

Abstract
A new strategy is described for functionalizing the ω-terminal end of polymers synthesized by reversible addition–fragmentation chain transfer (RAFT) polymerization that provides spatially controlled bioconjugation sites. Traditional methods for preparing ω-functional polymers require the reduction of the RAFT chain-transfer agent to yield secondary or tertiary thiols of low reactivity or the synthesis of novel chain-transfer agents that contain reactive groups. As an additional strategy, N-substituted maleimido monomers have been used in a modified block polymerization to add a single maleimido unit onto the RAFT polymer with nearly quantitative efficiency. Unique reactive groups contained in the N-substituent are thereby added to the ω-terminal end of the polymer and are subsequently available for conjugation reactions. This technique has been demonstrated using N-(2-aminoethyl)maleimide trifluoroacetate to introduce a single primary amine to the ω-terminus of poly(dimethy-laminoethyl methacrylate) and poly(N-isopropyl acrylamide) and to a specialized block copolymer for siRNA delivery. Evidence for retention of functional RAFT endgroups is provided by synthesis results where chain-extended polyDMAEMA (Mn = 10 600 g/mol, Mw/Mn = 1.14) was used as a macro chain transfer agent for the polymerization of styrene, yielding a diblock polymer of low polydispersity (Mn = 20 300 g/mol, Mw/Mn = 1.11). It is thus also possible to construct diblock copolymers with a bioconjugation site precisely located at the junction between the two blocks. The chain-extended polymers are functionalized with an amine-reactive fluorescent dye or folic acid at conjugation efficiencies of 86 and 94%, respectively. The versatile chain-extension technique described here offers unique opportunities for the synthesis of well-defined polymeric conjugates to molecules of biological and targeting interest.
INTRODUCTION
Controlled radical polymerization (CRP) techniques such as nitroxide-mediated polymerization (NMP) (1), atom transfer radical polymerization (ATRP) (2), and reversible addition–fragmentation chain transfer polymerization (RAFT) (3, 4) offer the ability to produce polymers with predetermined molecular weights, narrow molecular weight distributions, and advanced architectures. For RAFT polymerizations, numerous groups have reported the incorporation of diverse monomers in both organic and aqueous conditions (5–11). One particularly significant feature of RAFT is that it allows a facile route to prepare telechelic polymers with distinct α and ω functionalities. This feature provides a useful tool for the tailored design of novel polymeric conjugates via coupling to fluorescent dyes, nanoparticles, drugs, peptides, proteins, or targeting moieties (12–17). In particular, reactive α and ω endgroups offer unique opportunities for the synthesis of well-defined polymeric drug carriers that incorporate both targeting and therapeutic moieties.
Functional endgroups can be prepared using ATRP by modifying the initiator (18, 19) or by RAFT through modifications to the chain transfer agent (CTA). The majority of literature reports have focused on RAFT CTAs with functional R groups (16, 17, 20–23), largely due to the greater stability of the R group following polymerization. Notable examples of RAFT agents with functional Z groups also exist. Liu et al. (24) prepared a novel trithiocarbonate CTA containing a pyridyl disulfide group in the Z position that allows for direct coupling to thiol-bearing molecules without postpolymerization modifications. Similarly, Carter et al. prepared a highly branched polymer with imidazole endgroups by employing a Z-functional dithioester (25). However, preparation of Z-functionalized RAFT CTAs often requires more difficult synthetic protocols, and many desirable chemical moieties are unsuitable for incorporation into CTA moieties.
RAFT CTAs based on xanthates, dithioesters, dithiocarabamates, and trithiocarbonates can be reduced with primary amines (26–28) or NaBH4 (12, 13) to yield a polymeric thiol endgroup that can be used for conjugation reactions (29–33). While conjugations to secondary thiols (from styrenic and non-α-substituted monomers) or tertiary thiols (from α-substituted monomers) have been described (11, 13, 14, 26, 34, 35), these reactions typically require elevated temperatures, long reaction times, and stoichiometric ratios that can be problematic for many molecules of biological interest due to their expense or instability. A route around this reactivity barrier was described by Scales et al. (13), who showed that a secondary thiol could be reacted with a small linker-like molecule for subsequent conjugations. Similarly, York et al. (34) obtained 80% conjugation of a fluorescent dye to an α-substituted N-(2-hydroxypropyl) methacrylamide block copolymer following reduction of the CTA and subsequent conversion of the tertiary thiol to a primary amine. However, these conversions require reaction conditions that may prove impractical for many polymer compositions. Additional routes to the ω-functionalization of RAFT polymers are needed that are compatible with a wide range of monomers and can be conducted at the time of synthesis under mild conditions.
Our research group has been interested in applications of stimuli-responsive “smart” polymers for therapeutic and diagnostic/bioanalytical applications. The therapeutic applications utilize pH-responsive polymers for intracellular delivery of macromolecular biologics such as proteins, peptides, antisense DNA, and small interfering RNA (siRNA) (36–42). Additionally, we have worked on the development of stimuli-responsive polymers for bioseparations and diagnostic applications (43–45). These applications would benefit from more efficient chemistries for spatially controlled ω (or Z)-end conjugations, and here, we describe a technique for functionalizing the ω-terminal end of RAFT polymers by chain extension using N-substituted maleimides. Maleimido monomers readily undergo copolymerization with other species but do not efficiently homopolymerize under typical RAFT conditions (46–50). We show that this property can be exploited in a block polymerization in order to add a single amine-substituted maleimido unit to the end of a RAFT polymer. Because a high percentage of the maleimido-terminated chains are living and a second block can be polymerized from this macro-CTA, the reactive amine can also be positioned precisely at the junction of AA or AB block segments, providing spatial control over the position of a branched bioconjugation site.
EXPERIMENTAL DETAILS
Materials
All reagents were purchased from Sigma-Aldrich and used without further purification unless otherwise noted. Dimethylaminoethyl methacrylate (DMAEMA), butyl methacrylate (BMA), and styrene were purified by vacuum distillation. N-Isopropylacrylamide (NIPAM) was recrystallized from hexanes. Propylacrylic acid (PAA) was synthesized as previously published (51). Unless noted otherwise, the primary radical source used in all polymerizations was 2,2-azobisisobutyronitrile (AIBN), which was recrystalized from methanol. The trithiocarbonate CTA ethyl cyanovaleric trithiocarbonate (ECT) was synthesized as previously described (41) and used as the CTA in all RAFT polymerizations. All polymerizations were conducted under N2 atmosphere.
