

Summary
The histone acetyltransferase (HAT) p300/CBP is a transcriptional coactivator implicated in many gene regulatory pathways and protein acetylation events. While p300 inhibitors have been reported, a potent, selective, and readily available active-site directed small molecule inhibitor is not yet known. Here we use a structure-based, in silico screening approach to identify a commercially available pyrazolone-containing small molecule p300 HAT inhibitor, C646. C646 is a competitive p300 inhibitor with a Ki of 400 nM and is selective versus other acetyltransferases. Studies on site-directed p300 HAT mutants and synthetic modifications of C646 confirm the importance of predicted interactions in conferring potency. Inhibition of histone acetylation and cell growth by C646 in cells validate its utility as a pharmacologic probe and suggest that p300/CBP HAT is a worthy anti-cancer target.
The reversible acetylation of histones and other proteins rivals protein phosphorylation as a major mechanism for cellular regulation (Walsh, 2006; Choudhary et al., 2009; Macek et al., 2009). Acetylation on protein lysine residues is catalyzed by histone acetyltransferases (HATs) and acetyl-Lys cleavage is performed by histone deacetylases (HDACs) (Hodawadekar and Marmorstein, 2007; Haberland et al., 2009; Cole 2008). These enzymes and the associated acetylation events have been implicated in a wide variety of physiological and disease processes. In this study, we focus on the paralog HATs p300 and CBP (referred to as p300/CBP), which were originally discovered as E1A oncoprotein binding partners and cyclic AMP effectors, respectively (Goodman and Smolik, 2000). p300/CBP often serves as a transcriptional coactivator and has been suggested to bind to a range of important transcription factors (Goodman and Smolik, 2000). In 1996, p300/CBP was reported to possess intrinsic HAT activity (Ogryzko et al., 1997; Bannister et al., 1996). Over the ensuing years, p300/CBP has been shown to be a rather promiscuous acetyltransferase, with more than 75 protein substrates described including p53, MyoD, and NFκB (Gu et al., 1997; Yang et al., 2008; Wang et al., 2008). Dissecting the importance of the enzymatic activity of p300/CBP as opposed to its protein recruitment functions in clarifying p300/CBP's biological roles would benefit from selective cell permeable HAT inhibitors. Recent studies suggest that the biologic functions of p300/CBP HAT activity may be associated with tumorigenesis, and it is therefore plausible that p300/CBP HAT inhibitors may serve as potential anti-cancer agents (Dekker et al., 2009; Iyer et al., 2007).
While studies on histone deacetylases have led to the discovery of highly potent compounds with clinical impact in cancer, the identification of histone acetyltransferase inhibitors has proved more challenging (Cole, 2008). Several reports of p300/CBP HAT inhibitors identified through screens or based on bisubstrate analogs have been reported (Lau et al., 2000; Thompson et al., 2001; Zheng et al., 2005; Guidez et al., 2005; Liu et al, 2008; Stimson et al., 2005; Balasubramanyam et al., 2003; Balasubramanyam et al., 2004; Mantelingu et al., 2007; Arif et al., 2009; Ravindra et al., 2009). The most potent and selective compound, Lys-CoA (Ki=20 nM), has been converted to a cell permeable form with Tat peptide attachment (Lys-CoA-Tat) and has been used in a variety of studies, but its complexity is somewhat limiting for pharmacologic applications (Lau et al., 2000; Thompson et al., 2001; Zheng et al., 2005; Guidez et al., 2005; Liu et al, 2008). High throughput screening experiments have led to several small molecule synthetic agents and natural product derivatives of moderate potency as p300 HAT inhibitors (micromolar Ki values) but their selectivity and mechanism of inhibition remains to be fully characterized (Stimson et al., 2005; Balasubramanyam et al., 2003; Balasubramanyam et al., 2004; Mantelingu et al., 2007; Arif et al., 2009; Ravindra et al., 2009).
A recent high resolution X-ray structure of the p300 HAT in complex with the bisubstrate analog Lys-CoA has revealed key aspects of substrate recognition and catalytic mechanism (Liu et al., 2008). A narrow tunnel in p300 accommodates Lys-CoA, and the inhibitor makes a range of hydrogen bonding and Van der Waals interactions with the HAT active site (Liu et al., 2008). Based on this structure and steady-state kinetic studies, a Theorell-Chance catalytic mechanism has been proposed (Liu et al., 2008). This “hit and run” kinetic mechanism involves initial, stable binding of acetyl-CoA followed by weak and transient interaction with histone substrate which permits acetyl transfer. The p300/CBP mechanism differs from that of another family of HATs, PCAF/GCN5 (Poux et al., 2002), which use a ternary complex mechanism.
The p300 HAT/Lys-CoA crystal structure affords the opportunity for computational docking of novel inhibitory scaffolds. In this regard, recent virtual ligand screening based on the X-ray structure of serotonin N-acetyltransferase (AANAT) complexed to a bisubstrate analog led to the identification of a novel, moderately potent and selective small molecule inhibitor for this circadian rhythm enzyme (Szewczuk et al., 2007). In this study, we have pursued the use of this Lys-CoA/p300 HAT structure in virtual ligand screening to identify novel small molecule p300 HAT inhibitors. Below, we describe an in silico search and experimental validation that has led to a commercially available, selective, sub-micromolar small molecule p300 HAT inhibitory agent with pharmacologic potential.
