;9:1108–1116. doi: 10.1038/nn1751. [DOI] [PubMed] [Google Scholar]
- 68.Sinnegger-Brauns MJ, Hetzenauer A, Huber IG, Renstrom E, Wietzorrek G, Berjukov S, Cavalli M, Walter D, Koschak A, Waldschutz R, Hering S, Bova S, Rorsman P, Pongs O, Singewald N, Striessnig JJ. Isoform-specific regulation of mood behavior and pancreatic beta cell and cardiovascular function by L-type Ca2+ channels. J Clin Invest. 2004;113:1430–1439. doi: 10.1172/JCI20208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sipos I, Jurkat-Rott K, Harasztosi C, Fontaine B, Kovacs L, Melzer W, Lehmann-Horn F. Skeletal muscle DHP receptor mutations alter calcium currents in human hypokalaemic periodic paralysis myotubes. J Physiol. 1995;483(Pt 2):299–306. doi: 10.1113/jphysiol.1995.sp020586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sokolov S, Scheuer T, Catterall WA. Gating pore current in an inherited ion channelopathy. Nature. 2007;446:76–78. doi: 10.1038/nature05598. [DOI] [PubMed] [Google Scholar]
- 71.Splawski I, Timothy KW, Decher N, Kumar P, Sachse FB, Beggs AH, Sanguinetti MC, Keating MT. Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. Proc Natl Acad Sci U S A. 2005;102:8089–8096. doi: 10.1073/pnas.0502506102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, Tager-Flusberg H, Priori SG, Sanguinetti MC, Keating MT. Cav1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119:19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 73.Starace DM, Bezanilla F. Histidine scanning mutagenesis of basic residues of the S4 segment of the Shaker K+ channel. J Gen Physiol. 2001;117:469–490. doi: 10.1085/jgp.117.5.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Starace DM, Bezanilla F. A proton pore in a potassium channel voltage sensor reveals a focused electric field. Nature. 2004;427:548–553. doi: 10.1038/nature02270. [DOI] [PubMed] [Google Scholar]
- 75.Stary A, Kudrnac M, Beyl S, Hohaus A, Timin EN, Wolschann P, Guy HR, Hering S (2008) Molecular dynamics and mutational analysis of a channelopathy mutation in the IIS6 helix of Cav1.2. Channels (Austin) 2:216–223 [DOI] [PMC free article] [PubMed]
- 76.Stary A, Shafrir Y, Hering S, Wolschann P, Guy HR (2008) Structural model of the Cav1.2 pore. Channels (Austin) 2:210–215 [DOI] [PubMed]
- 77.Striessnig J, Koschak A. Exploring the function and pharmacotherapeutic potential of voltage-gated Ca2+ channels with gene knockout models. Channels (Austin) 2008;2:233–251. doi: 10.4161/chan.2.4.5847. [DOI] [PubMed] [Google Scholar]
- 78.Striessnig J, Koschak A, Sinnegger-Brauns MJ, Hetzenauer A, Nguyen NK, Busquet P, Pelster G, Singewald N. Role of voltage-gated L-type Ca2+ channel isoforms for brain function. Biochem Soc Trans. 2006;34:903–909. doi: 10.1042/BST0340903. [DOI] [PubMed] [Google Scholar]
- 79.Struyk AF, Cannon SC. Paradoxical depolarization of Ba2+-treated muscle exposed to low extracellular K+: insights into resting potential abnormalities in hypokalemic paralysis. Muscle Nerve. 2008;37:326–337. doi: 10.1002/mus.20928. [DOI] [PubMed] [Google Scholar]
- 80.Thoreson WB, Rabl K, Townes-Anderson E, Heidelberger R. A highly Ca2+-sensitive pool of vesicles contributes to linearity at the rod photoreceptor ribbon synapse. Neuron. 2004;42:595–605. doi: 10.1016/S0896-6273(04)00254-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tombola F, Pathak MM, Isacoff EY. Voltage-sensing arginines in a potassium channel permeate and occlude cation-selective pores. Neuron. 2005;45:379–388. doi: 10.1016/j.neuron.2004.12.047. [DOI] [PubMed] [Google Scholar]