RAFT polymerization of pDMAEMA and p(DMAEMA-b-DMAEMA/BMA/PAA)
Poly(DMAEMA) was obtained by RAFT polymerization using experimental conditions appropriate to target the various molecular weights used in this study. As an example of typical conditions, the [CTAo]/[Io] ratio was10: 1, the [CTAo]/[Mo] ratio was 1:150, and the monomer was 50 wt % in N,N′-dimethylformamide (DMF). The polymerization was conducted at 60 °C for 7 h. The molecular weight and polydispersity were 8100 g/mol and 1.21, respectively. All DMAEMA polymers were isolated by precipitation in an 80/20 v/v pentane/ether mixture, followed by repeated cycles of dissolution in ether and precipitation in pentane. The polymer was dried under vacuum overnight. Molecular weight distributions were determined by gel permeation chromatography (GPC).
The block copolymer p(DMAMEA-b-DMAEMA/BMA/PAA) was prepared following previously described protocols (41). Briefly, a pDMAEMA macro CTA (Mn = 9000 g/mol, Mw/Mn = 1.30) was employed for the polymerization of the copolymer block using a [CTAo]/[Io] ratio of 10:1 and a [CTAo]/[Mo] ratio of 1:250 in DMF at 40 wt % monomer. The composition of the feed was 40 mol % butylmethacrylate (BMA), 30 mol % propylacrylic acid (PAA), and 30 mol % DMAEMA. 2,2′-Azobis(4-methoxy-2,4-dimethylvaleronitrile) (V70) was the radical source, and the polymerization time was 18 h at 30 °C. The block copolymer was isolated by multiple precipitations in an 80/20 v/v pentane and ether mixture, followed by precipitation in pentane.
RAFT Polymerization of pNIPAM
Poly(NIPAM) was obtained by RAFT polymerization based on previously published conditions (52) using a [CTAo]/[Io] ratio of 20:1, a [CTAo]/[Mo] ratio of 1:100, and the monomer was 33 wt % in p-dioxane. The polymerization was conducted at 70 °C for 120 min. pNIPAM was isolated by repeated cycles of precipitation into ethyl ether, followed by dissolution in acetone, and dried under vacuum overnight.
Maleimido Chain Extension
Maleimide chain extension was performed at 60 °C in DMF for 16 h using N-(2-aminoethyl)-maleimide trifluoroacetate (AM) as the monomer and poly(D-MAEMA), poly(NIPAM), or poly(DMAEMA-b-DMAEMA/BMA/PAA) as the macro-CTA (Scheme 1). The polymerizations were conducted with a [CTAo]/[Io] ratio of 10:1 and a [CTAo]/[Mo] ratio of 1:100. The polymerization solution was 60 wt % solvent. As an example of polymerization, 400 mg pDMAEMA (49 μmol) and 1.26 g AM (4.9 mmol) were polymerized with 0.81 mg AIBN (4.9 μmol) in 2.5 g DMF. 1H NMR was used to confirm that the macro-CTAs were free of residual monomer prior to chain extension in order to eliminate the possibility of copolymerization during maleimide chain extension.
Scheme 1.
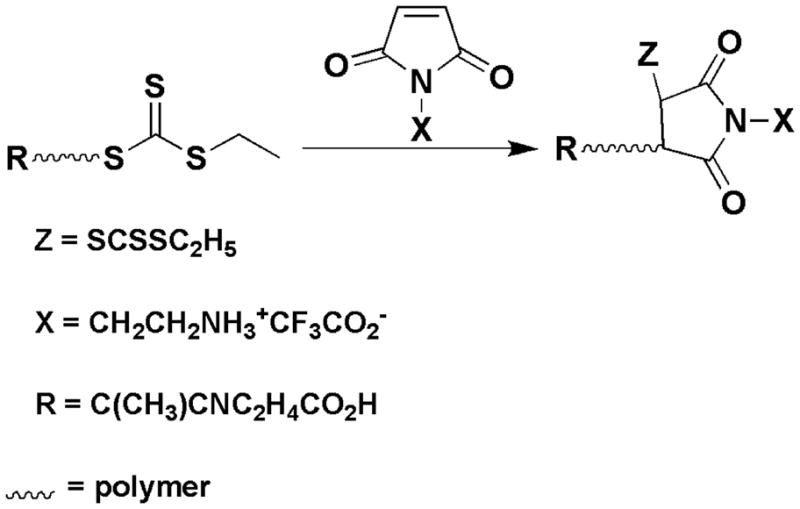
Synthetic Pathway for Maleimido Chain Extension of RAFT Polymers
Chain-extended pDMAEMA-AM was isolated by six cycles of precipitation in an 80/20 solution of pentane/ether. Between precipitations, the polymer was redissolved in acetone and separated from any insoluble AM by centrifugation and filtration through 0.2 μm syringe filters. Following precipitation, a solution of pDMAEMA-AM was prepared in deionized water (DI) and the polymer was further purified from residual AM monomer using two PD-10 desalting columns (Amersham, Piscataway NJ) in series and lyophilized. Chain-extended pNIPAM-AM was purified from AM by running the reaction mixture through two PD-10 columns following 1:2 dilution in DI, then lyophilized. The extent of AM chain extension was determined using 1H NMR, and the molecular weight distribution was characterized by GPC.
Styrene Polymerization from pDMAEMA-AM
Chain-extended pDMAEMA-AM was used as a macro-CTA for the polymerization of styrene (pDMAEMA-AM-STY) in order to demonstrate retention of functional RAFT chain ends. The [CTAo]/[Io] ratio was10:1, the [CTAo]/[Mo] ratio was 1:250, and the monomer was 50 wt % in DMF. The polymerization was conducted at 60 °C for 24 h, and the polymer was isolated by precipitation in 80/20 v/v pentane/ether as previously described.