Results
Ranking and Selection of Compounds from Virtual Screening of the p300 HAT
Compounds from a screening set of nearly 500,000 commercially available small molecules were docked into the p300 HAT structure in the Lys-CoA binding pocket and assigned a score using the ICM-VLS software version 3.5. The components of the ICM (Internal Coordinate Mechanics) score include the internal force-field energy of the ligand, conformational entropy loss of the ligand, receptor-ligand hydrogen-bond interaction, solvation electrostatic energy change, hydrogen-bond donor/acceptor desolvation and hydrophobic energy (Totrov et al., 1999). The score is trained to separate binders and non-binders and to rank the interaction of the compound with the receptor. The best score from each of the three docking runs was recorded. The screened compounds were then ranked by ICM score and the 194 highest scoring compounds (representing 0.04% of the total compound database) were visually inspected and selected based on their availability and interactions with the receptor. Each of the 194 compounds was purchased from ChemBridge and tested experimentally as described below.
Experimental p300 HAT assays and inhibitor validation
After completing the computational docking screen, we tested the top 194 inhibitors of the p300 HAT by employing a convenient spectrophotometric assay (Kim et al., 2000) followed by a series of secondary assays (Supplementary Table 1). In the coupled spectrophotometric assay, the acetyltransferase reaction product CoASH becomes a substrate for alpha-ketoglutarate dehydrogenase, which converts NAD to NADH, resulting in an increase of UV absorbance at 340 nm (Kim et al., 2000). Screening conditions for these p300 assays included semisynthetic HAT enzyme (Thompson et al., 2004), substrate concentrations of 200 μM histone H4-15 peptide and 50 μM acetyl-CoA, and potential small molecule inhibitors tested at 100 μM. Thirty of the original 194 compounds showed at least 50% inhibition under these conditions. To distinguish inhibitory effects related to blockade of coupling enzyme rather than p300, assays were repeated using increased (2-fold) alpha-ketoglutarate dehydrogenase. This eliminated six of the thirty hits. To test for protein aggregation-related inhibitory phenomena (Feng et al., 2005), assays with the remaining compounds were next performed in the presence of 0.01% Triton X-100, which attenuated inhibition of eleven of 24 compounds. NMR and mass spectrometry confirmed the structure and purity of the thirteen potential p300 inhibitors which were subsequently tested in a direct, radioactive assay for IC50 measurements. In this radioactive p300 HAT assay, 14C transfer from 14C-acetyl-CoA into peptide substrate is directly measured using Tris-Tricine SDSPAGE and phosphorimage analysis. Three compounds, C646 (ChemBridge#5838646), C146 (ChemBridge#5202146), and C375 (ChemBridge#6643375) were ultimately shown to be relatively potent p300 HAT inhibitors (Ki<5 μM) (Figure 1A). Interestingly, though C646, C146, and C375 possess distinct scaffolds, these three compounds all contain a linear arrangement of 3-4 aromatic rings terminating in a benzoic acid.
Figure 1.
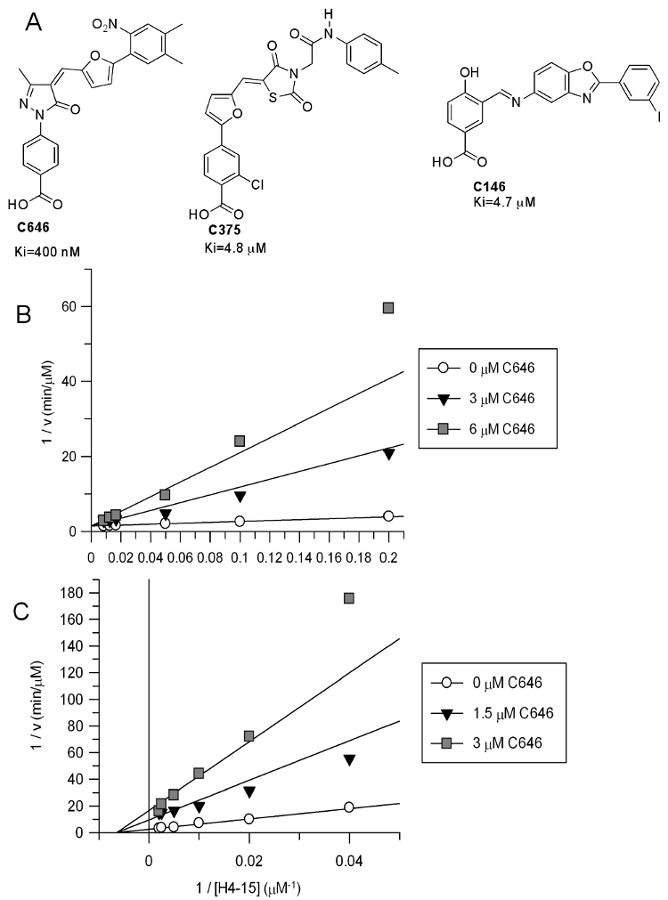
A. Three validated p300 HAT small molecule inhibitors C646, C375, and C146 and their inhibitory constants measured as described in B and Supplementary Figure 1. B. Inhibitory characteristics of C646 toward p300. A. Plot of 1/v vs. 1/[acetyl-CoA] at fixed H4-15 peptide substrate (100 μM) and three concentrations of C646 shows competitive inhibition. C646 Ki=400±60 nM, apparent acetyl-CoA Km=8.5±1.4 μM, apparent kcat=18±1 min-1. C. Plot of 1/v vs. 1/[peptide substrate (H4-15)] at fixed acetyl-CoA (10 μM) and three concentrations of C646 shows noncompetitive inhibition. C646 Ki=530±40 nM, apparent H4-15 Km=155±19 μM, apparent kcat=40±2 min-1.