- 82.Tombola F, Ulbrich MH, Isacoff EY. Architecture and gating of Hv1 proton channels. J Physiol. 2009;587:5325–5329. doi: 10.1113/jphysiol.2009.180265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tremblay F, Laroche RG, De Becker I. The electroretinographic diagnosis of the incomplete form of congenital stationary night blindness. Vision Res. 1995;35:2383–2393. doi: 10.1016/0042-6989(95)00006-L. [DOI] [PubMed] [Google Scholar]
- 84.Wahl-Schott C, Baumann L, Cuny H, Eckert C, Griessmeier K, Biel M. Switching off calcium-dependent inactivation in L-type calcium channels by an autoinhibitory domain. Proc Natl Acad Sci U S A. 2006;103:15657–15662. doi: 10.1073/pnas.0604621103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wilkinson MF, Barnes S. The dihydropyridine-sensitive calcium channel subtype in cone photoreceptors. J Gen Physiol. 1996;107:621–630. doi: 10.1085/jgp.107.5.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Witkovsky P, Schmitz Y, Akopian A, Krizaj D, Tranchina D. Gain of rod to horizontal cell synaptic transfer: relation to glutamate release and a dihydropyridine-sensitive calcium current. J Neurosci. 1997;17:7297–7306. doi: 10.1523/JNEUROSCI.17-19-07297.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wycisk KA, Budde B, Feil S, Skosyrski S, Buzzi F, Neidhardt J, Glaus E, Nurnberg P, Ruether K, Berger W. Structural and functional abnormalities of retinal ribbon synapses due to Cacna2d4 mutation. Invest Ophthalmol Vis Sci. 2006;47:3523–3530. doi: 10.1167/iovs.06-0271. [DOI] [PubMed] [Google Scholar]
- 88.Wycisk KA, Zeitz C, Feil S, Wittmer M, Forster U, Neidhardt J, Wissinger B, Zrenner E, Wilke R, Kohl S, Berger W. Mutation in the auxiliary calcium-channel subunit CACNA2D4 causes autosomal recessive cone dystrophy. Am J Hum Genet. 2006;79:973–977. doi: 10.1086/508944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Xu W, Lipscombe D. Neuronal Cav1.3α1 L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J Neurosci. 2001;21:5944–5951. doi: 10.1523/JNEUROSCI.21-16-05944.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yarov-Yarovoy V, Baker D, Catterall WA. Voltage sensor conformations in the open and closed states in ROSETTA structural models of K+ channels. Proc Natl Acad Sci U S A. 2006;103:7292–7297. doi: 10.1073/pnas.0602350103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zanazzi G, Matthews G. The molecular architecture of ribbon presynaptic terminals. Mol Neurobiol. 2009;39:130–148. doi: 10.1007/s12035-009-8058-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang H, Fu Y, Altier C, Platzer J, Surmeier DJ, Bezprozvanny I. Cav1.2 and Cav1.3 neuronal L-type calcium channels: differential targeting and signaling to pCREB. Eur J NeuroSci. 2006;23:2297–2310. doi: 10.1111/j.1460-9568.2006.04734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang H, Maximov A, Fu Y, Xu F, Tang TS, Tkatch T, Surmeier DJ, Bezprozvanny I. Association of Cav1.3 L-type calcium channels with Shank. J Neurosci. 2005;25:1037–1049. doi: 10.1523/JNEUROSCI.4554-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zuhlke RD, Reuter H. Identification of a single amino acid residue as molecular determinant of calcium-dependet inactivation and facilitation of L-type calcium channels. Biophys J. 1999;76:343. [Google Scholar]