Fluorescent Labeling
Amine functionalized pDMAEMA-AM and pDMAEMA-AM-STY were labeled with 1-pyrenebutanoic acid succimidyl ester (PNHS) (Invitrogen). pDMAEMA-AM (75 mg, 10 μmol) was dissolved in 1 mL of anhydrous DMF. Subsequently, diisopropylethylamine (123 mg, 0.96 mmol) was added at 2-fold molar excess relative to the number of DMAEMA residues to ensure removal of the triflate salt from the pDMAEMA-AM primary amine. After 60 min, PNHS (20 mg, 70 μmol) was added to the solution at 7-fold molar excess relative to the number of pDMAEMA-AM chains, and the reaction was allowed to proceed in the dark at room temperature for 18 h under anhydrous conditions. At the end of the reaction time, free PNHS was precipitated from solution by the addition of 4 mL of DI water and separated by centrifugation. The supernatant was filtered through a 0.2 μm syringe filter, eluted through 2 PD-10 desalting columns, and dried. The sample was then dissolved in 2.5 mL of 50% v/v DI/DMF and eluted through two additional PD-10 columns in series, and frozen and lyophilized. The percent conjugation was determined by GPC using online UV and RI detectors.
pDMAEMA-AM-STY was labeled using a similar procedure, but PNHS was added at 50-fold molar excess to compensate for the increased steric hindrance of the amine caused by the presence of the styrene block. Following labeling, the reaction mixture was added to 50 mL anhydrous ether causing the precipitation of PNHS, which was removed by centrifugation. pDMAEMA-AM-STY was subsequently precipitated from the ether by the addition of 50 vol % pentane and recovered by centrifugation.
To determine the concentration of pyrene associated with pDMAEMA-AM or pDMAEMA-AM-STY, a linear pyrene calibration curve was generated by injecting multiple known concentrations of pyrene on the GPC column and plotting their corresponding UV peak areas at 338 nm. The concentration of pyrene associated with the polymers was subsequently determined by comparison of the polymer conjugate UV peak area (λ = 338) to the pyrene calibration curve. The UV peak area of the unlabeled polymers was used to account for the contribution of the RAFT CTA to the absorption at 338 nm while maintaining constant RI peak area between the two samples.
Folic Acid Conjugation
Folic acid was activated with dicyclocarbodiimide (DCC) in dry dimethylsulfoxide (DMSO) at a molar ratio of 1:1.1 for 60 min at 4 °C. A solution of pDMAEMA-b-DMAEMA/BMA/PAA that had been chain-extended with N-(2-aminoethyl)maleimide and TEA (1:1 mol ratio with PAA and AM) in DMSO was added to the activated folate solution (10:1 folate/polymer ratio) and reacted overnight at room temperature. The polymer–folate conjugate was purified by extensive dialysis in water, then lyophilized. The remaining folic acid was separated from the lyophilized conjugate using a series of 5 PD-10 desalting columns until no further reduction in folic acid absorbance (λ = 363 nm) was observed in the column flow-through. The purified polymer–folate conjugate was dried by lyophilization, and the amount of folate present in the conjugate was determined by UV absorbance relative to a standard curve of free folic acid. No shift in the folate absorbance spectrum was observed as a result of conjugation to the polymer. UV measurements were done in phosphate buffered saline (PBS), pH = 7.4. Concentrations of the polymer–folate conjugate were between 0.1 and 1 mg/mL.
Instrumentation
The polymers in this study were characterized by gel permeation chromatography using three Tosoh TSK-GEL columns (TSK-α3000, α3000, α4000) connected in series to a Viscotek GPCmax VE2001 and Viscotek RI and UV detectors (VE3580 and VE3210, respectively) (Viscotek, Houston, TX). The mobile phase was HPLC-grade DMF containing 0.1 wt % LiBr. Molecular weight distributions were determined relative to a series of poly(methyl methacrylate) standards. Characterization of polymer–folate conjugates was performed on an HP Agilent 8453 UV–vis spectrophotometer. All NMR spectroscopy was done on a Bruker DRX499 system in D2O.
RESULTS AND DISCUSSION
A single N-substituted maleimido repeat unit can be added to the ω-terminal end of a polymer using RAFT and the common techniques of block polymerization. By varying the identity of the N-substituent, maleimido chain extension provides a method to introduce highly reactive chemical groups at the ω (Z)-end (Scheme 1). The technique is versatile in that it is compatible with a wide variety of polymers, does not require post-polymerization processing to obtain reactive moieties, and yields functional groups for bioconjugations that are significantly more reactive than the polymeric thiols that can be obtained by CTA reduction. Using the chain extension technique, we have successfully introduced primary amine ω-endgroups to acrylamido and α-substituted methacrylic polymers and at the junction between diblock segments (Figure 1). The utility of the amines has been shown by conjugations to an optical dye and to the targeting moiety folic acid.
Figure 1.
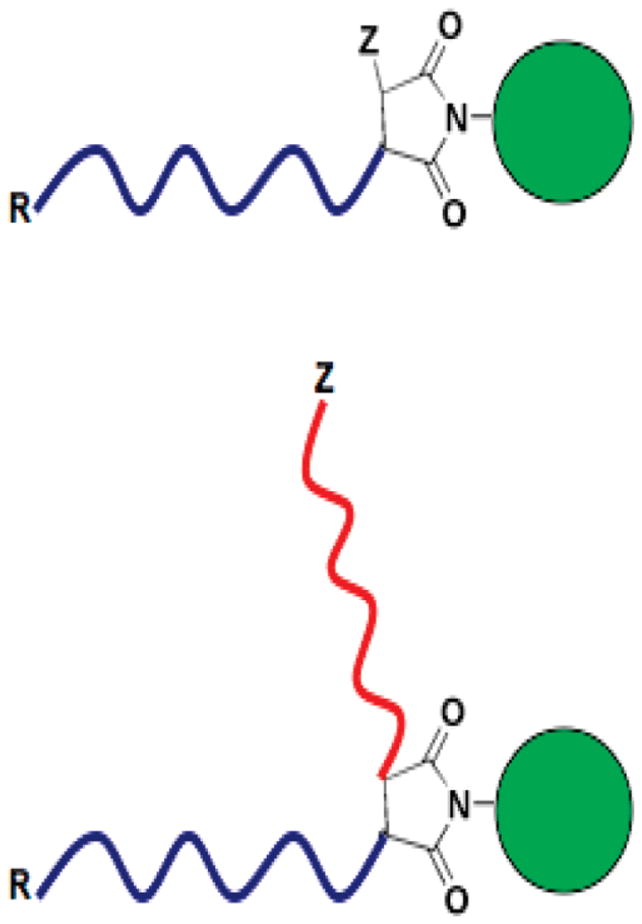
Schematic design of functionalized telechelic RAFT-polymers that provide a convenient route to controlled bioconjugations at the ω(Z)-chain end, or at the branched junction between two block segments. Using the CTA described in the text, the R group is a reactive carboxylate and the Z-group is an unreduced trithiocarbonate or reduced polymeric thiol.