Acetyltransferase inhibitor selectivity
Each of the three inhibitors C646, C146, and C375 was further analyzed versus other acetyltransferases for specificity (Supplementary Table 2). We looked at serotonin N-acetyltransferase (Szewczuk et al., 2007), PCAF histone acetyltransferase (Lau et al., 2000), GCN5 histone acetyltransferase (Poux et al., 2002), Rtt109 histone acetyltransferase (Tang et al., 2008), Sas histone acetyltransferase (Shia et al., 2005), and MOZ histone acetyltransferase (Holbert et al., 2007). While compound C646 at 10 μM was highly selective in inhibiting p300 (86% inhibition) versus the other six acetyltransferases (less than 10% inhibition), C146 and C375 were less selective, inhibiting at least one of these enzymes with comparable potency to their p300 blockade (Supplementary Table 2). Thus compound C646 looked most promising for applications requiring selective inhibition of p300/CBP.
Mechanism of p300 inhibition
We evaluated the steady-state kinetic mechanism of inhibition of p300 by compounds C646, C146, and C375 by exploring inhibitory effects over a range of acetyl-CoA concentrations. Compounds C646 proved to be a linear competitive inhibitor of p300 versus acetyl-CoA with a Ki of 400 nM (Figure 1B). Compound C146 showed pseudo-competitive inhibition versus acetyl-CoA with Ki-slope=4.7 μM, although this compound exhibited a measurable Ki-intercept (35 μM) suggesting that C146 still had affinity for the acetyl-CoA-bound form of p300 (Supplementary Figure 1A) (Copeland, 2000). C375 showed classical noncompetitive inhibition versus p300 (Ki=4.8 μM) indicating that it could bind efficiently to the acetyl-CoA bound form of p300 (Supplementary Figure 1B). Computational models of the complex of these compounds bound to p300 show extensive overlap with the CoA-p300 interaction, so we predicted that a pure competitive kinetic model would be observed. This was only true for C646. In further analysis, compound C646 showed a noncompetitive pattern of p300 inhibition versus H4-15 peptide substrate (Figure 1C), consistent with the expected behavior of a bisubstrate analog interacting with an enzyme with ordered substrate binding, like that of p300 (Yu et al., 2006). Compound C146 was found to be a competitive inhibitor of p300 versus H4-15 peptide substrate (Supplementary Figure 1C), suggesting a more complex mode of interaction with the enzyme than a simple bisubstrate analog.
C646 is not a time-dependent inactivator of p300
The conjugated pyrazolone exomethylene vinyl functionality in C646 appeared to us to be potentially electrophilic, serving as a possible Michael acceptor, which could covalently modify its protein target. The nucleophilic compound dithiothreitol (DTT) was added to p300 HAT assays, and we showed that C646 inhibitory potency was similar (within 2-fold) if DTT was replaced with beta-mercaptoethanol or glutathione. We investigated whether DTT or beta-mercaptoethanol could generate adducts with C646, however NMR and chromatography revealed no evidence of reaction under the buffer conditions of the enzyme assay. We deduce that this resistance to thiol adduct formation results from the extended conjugation of the polyaromatic system in C646.
Irreversible enzyme inhibitors typically exhibit time-dependent enzyme inactivation (Copeland, 2000; Kitz and Wilson, 1962). In contrast, acetyl transfer catalyzed by p300 HAT in the presence of C646 remained linear over time, consistent with the behavior of a reversible inhibitor (Supplementary Figure 2A). Moreover, incubation of C646 with p300 for 2, 4, 6, 8 or 10 min prior to acetyltransferase assays showed that the level of inhibition was independent of preincubation time (Supplementary Figure 2B). Taken together, these results suggest that C646 is a classical reversible p300 inhibitor, consistent with the steady-state kinetic analysis described above.
Conformational analysis of C646
Prior studies suggested that pyrazolone-furan compounds such as C646 could exist in two double bond stereoisomers involving the exomethylene (Moreau, 2008) (Supplementary Figure 3A). Since the computational docking model of C646 bound to p300 suggests that the compound binds as the Z-isomer, we investigated C646 stereochemistry in solution using 1H NMR and HPLC. 1H NMR analysis of C646 in DMSO suggests a 70:30 Z:E-mixture of the olefinic isomers based on the pyrazolone methyl protons (Supplementary Figure 3A). Using 1H-1H 2D NOESY NMR analysis, the major peak can be assigned as the E-isomer based on the NOE between the vinyl proton and the methyl protons in the Z-isomer. We also show that these isomers can be separated by reversed phase HPLC (Supplementary Figure 3B) but that they facilely interconvert, since re-injection of samples derived from the individual peaks shows that re-equilibration of peaks is established within a few hours. These data suggest that the Z-isomer is readily accessible and likely to be favored in solution.