Primary amines are highly nucleophilic and have significant synthetic utility in the preparation of polymeric bioconjugates. However, primary amines are commonly used for the reduction of trithiocarbonates and dithiocarbamates, and therefore difficult to incorporate into RAFT polymers or RAFT CTAs. As shown in Scheme 1, we have used the triflate salt of N-(2-aminoeth-yl)maleimide (AM) to introduce primary amine functionality on the ω-end of polymer chains. The triflate salt prevents reduction of the RAFT CTA during chain extension with the maleimide, but does not interfere with subsequent conjugation reactions. The success of this technique can be shown using 1H NMR. Figure 2 shows the spectrum of a chain extended pDMAEMA-AM macro-CTA. The emergence of the triplet at δ = 3.77 corresponding to the (NCH2CH2) group in AM and the absence of free vinyl peaks at δ = 6.8 shows that AM is incorporated into the pDMAEMA polymer and not present as a contaminating monomer. By comparing the pDMAEMA (CO2CH2) peak area at δ = 4.1 to the AM peak area at δ = 3.77, and using the molecular weight of pDMAEMA determined by GPC, the number of AM residues incorporated into each polymer chain is calculated at 1.02 (P1, Table 1). This result clearly shows the addition of a single functional maleimide to the ω-end of the polymer chain.
Figure 2.
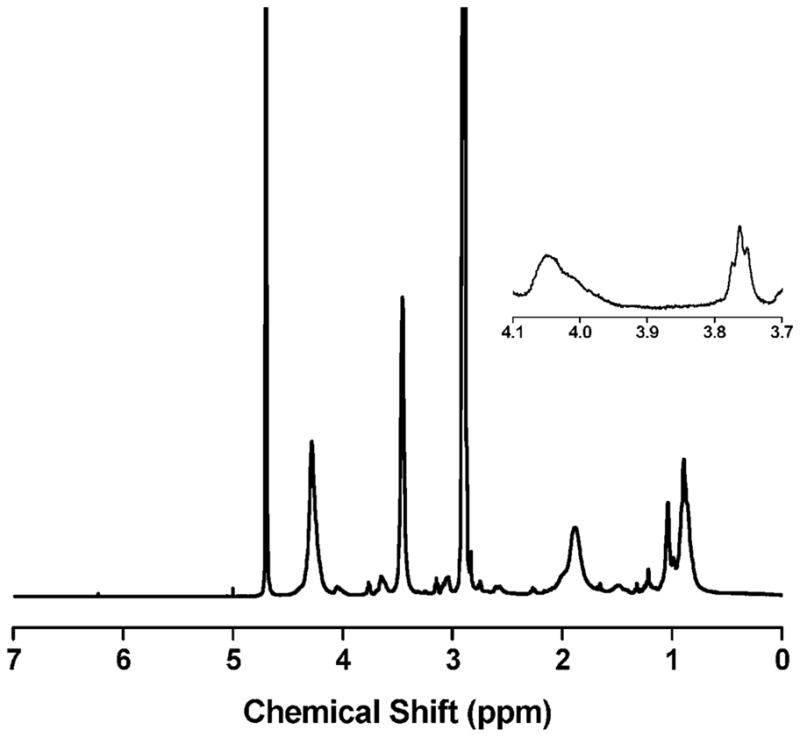
1H NMR of pDMAEMA after chain extension with N-(2-aminoethyl)maleimide trifluoroacetate (AM). The inset shows the triplet at δ = 3.77 that results from the addition of AM to the polymer chain.
Table 1.
Molecular Weight Distributions, Degree of Chain Extension, and Degree of Conjugation
| # | polymer | Mna g/mol | Mw/Mna | % AM extensionb | % conjugation pyrenea | % conjugation folatec |
|---|---|---|---|---|---|---|
| P1 | pDMAEMA-A | 8100 | 1.26 | 102 | 86 | -- |
| P2 | pDMAEMA-AM-STY | 20300 | 1.11 | 82 | 44 | -- |
| P3 | pNIPAM-AM | 20300 | 1.17 | 97 | -- | -- |
| P4 | pDMAEMA-b-DMAEMA/BMA/PAA-AM | 19500 | 1.45 | -- | -- | 94 |
Determined by GPC.
Determined by 1H NMR.
Determined by UV spectroscopy.
The chain extension technique is not limited to the preparation of ω-functional methacrylic polymers. Using the thermally responsive polymer poly(N-isopropylacrylamide) (pNIPAM), we have shown that acrylamido polymers may be functionalized by maleimido chain extension. Figure 3 shows the 1H NMR spectrum of a chain-extended pNIPAM-AM macro-CTA. The triplet at δ = 3.15 corresponds to the (CH2CH2NH2) group in AM and the spectrum is free of vinyl peaks, indicating incorporation of AM into the pNIPAM polymer. The number of AM residues incorporated into each polymer was determined to be 0.97 (P3, Table 1) by comparing the pNIPAM methine peak area (NHCH(CH3)2) at δ = 3.81 to the AM peak area at δ = 3.15 and using the molecular weight of pNIPAM determined by GPC. Moreover, the AM peak area can be compared directly to the (SCH2CH3) peak from the Z-group of the CTA at δ = 3.31, indicating that the CTA and AM are present on the polymer in a 1:1 ratio. Calculation of the degree of polymerization based on the pNIPAM methine peak and the CTA peak at δ = 3.31 yields the degree of polymerization expected from GPC measurements, providing further evidence that pNIPAM has been successfully functionalized by a single AM residue and that functional CTA moieties are retained on the polymer chain end. Together with the results of the pDMAEMA chain extension, these results indicate that maleimido chain extension may not be restricted by monomer class and will have synthetic utility with a wide array of commonly used polymers (e.g., (meth)acrylic and (meth)acrylamido).
Figure 3.
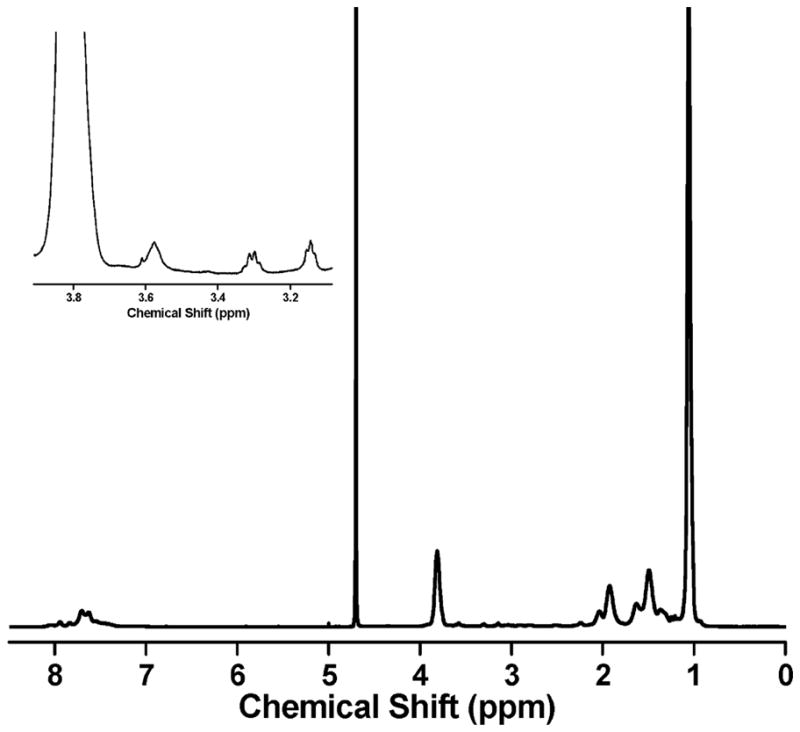
The 1H NMR spectrum of pNIPAM following chain extension with AM. The inset shows the triplet at δ = 3.15 that results from the addition of AM to the polymer chain and the quartet at δ = 3.31 that indicates the presence of intact RAFT Z groups.
The chain extension conditions we describe do not result in changes to the molecular weight distribution of the polymer undergoing extension. Figure 4A shows the RI traces for pDMAEMA and pDMAEMA-AM (P1, Table 1) following chain extension. No apparent shift in the peak elution volume results from chain extension, consistent with the addition of a single monomer to the chain end, and no change was observed in the calculated Mn (8100 g/mol) upon chain extension. Moreover, no peak broadening is exhibited in the RI traces, as would be expected if significant polymer–polymer coupling or formation of homomaleimide polymer fragments were to occur (Mw/Mn =.21 and 1.26 before and after chain extension, respectively). Similarly, no significant changes in molecular weight distribution were observed after chain extension of pNIPAM (Mn = 8000 g/mol Mw/Mn = 1.16, and Mn = 8000 g/mol Mw/Mn= 1.17) or the block copolymer pDMAEMA-b-DMAEMA/BMA/PAA (Mn = 19 400 g/mol Mw/Mn = 1.42, and Mn = 19 500 g/mol Mw/Mn = 1.45) (P4, Table 1).
Figure 4.

(A) GPC traces showing the molecular weight distribution of pDMAEMA and pDMAEMA-AM following chain extension. (B) GPC traces of a pDMAEMA-AM-STY triblock polymer and the corresponding pDMAEMA-AM macro-CTA.
A possible complication of chain extension with a primary amine containing monomer is destruction of the RAFT moiety through aminolysis. If this undesirable reaction were to occur, it would yield polymeric thiols that might result in polymer–polymer coupling by disulfide formation or reaction with maleimide monomers. Although GPC traces provided no evidence of polymer coupling following chain extension, we prepared an additional chain-extended pDMAEMA-AM to serve as a macro-CTA in a subsequent styrene polymerization (Mn = 10 600 g/mol, Mw/Mn = 1.14). If destruction of some or all of the RAFT CTA were to occur during chain extension, subsequent block formation using the pD-MAEMA-AM macro-CTA would be impossible or would yield polymers with broad or multimodal molecular weight distributions due to the presence of nonfunctional macro-CTA. Figure 4B shows RI traces for the pDMAEMA-AM macro-CTA and for the triblock polymer obtained by block polymerization, pDMAEMA-AM-STY (P2, Table 1). A clear shift in the retention volume of the polymer is observed following polymerization, and the molecular weight distribution of the triblock polymer is characteristically narrow (Mn= 20 300 g/mol and Mw/Mn = 1.11). These results strongly suggest that significant aminolysis of the CTA is not occurring. Further evidence in support of this point can be seen by examining the 1H NMR spectrum of pNIPAM-AM, shown in Figure 3.
This spectrum clearly shows the methylene peak from the Z-group of the CTA at δ = 3.31. The ratio of this peak area to the methine peak area in chain-extended pNIPAM-AM is the same as the corresponding ratio in the parent pNIPAM, indicating that no detectable aminolysis of the CTA occurs during chain extension.
The retention of functional RAFT moieties on the polymer following chain extension offers unique synthetic opportunities. The maleimido-terminated chains are living and can be used in subsequent block polymerizations, as illustrated by our polymerization of pDMAEMA-AM-STY. Such polymerizations have the effect of positioning a bioconjugation site precisely at the junction of the AA or AB blocks, whose length and composition can also be readily controlled. Moreover, multiple rounds of block polymerizations could potentially be used to position multiple bioconjugation sites at defined intervals along a polymer backbone of hetero- or homogeneous composition. These materials could be used to prepare therapeutic and diagnostic polymer conjugates with uniquely defined architectures.
To confirm that the amines introduced to the polymers by chain extension were reactive and available, a fluorescent dye was used as a model compound for imaging agent conjugations. Amine-reactive 1-pyrenebutanoic acid succimidyl ester (PNHS) was used to label pDMAEMA-AM, and the amount of pyrene conjugated was evaluated by GPC and UV spectroscopy following purification. Figure 5A shows normalized RI and UV traces for pyrene-labeled pDMAEMA-AM. Upon reaction with PNHS, an intense UV signal is observed with polymer elution that is not present in unconjugated pDMAEMA-AM, indicating that the pDMAEMA-AM chains were successfully labeled with pyrene. Figure 5B shows the UV absorbance spectra of pyrene-labeled pDMAEMA-AM and unlabeled pDMAEMA-AM, illustrating the significant increase in absorptivity that occurs upon pyrene addition. The percentage of pDMAEMA-AM chains conjugated to pyrene was determined to be 86.1% (P1, Table 1) by comparing the UV peak area associated with polymer elution to a calibration curve. Similarly, PNHS was also used to label the primary amine located at the maleimido junction of the DMAEMA and STY block segments in pD-MAEMA-AM-STY (P2, Table 1). The degree of conjugation to this polymer was 44%, suggesting that the steric effects of the large styrene block reduce the conjugation efficiency.
Figure 5.