Exploring the interactions of C646 and p300 using site-directed mutagenesis
While we have not yet been successful in obtaining an X-ray structure of C646 complexed to p300 HAT, we have used site-directed mutagenesis to evaluate specific interactions predicted by the computational model. In this model, a series of hydrogen bonding donor interactions from the side chains of Thr1411, Tyr1467, Trp1466, and Arg1410 to oxygen atoms of C646 are proposed (Figure 2A). These sidechains also make interactions with Lys-CoA based on the X-ray structure (Figure 2A) (Liu et al., 2008). We thus tested C646 against p300 HAT mutants T1411A, Y1467F, W1466F, and R1410A and the corresponding IC50 values are shown in Figure 2B. Since each of these mutants can alter acetyl-CoA interactions as well, we measured the corresponding Kms for acetyl-CoA for the four mutant proteins and used the equation Ki = IC50/(S/Km+1) to calculate the apparent Ki values based on a competitive inhibition model (Copeland, 2000). As shown in Figure 2B, each mutation increased the apparent Ki of C646 by at least 2-fold, and the most significant effect was seen with W1466F which showed a 7-fold increase in Ki. This increase may reflect a combination of the loss of the hydrogen bond from the indole nitrogen as well as altered Van der Waals contacts. It is noteworthy that the kcat/Km for W1466F p300 is essentially identical to the wild type enzyme (Figure 2C), suggesting that inhibition by C646 and catalysis rely on subtly different forces.
Figure 2.

Interactions of C646 with p300. A. In silico model of C646 bound to p300 HAT active site (upper) shows overlapping interactions with the X-ray crystal structure of p300 HAT complexed with Lys-CoA (lower). B. Kinetic analysis of selected active site mutants
Structure-Activity-Relationship Analysis of C646 in p300 inhibition
To explore structural elements of C646 responsible for inhibition of p300, we developed modular synthetic approaches to a series of analogs that could probe the benzoate, pyrazolone, exomethylene, and arylnitro moieties (see Fig. 3 and Supplementary Schemes 1-9). The general synthetic approach to prepare many of these analogs involved initially producing the building block phenyl-pyrazolones (3) and the aryl-furan aldehydes (Fig. 3). Production of the phenyl-pyrazolones (3) (Kim and Lee, 1991) was achieved from the corresponding anilines 1 which could be diazotized and transformed to the aryl hydrazines (2). Reaction of intermediates 2 with ethyl acetylacetate led to pyrazolones (3) formation. Palladium-catalyzed Suzuki coupling of 5-formyl-2-furanboronic acid with aryl halides (4) led to aryl-furan aldehydes (5) (Langner et al., 2005; Hosoya et al., 2003). Knoevenagel condensation between 3 and 5 generated the desired analogs of C646 (Vasyunkina et al., 2005).
Figure 3.

Synthetic analogs of C646 and their relative IC50s for p300 inhibition referenced to C646.
Each of the analogs 6a-r along with two other related commercially available derivatives were tested for p300 HAT inhibition using the direct, 14C-acetyl-CoA transfer assay at a range of concentrations and the relative IC50s are shown in Figure 3. Derivatization of the carboxylic acid of C646 as the methyl ester affording 6h led to a dramatic weakening of inhibitory potency (>15-fold), establishing the likely importance of this functionality in hydrogen bonding. Reduction of the C646 enone with sodium borohydride gives rise to C37 (6c) and this compound did not detectably block p300 HAT activity (Figure 3). The loss of inhibitory potency of C37 suggests that shape (planarity) and/or electronic properties of the conjugated system in C646 is critical. Replacement of the C646 nitro group with hydroxymethylene (6l) also led to a substantial reduction in potency supporting the importance of hydrogen bonding of the nitro predicted by the computational model.
Other hydrogen bond acceptors replacing the C646 para-carboxylic acid were well tolerated at both the para and meta positions including a para-carboxamide (6a), a para-sulfonic acid (6f), a para-sulfonamide (6g), and a meta-methyl ester (6i) or carboxylic acid (6e). Molecular modeling reveals considerable p300 active site flexibility in accommodating these other para- and meta-substituted compounds (Supp. Figure 4). Molecular recognition versatility with regard to nitrophenyl interactions is indicated by the fact that replacement of the nitro group by methylbenzoate (6m) or cyanophenyl (6o) functionalities is also well tolerated (Figure 3). That the nitrophenyl group could be effectively replaced by a methylbenzoate might offer pharmacokinetic advantages for in vivo studies (Hodgkiss et al., 1991). However, replacement of the nitrophenyl ring with a pyridine ring (6p) eliminated p300 inhibitory action.