Chain-extended polymers were labeled with an amine-reactive pyrene. (A) RI and UV traces of pDMAEMA-AM-P following pyrene conjugation. (B) Unmodified pDMAEMA-AM shows limited UV absorbance due to the trithiocarbonate moiety on the chain end, while pyrene-labeled pDMAEMA-AM-P exhibits significantly increased absorption.
Recently, our group described the use of the block copolymer pDMAEMA-b-DMAEMA/BMA/PAA as a siRNA delivery vehicle (41). The positively charged pDMAEMA block functions to condense nucleic acid residues, while the pH-responsive and membrane-disruptive copolymer block mediates intracellular delivery of the complexes following endosomal uptake. Though the block copolymer mediated excellent gene knockdown (up to 80%) (41), incorporation of targeting ligands can further enhance knockdown in vitro and facilitate more efficient in vivo delivery (53, 54). Folic acid is a commonly used targeting ligand, mediating greater internalization for cells that express or overexpress the folic acid receptor (55, 56). As a demonstration of the utility of maleimido chain extension, we conjugated folic acid to the AM chain-extended pDMAEMA-b-DMAEMA/BMA/PAA block copolymer. Figure 6A shows the RI and UV GPC traces of the unmodified copolymer, while Figure 6B shows the RI and UV GPC traces of the copolymer–folate conjugate. The unmodified copolymer shows negligible UV absorbance at 363 nm (Figure 6A), while an intense UV signal is associated with polymer elution in the case of the copolymer–folate conjugate (Figure 6B). The degree of folate conjugation to the copolymer was determined by comparison to a calibration curve of free folic acid and found to be 94% (P4, Table 1).
Figure 6.

(A) RI and UV traces of pDMAEMA-b-DMAEMA/BMA/PAA before conjugation to folic acid. (B) RI and UV traces of pDMAEMA-b-DMAEMA/BMA/PAA after the addition of folic acid.
It is important to note that the highly efficient conjugation reactions demonstrated here were conducted for less than 24 h at room temperature and relatively low stoichiometric ratios. These reaction conditions are possible because maleimide chain extension provides a functional group with far greater reactivity than the tertiary thiols that can be obtained following reduction of the CTA. For this reason, chain extension is particularly well suited for functionalizing polymers with molecules of biological interest that may be insufficiently stable or prohibitively expensive for use with other ω-functionalization strategies.
CONCLUSION
A facile method to achieve end-functionalization of RAFT polymers with moieties for bioconjugation was demonstrated. Using an N-substituted maleimido monomer, a unique functional group was introduced to the ω-terminal end of RAFT polymers. Specifically, N-(2-aminoethyl)maleimide trifluoroacetate (AM) was used to introduce a single primary amine to an α-substituted polymer, dimethylaminoethyl methacrylate (pDMAEMA), N-isopropyl acrylamide (pNIPAM) and a block copolymer of DMAEMA, butyl methacrylate (BMA), and propylacrylic acid (PAA), (pDMAEMA-block-DMAEMA/BMA/PAA). The primary amine was active, and conjugation efficiencies of 86% to pyrene and 94% to folic acid were readily achieved at room temperature and low stoichiometric ratios. This synthetic approach should provide a versatile method for ω-end functionalization of RAFT polymers, since alternate ω-end functionalities could be readily prepared by altering the identity of the maleimido N-substituent.
Acknowledgments
This work was funded by the National Institutes of Health (R01EB002991). Danielle S.W. Benoit is a Merck Fellow supported by the Damon Runyon Cancer Research Foundation (DRG-1948-07).
LITERATURE CITED
- 1.Georges MK, Veregin RPN, Kazmaier PM, Hamer GK. Narrow molecular-weight resins by a free-radical polymerization process. Macromolecules. 1993;26:2987–2988. [Google Scholar]
- 2.Wang JS, Matyjaszewski K. Controlled living radical polymerization - atom-transfer radical polymerization in the presence of transition-metal complexes. J Am Chem Soc. 1995;117:5614–5615. [Google Scholar]
- 3.Maydunne RTA, Rizzardo E, Chiefari J, Chang YK, Moad G, Thang SH. A more versatile route to block copolymer and other polymers of complex architecture by living radical polymerization: the RAFT process. Macromolecules. 1999;32:6977–6980. [Google Scholar]
- 4.Chiefari J, Chang YK, Ercole F, Krstina J, Jeffery J, Le TPT, Mayadunne RTA, Meijs GF, Moad CL, Moad G, Rizzardo E, Thang SH. Living free-radical polymerization by reversible addition-fragmentation transfer: the RAFT process. Macromolecules. 1999;31:5559–5562. [Google Scholar]
- 5.Convertine AJ, Ayres N, Scales CW, Lowe AB, McCormick CL. Facile, controlled, room-temperature RAFT polymerization of N-isopropylacrylamide. Biomacromolecules. 2004;5:1177–1180. doi: 10.1021/bm049825h. [DOI] [PubMed] [Google Scholar]
- 6.Moad G, Rizzardo E, Thang SH. Living radical polymerization by the RAFT process. Aust J Chem. 2005;58:379–410. [Google Scholar]
- 7.Sumerlin BS, Donovan MS, Mitsukami Y, Lowe AB, McCormick CL. Water-soluble polymers. 84 Controlled polymerization in aqueous media of anionic acrylamido monomers via RAFT. Macromolecules. 2001;34:6561–6564. [Google Scholar]
- 8.Schilli C, Lanzendorfer MG, Muller AHE. Benzyl and cumyl dithiocarbamates as chain transfer agent in the RAFT polymerization of N-isopropylacrylamide. In situ FT-NIR and MALDI-TOF MS investigation. Macromolecules. 2002;35:6819–6827. [Google Scholar]