Replacing the pyrazolone of C646 with a succinimide ring in the context of a methyl benzoate substituent replacing the nitro group (6r) retains potency for p300 inhibition (Figure 3). Since succinimides related to 6r are believed to exist with a strong preference for E-exomethylene stereochemistry (Haval and Argade, 2008), this result further supports the conformational arrangement predicted by the docking model for C646. We have found that a propionate group (6k) cannot successfully replace the benzoate ring of C646 and still retain p300 inhibitory potency. Interestingly, carbonyl substitution of the exomethylene vinyl (6q) shows 6-fold weaker potency compared with C646.
Effects of C646 on histone acetylation in mouse cells
We next investigated the effects of compound C646 on cellular histone acetylation using C3H 10T1/2 mouse fibroblasts. We examined histone acetylation using modification-specific antibodies against H3K9ac and H4K12ac (Figure 4). In addition to analysis of total acetylation levels by western blots of SDS gels (Figure 4A) we used acid-urea (AU) gels (Figure 4B) which separate histones on the basis of charge. Loss of a positive charge upon lysine acetylation retards migration, producing a ladder of bands corresponding to integral changes in acetylation (Shechter et al., 2007).
Figure 4.
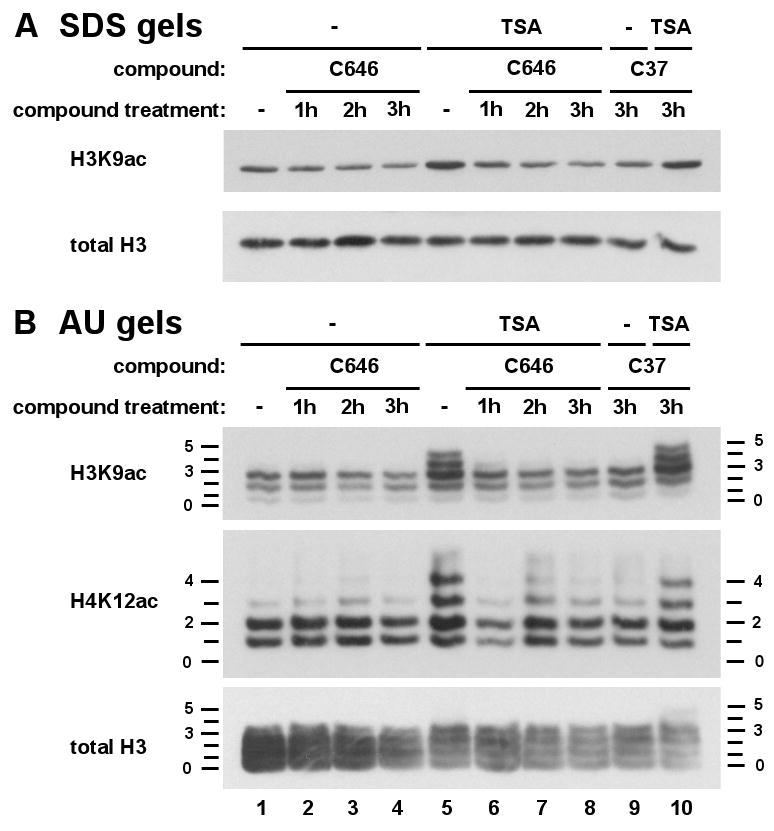
C646 treatment reduces histone H3 and H4 acetylation levels and abrogates TSA-induced acetylation in cells. Quiescent C3H 10T1/2 mouse fibroblasts were pretreated with C646 (25 uM, lanes 2-4, 6-8) for the indicated times (1, 2 or 3h) or the control compound, C37 (25 uM, 3h, lanes 9, 10), and TSA (33 nM) added where indicated (lanes 6-8, 10) for the final 30 min of incubation. Histones were extracted and analysed by western blotting to visualize total H3 and H3K9ac using SDS (Panel A) or H3, H3K9ac, and H4K12ac using acid-urea (Panel B) gel electrophoresis. Acid-urea gels separate histones on the basis of charge. Each acetylation event incrementally retards migration to produce a ‘ladder’ of bands, numbers on either side of the blot indicating extent of modification. (lane 1, untreated control cells; lane 5, TSA-treated cells).
In control cells, basal acetylation levels of H3 and H4 are slightly diminished by C646 treatment over the 1-3 h time-course tested (Figures 4A and B; lanes 1-4). To further investigate this effect, we used a 30 min treatment with the HDAC inhibitor trichostatin A (TSA; Yoshida et al., 1990) which produces increased H3 and H4 acetylation, visualized by the appearance of higher migrating forms in AU gels (Figures 4A and B; compare lanes 1 and 5). At all time-points tested (1-3 h; lanes 6-8), incubation with 25 μM C646 prior to addition of TSA virtually completely inhibits this inducible acetylation, both in SDS and AU gels.
By contrast 3 h treatment with the C646 analog C37, which is devoid of p300 HAT inhibitory activity in vitro, produces no effect on acetylation of H3 or H4 (Figures 4A and B; lanes 9, 10). Thus, blockade of p300/CBP by C646 treatment of intact cells results in inhibition of both basal and TSA-inducible acetylation of histones H3 and H4, demonstrating its efficacy in live cells. Similar inhibition was observed in cells subject to RNA interference targeting this enzyme (Crump et al; data not shown).