- 9.Vasilieva YA, Thomas DB, Scales CW, McCormick CL. Direct controlled polymerization of a cationic methacrylamido monomer in aqueous media via the RAFT p(r)ocess. Macromolecules. 2004;37:2728–2737. [Google Scholar]
- 10.Ganachaud F, Monteiro MJ, Gilbert RG, Dourges MA, Thang SH, Rizzardo E. Molecular weight characterization of poly(N-isopropylacrylamide) prepared by living free-radical polymerization. Macromolecules. 2000;33:6738–6745. [Google Scholar]
- 11.Segui F, Qiu XP, Winnik FM. An efficient synthesis of telechelic poly(N-isopropylacrylamides) and its application to the preparation of alpha, omega-dicholesteryl and alpha, omega-dipyrenyl polymers. J Polym Sci Polym Chem. 2008;46:314–326. [Google Scholar]
- 12.Lowe AB, Sumerlin BS, Donovan MS, McCormick CL. Facile preparation of transition metal nanoparticles stabilized by well-defined (co)polymers synthesized via aqueous reversible addition-fragmentation chain transfer polymerization. J Am Chem Soc. 2002;124:11562–3. doi: 10.1021/ja020556h. [DOI] [PubMed] [Google Scholar]
- 13.Scales CW, Convertine AJ, McCormick CL. Fluorescent labeling of RAFT-generated poly(N-isopropylacrylamide) via a facile maleimidethiol coupling reaction. Biomacromolecules. 2006;7:1389–1392. doi: 10.1021/bm060192b. [DOI] [PubMed] [Google Scholar]
- 14.Kulkarni S, Schilli C, Muller AHE, Hoffman AS, Stayton PS. Reversible meso-scale smart polymer-protein particles of controlled sizes. Bioconjugate Chem. 2004;15:747–753. doi: 10.1021/bc034215k. [DOI] [PubMed] [Google Scholar]
- 15.Boyer C, Bulmus V, Liu JQ, Davis TP, Stenzel MH, Barner-Kowollik C. Well-defined protein-polymer conjugates via in situ RAFT polymerization. J Am Chem Soc. 2007;129:7145–7154. doi: 10.1021/ja070956a. [DOI] [PubMed] [Google Scholar]
- 16.De P, Gondi SR, Sumerlin BS. Folate-conjugated thermoresponsive block copolymers: Highly efficient conjugation and solution self-assembly. Biomacromolecules. 2008;9:1064–1070. doi: 10.1021/bm701255v. [DOI] [PubMed] [Google Scholar]
- 17.Heredia KL, Nguyen TH, Chang C, Bulmus V, Davis TP, Maynard HD. Reversible siRNA-polymer conjugates by RAFT polymerization. Chem Commun. 2008;28:3245–3247. doi: 10.1039/b804812f. [DOI] [PubMed] [Google Scholar]
- 18.Maynard HD, Heredia KL, Li RC, Parra DP, Vazquez-Dorbatt V. Thermoresponsive biohybrid materials synthesized by ATRP. J Mater Chem. 2007;17:4015–4017. [Google Scholar]
- 19.Venkataraman S, Wooley KL. ATRP from an amino acid-based initiator: A facile approach for alpha-functionalized polymers. Macromolecules. 2006;39:9661–9664. doi: 10.1021/ma0619583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bathfield M, D’Agosto F, Spitz R, Charreyre MT, Delair T. Versatile precursors of functional RAFT agents. Application to the synthesis of bio-related end-functionalized polymers. J Am Chem Soc. 2006;128:2546–2547. doi: 10.1021/ja057481c. [DOI] [PubMed] [Google Scholar]
- 21.Hong CY, Pan CY. Direct synthesis of biotinylated stimuli-responsive polymer and diblock copolymer by RAFT polymerization using biotinylated trithiocarbonate as RAFT agent. Macromolecules. 2006;39:3517–3524. [Google Scholar]
- 22.Gondi SR, Vogt AP, Sumerlin BS. Versatile pathway to functional telechelics via RAFT polymerization and click chemistry. Macromolecules. 2007;40:474–481. [Google Scholar]
- 23.Ranjan R, Brittain WJ. Combination of living radical polymerization and click chemistry for surface modification. Macromolecules. 2007;40:6217–6223. [Google Scholar]
- 24.Liu J, Bulmus V, Barner-Kowollik C, Stenzel MH, Davis TP. Direct synthesis of pyridyl disulfide-terminated polymers by RAFT polymerization. Macromol Rapid Commun. 2007;28:305–314. [Google Scholar]
- 25.Carter S, Hunt B, Rimmer S. Highly branched poly(N-isopropylacrylamide)s with imidazole end groups prepared by radical polymerization in the presence of a styryl monomer containing a dithioester group. Macromolecules. 2005;38:4595–4603. [Google Scholar]
- 26.Patton DL, Mullings M, Fulghum T, Advincula RC. A facile synthesis route to thiol-functionalized alpha, w-telechelic polymers via reversible addition fragmentation chain transfer polymerization. Macromolecules. 2005;38:8597–8602. [Google Scholar]
- 27.Deletre M, Levesque G. Kinetics and mechanism of polythioamidation in solution 0.1. reaction of mono(dithioester)s and bis(dithioester)s with excess amine. Macromolecules. 1990;23:4733–4741. [Google Scholar]
- 28.Qiu XP, Winnik FM. Synthesis of alpha-, omega-dimercapto poly(N-isopropylacrylamides) by RAFT polymerization with a hydrophilic difunctional chain transfer agent. Macromolecules. 2007;40:872–878. [Google Scholar]
- 29.Roberts MJ, Bentley MD, Harris JM. Chemistry for peptide and protein PEGylation. AdV Drug Deli Very ReV. 2002;54:459–476. doi: 10.1016/s0169-409x(02)00022-4. [DOI] [PubMed] [Google Scholar]
- 30.Toutchkine A, Nalbant P, Hahn KM. Facile synthesis of thiol-reactive Cy3 and Cy5 derivatives with enhanced water solubility. Bioconjugate Chem. 2002;13:387–391. doi: 10.1021/bc015558q. [DOI] [PubMed] [Google Scholar]
- 31.Dufresne MH, Gauthier MA, Leroux JC. Thiol-functionalized polymeric micelles: From molecular recognition to improved mucoadhesion. Bioconjugate Chem. 2005;16:1027–1033. doi: 10.1021/bc050007b. [DOI] [PubMed] [Google Scholar]
- 32.Wang LX, Kristensen J, Ruffner DE. Delivery of antisense oligonucleotides using HPMA polymer: Synthesis of a thiol polymer and its conjugation to water-soluble molecules. Bioconjugate Chem. 1998;9:749–757. doi: 10.1021/bc980034k. [DOI] [PubMed] [Google Scholar]