Cell growth effects of C646 on melanoma and lung cancer lines
Given the broad and important effects of p300/CBP on key genes and pathways involved in cell growth, we explored the pharmacologic effects of C646 on 3H-thymidine incorporation at 24h in three melanoma (Figure 5A) and three non-small cell lung cancer lines (Figure 5B). We compared the effects of C646 (10 μM) and the peptide-based bisubstrate p300/CBP HAT inhibitor Lys-CoA-Tat (25 μM) and found that both were capable of reducing 3H-thymidine incorporation in several of these human cancer lines to varying degrees (Figure 5A and 5B). Control compounds, C37 and Ac-DDDD-Tat, which lack p300 inhibitory properties, had no effects on 3H-thymidine incorporation as shown in Figure 5A and 5B. In general, reduction of 3H-thymidine incorporation was correlated for Lys-CoA-Tat and C646 among the different cell lines (Figure 5A,B), consistent with a common protein target presumed to be the p300/CBP HAT, although C646 generally showed greater effects at the doses used. For example, the melanoma line WM35 and the lung cancer line H23 were quite susceptible to C646 treatment with Lys-CoA-Tat showing modest inhibitory effects in these cells. Notably, the immortalized murine fibroblast cell line, NIH3T3, was not inhibited by C646 at 20 μM suggesting a differential effect on the malignant cells evaluated (Figure 5C).
Figure 5.
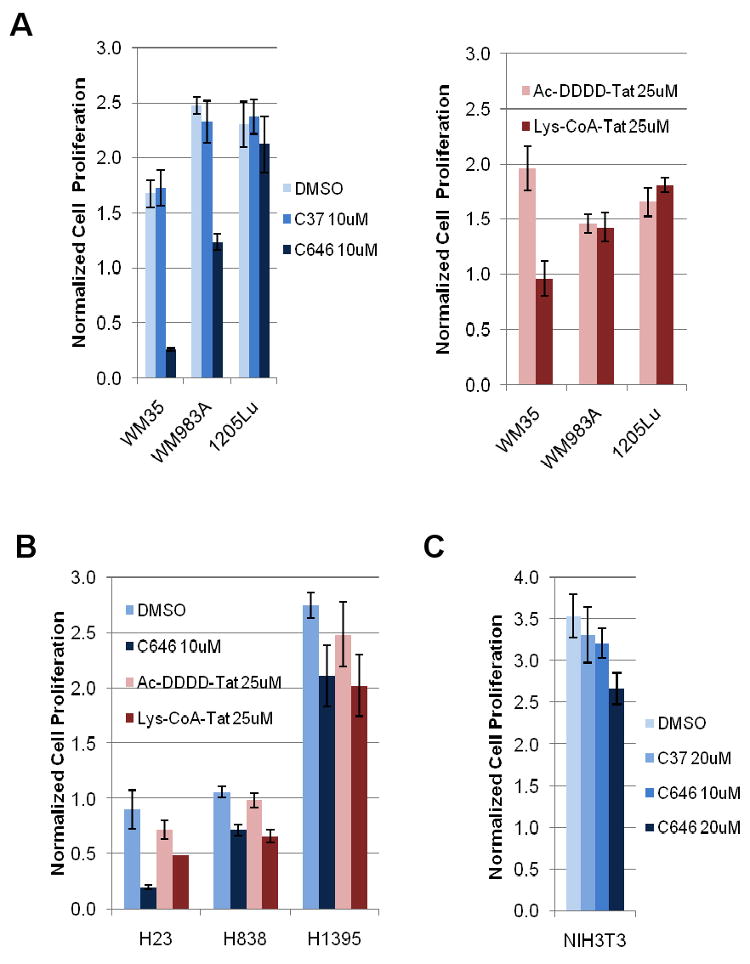
(A) C646 has a more potent effect on cell growth than Lys-CoA-Tat does. Cells were treated for 24 h and proliferation was measured via 3H-thymidine incorporation pre- and post-treatment. Data for each cell line were normalized to 3H counts measured pre-treatment. C37 is an inactive derivative of C646. DMSO serves as a vehicle control for C37 and C646. Ac-DDDD-Tat is a control peptide for Lys-CoA-Tat. WM35, WM983A, and 1205Lu are melanoma cell lines representing three stages of cancer progression: radial, vertical, and metastatic, respectively. (B) Two out of three non-small cell lung adenocarcinoma cell lines demonstrate significant growth inhibition after treating with C646 and Lys-CoA-Tat for 24 h. (C) C646 does not have a significant inhibitory effect on NIH3T3 cell proliferation.
We pursued more detailed studies on the moderately sensitive melanoma line WM983A and showed that the IC50 for C646 was in the range of 10-20 μM, but that this cell line was resistant to the C646 analog control compound C37 at 20 μM (Figure 6A). Correlating with this growth effect on WM983A cells was a dose-dependent reduction in global histone H3 acetylation revealed by western blotting that were treated for 6 h (Figure 6B). Based on FACS analysis, the effects of C646 (20 μM, 24 h treatment) on WM983A and the more sensitive melanoma line WM35 were consistent with a G1-S cell cycle arrest, predicted to be related to p300 modulation (Iyer et al., 2007), since the S-phase dropped under these conditions (Figure 6C).
Figure 6.
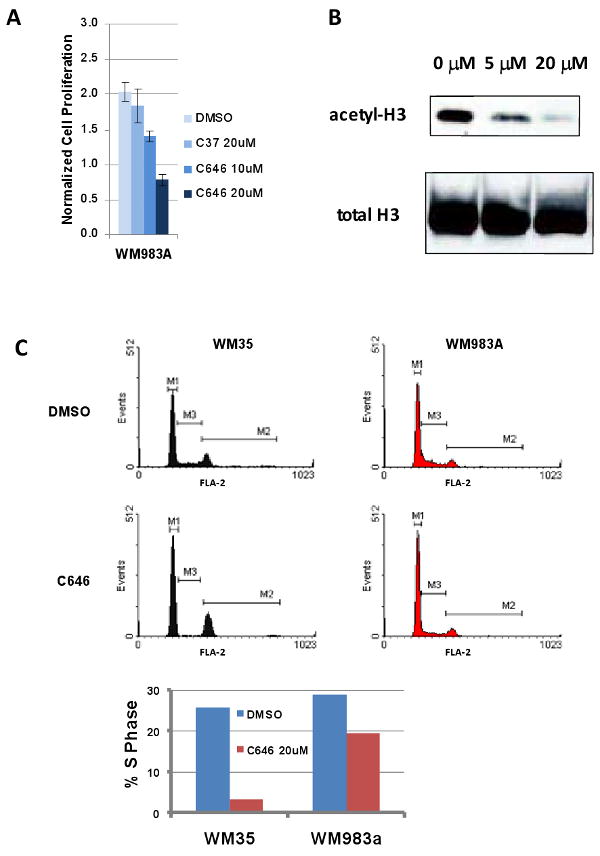
(A) C646 inhibits WM983A melanoma cell proliferation but C37 does not. Cells were treated with each compound for 24 h. Proliferation was measured via 3H-thymidine incorporation. (B) C646 blocks H3 acetylation in WM983A cells. Cells were treated for 6 h with increasing concentrations of C646. Nuclear lysates were subjected to western blot analysis for acetylated H3 (Upstate 06-599). Total H3 (Abcam ab1791) was blotted as a loading control. (C) C646 causes growth arrest in melanoma cell line WM35 and WM983A, indicated by a decrease in %S phase. Cells were treated for 24 h with 20 μM C646 or DMSO, then collected and stained with propidium iodide, followed by FACS analysis.
Discussion
This study has led to the identification of a relatively potent and selective small molecule p300 HAT inhibitor which was discovered by using virtual ligand screening. The docking model used the ICM approach which has been successful in identifying ligands for a range of other protein targets (Szewczuk et al., 2007; Mallya et al., 2007; Bisson et al., 2009; Cavasotto et al., 2008). An X-ray crystal structure of the complex of p300 HAT with the bisubstrate analog Lys-CoA revealed a pocket which included a series of hydrogen bonding residues that interact with the pantetheine moiety of Lys-CoA as well as two phosphate groups. The pyrazolone C646 was predicted to dock into this site, utilizing several of the same residues. Such active site interactions between C646 and p300 were projected to result in exclusion of acetyl-CoA. Consistent with this model, C646 was a competitive p300 inhibitor versus acetyl-CoA using steady-state kinetic analysis. No evidence of time-dependent inactivation of p300 by C646 was observed, arguing against the idea that C646 was undergoing covalent reaction with the enzyme. For an ordered binding mechanism like p300's, bisubstrate analogs are predicted to be noncompetitive inhibitors versus the second substrate, as found for C646.
The nature of the interaction between C646 and p300 was also probed by altering enzyme residues and functional groups in the inhibitor. These studies showed a maximal effect of a 7-fold drop in affinity by mutagenesis of Trp1466 to Phe in p300 and >15-fold effects for several synthetic modifications of C646. Taken together, these results further support active site targeting of p300 by C646 although ultimately an X-ray crystal structure of the complex will be needed to most rigorously test the computational model proposed. The relative resistance of the W1466F p300 mutant to C646 but the retention of wild type catalytic activity suggest that it may ultimately be possible to pursue chemical-genetic complementation studies on p300/CBP analogous to those done on protein kinases (Shogren-Knaak et al., 2001; Qiao et al, 2006).
Compound C646 appears selective versus the other acetyltransferases so far examined. Compared to HDACS, HATs show greater sequence and structural divergence and specific targeting is more tractable (Hodawadekar and Marmorstein, 2007; Haberland et al., 2009; Cole, 2008). Although p300 and Rtt109 are similar in fold, these two proteins show very little or no sequence homology to other HATs and very modest structural detailed homology in active site interactions (Wang et al., 2008). Moreover, the GNATs PCAF and serotonin N-acetyltransferase and the MYST family member MOZ make distinctive three-dimensional interactions with CoA (Hodawadekar and Marmorstein, 2007).