- 33.Segura T, Hubbell JA. Synthesis and in vitro characterization of an ABC triblock copolymer for siRNA delivery. Bioconjugate Chem. 2007;18:736–745. doi: 10.1021/bc060284y. [DOI] [PubMed] [Google Scholar]
- 34.York AW, Scales CW, Huang FQ, McCormick CL. Facile synthetic procedure for omega, primary amine functionalization directly in water for subsequent fluorescent labeling and potential bioconjugation of RAFT-synthesized (Co)polymers. Biomacromolecules. 2007;8:2337–2341. doi: 10.1021/bm700514q. [DOI] [PubMed] [Google Scholar]
- 35.Nakayama M, Okano T. Polymer terminal group effects on properties of thermoresponsive polymeric micelles with controlled outer-shell chain lengths. Biomacromolecules. 2005;6:2320–2327. doi: 10.1021/bm050232w. [DOI] [PubMed] [Google Scholar]
- 36.Henry SM, El-Sayed MEH, Pirie CM, Hoffman AS, Stayton PS. pH-responsive poly(styrene-alt-maleic anhydride) alkylamide copolymers for intracellular drug delivery. Biomacromolecules. 2006;7:2407–2414. doi: 10.1021/bm060143z. [DOI] [PubMed] [Google Scholar]
- 37.El-Sayed MEH, Hoffman AS, Stayton PS. Rational design of composition and activity correlations for pH-sensitive and glutathione-reactive polymer therapeutics (vol 101, pg 47, 2005) J Controlled Release. 2005;104:415. doi: 10.1016/j.jconrel.2004.08.032. [DOI] [PubMed] [Google Scholar]
- 38.Cheung CY, Stayton PS, Hoffman AS. Poly(propylacrylic acid)-mediated serum stabilization of cationic lipoplexes. J Biomater Sci, Polym Ed. 2005;16:163–179. doi: 10.1163/1568562053115390. [DOI] [PubMed] [Google Scholar]
- 39.Kyriakides TR, Cheung CY, Murthy N, Bornstein P, Stayton PS, Hoffman AS. pH-sensitive polymers that enhance intracellular drug delivery in vivo. J Controlled Release. 2002;78:295–303. doi: 10.1016/s0168-3659(01)00504-1. [DOI] [PubMed] [Google Scholar]
- 40.Johns RE, Hoffman AS, Stayton PS. Expression analysis of LPS stimulated THP-1 cells treated with antisense IRAK1 delivered using a pH-sensitive, membrane disruptive polymer. Mol Ther. 2004;9:S242–S242. [Google Scholar]
- 41.Convertine AJ, Benoit DSW, Duvall CL, Hoffman AS, Stayton PS. Novel endosomolytic diblock copolymer for siRNA delivery. J Controlled Release. 2008 doi: 10.1016/j.jconrel.2008.10.004. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Flanary S, Hoffman AS, Stayton PS. Antigen delivery with poly(propylacrylic acid) conjugation enhances MHC-1 presentation and T-cell activation. Bioconjugate Chem. 2008 doi: 10.1021/bc800317a. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Narain R, Gonzales M, Hoffman AS, Stayton PS, Krishnan KM. Synthesis of monodisperse biotinylated p(NIPAAm)-coated iron oxide magnetic nanoparticles and their bioconjugation to streptavidin. Langmuir. 2007;23:6299–6304. doi: 10.1021/la700268g. [DOI] [PubMed] [Google Scholar]
- 44.Lai JJ, Hoffman JM, Ebara M, Hoffman AS, Estournes C, Wattiaux A, Stayton PS. Dual magnetic-/temperature-responsive nanoparticles for microfluidic separations and assays. Langmuir. 2007;23:7385–7391. doi: 10.1021/la062527g. [DOI] [PubMed] [Google Scholar]
- 45.Ebara M, Hoffman JM, Stayton PS, Hoffman AS. Surface modification of microfluidic channels by UV-mediated graft polymerization of non-fouling and ‘smart’ polymers. Radiat Phys Chem. 2007;76:1409–1413. [Google Scholar]
- 46.Amou S, Nishimura S, Takahashi A, Hagiwara T, Hamana H, Narita T. Synthesis and polymerization of N-(4-tetrahydropyranyloxyphenyl)maleimide. J Polym Sci Polym Chem. 1999;37:341–347. [Google Scholar]
- 47.Hill DT, Shao LY, Pomery PJ, Whittaker AK. The radical homopolymerization of N-phenylmaleimide, N-n-hexylmaleimide and N-cyclohexylmaleimide in tetrahydrofuran. Polymer. 2001;42:4791–4802. [Google Scholar]
- 48.Zhao B, Wang Y, Lu C, Shen Q, Deng M. Polymerization of N-phenyl maleimide by lanthanide complexes. J Polym Sci Polym Chem. 2005;43:3966–3972. [Google Scholar]
- 49.Sandreczki TC, Brown IM. Characterization of the free-radical homopolymerization of N-methylmaleimide. Macromolecules. 1990;23:1979–1983. [Google Scholar]
- 50.Caulfield MJ, Solomon DH. Studies on polyimides: 2. Formation of high molecular weight poly(N-(hydroxyphenyl) maleimides) Polymer. 1999;40:1251–1260. [Google Scholar]
- 51.Ferritto M, Tirrell DA. Poly(2-ethylacrylic acid) Macromol Synth. 1992;11:59–62. [Google Scholar]
- 52.Li M, De P, Gondi SR, Sumerlin BS. End group transformations of RAFT-generated polymers with bismaleimides: Functional telechelics and modular block copolymers. J Polym Sci Polym Chem. 2008;46:5093–5100. [Google Scholar]
- 53.Jeong JH, Kim SH, Kim SW, Park TG. In vivo tumor targeting of ODN-PEG-folic acid/PEI polyelectrolyte complex micelles. J Biomater Sci, Polym Ed. 2005;16:1409–19. doi: 10.1163/156856205774472335. [DOI] [PubMed] [Google Scholar]
- 54.Song E, Zhu P, Lee SK, Chowdhury D, Kussman S, Dykxhoorn DM, Feng Y, Palliser D, Weiner DB, Shankar P, Marasco WA, Lieberman J. Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat Biotechnol. 2005;23:709–17. doi: 10.1038/nbt1101. [DOI] [PubMed] [Google Scholar]
- 55.Hilgenbrink AR, Low PS. Folate receptor-mediated drug targeting: From therapeutics to diagnostics. J Pharm Sci. 2005;94:2135–2146. doi: 10.1002/jps.20457. [DOI] [PubMed] [Google Scholar]
- 56.Bae KH, Lee Y, Park TG. Oil-encapsulating PEO-PPO-PEO/PEG shell cross-linked nanocapsules for target-specific delivery of paclitaxel. Biomacromolecules. 2007;8:650–6. doi: 10.1021/bm0608939. [DOI] [PubMed] [Google Scholar]