The most potent and selective small molecule p300/CBP HAT inhibitor prior to this study is a garcinol derivative, LTK14, with a Ki of 5.1 μM (Mantelingu et al., 2007; Arif et al., 2009), about 12-fold weaker than C646. The complex natural product analog LTK14 is suggested to be a noncompetitive p300 inhibitor versus both acetyl-CoA and peptide and the structural basis of inhibition remains uncertain (Arif et al., 2009). Other published, small molecule p300 inhibitors including anacardic acid, curcumin, plumbagin, and isothiazolones have been demonstrated to be non-specific or are predicted to be quite chemically reactive, limiting their utility (Stimson et al., 2005; Balasubramanyam et al., 2003; Balasubramanyam et al., 2004; Ravindra et al., 2009). Lys-CoA is 20-fold more potent than C646 but is not cell permeable. Lys-CoA-Tat is more potent in vitro than C646 but is apparently also less active in cells and is a much more complicated molecule to make and work with.
Virtual ligand screening offers a relatively rapid, economical approach to search for potent enzyme inhibitors. The complexity of coping with solvation and conformational changes in proteins and estimating energetics of atomic interactions makes small molecule in silico docking a continuing challenge. Indeed, only three of the top 194 computational p300 ligands were confirmed to be reasonably potent inhibitors and only one proved competitive versus acetyl-CoA as predicted. It is generally understood that the virtual screening against one of even several conformations of the binding pocket is not designed to identify all potential p300/CBP inhibitors because the conformations of the binding pocket for certain chemical classes of inhibitors may extend far beyond the pocket conformation we used. Nevertheless, we suspect that even identifying one relatively potent and selective active site compound for p300 out of 194 experimentally tested compounds represents a higher ratio of success than expected from a high throughput screen of unselected small molecule libraries (Stimson et al., 2005).
We have also shown that C646 works rapidly in cell culture in blocking histone acetylation. The application of selective p300/CBP HAT inhibitors in vivo can help address a range of biological and pathophysiologic questions.
Significance
HDAC inhibitors, despite their limited selectivity, have been enormously important in dissecting the role of histone acetylation in gene expression pathways, and SAHA is clinically used for cancer treatment (Itoh et al., 2008). Our data here with the relatively potent, selective p300/CBP inhibitor C646 indicate the potential of p300/CBP HAT blockade in impeding cancer proliferation, though more work will be needed to clarify the multiple potential mechanisms for such growth effects. Other recently published studies underscore the potential value of blocking p300/CBP HAT in the treatment of diabetes mellitus, cardiac disease, and HIV (Cole, 2008).
Experimental Procedures
Molecular Modeling
The crystal structure of p300 HAT in complex with bisubstrate analog Lys-CoA (pdb: 3BIY) was prepared for virtual screening using the Internal Coordinate Mechanics (ICM-Pro) software version 3.5 (MolSoft LLC, San Diego CA) (Abagyan et al., 2005; Abagyan et al., 1994). The ligand and heteroatom molecules were removed from the structure and a continuous dielectric was used in place of the water molecules. Missing hydrogen and heavy atoms were added and atom types and partial charges were assigned. The protein model was adjusted so that the optimal positions of polar hydrogens were identified, the most isomeric form of each histidine was assigned and the correct orientation of the side-chains of glutamine and asparagine were found. Steric clashes were removed by an energy minimization procedure.
Virtual Screening
Virtual screening was undertaken with the ICM-VLS software version 3.5 (MolSoft LLC, San Diego CA) using dockScan version 4.4. This method uses an extension of the Empirical Conformational Energy Program for Peptides 3 (ECEPP/3) (Nemethy et al., 1992) force field parameters for proteins and the Merck Molecular Force Field (MMFF) (Halgren and Nachbar, 1996) for small molecules. Five continuously differentiable potential grid maps were calculated for the receptor using spline interpolation for efficient gradient minimization (Totrov and Abagyan, 1997). These maps include energy terms for electrostatics, directional hydrogen bond, hydrophobic interactions, and two for Van der Waals interactions for steric repulsion and dispersion attraction including a soft potential to limit the effect of minor steric clashes. A collection of 492,793 compounds from the ChemBridge small molecule database (ChemBridge Corp, San Diego CA) was screened to the p300 HAT bisubstrate inhibitor binding site. Each ligand was docked into the binding pocket three times. During docking the ligand is flexible and its position and internal torsions are sampled using the ICM biased probability Monte Carlo procedure which includes a local minimization after each random move. Each docked ligand is assigned a score according to the weighted components of the ICM-VLS scoring function (Totrov and Abagyan, 1999).
Peptide Synthesis
Synthetic peptides (H4-15, p300 peptides, Lys-CoA-Tat, Ac-DDDD-Tat) were prepared using automated solid phase peptide synthesis and the Fmoc strategy as described previously (Thompson et al., 2001; Zheng et al., 2005; Guidez et al, 2005; Liu et al., 2008; Liu et al., 2008). Peptides were purified using reversed phase HPLC and their structures confirmed by mass spectrometry.
Purification of semisynthetic p300 HAT domain
A variant of the HAT domain of p300 (residues 1287 to 1652) was expressed as a fusion with the VMA intein chitin binding domain as described previously (Thompson et al., 2004). Residues 1529-1560, comprising the regulatory autoacetylation loop, were deleted in the construct, rendering the enzyme constitutively active. The construct also contained an M1652G mutation. E. coli BL21(RIL)-DE3 cells containing the construct were grown to OD600 of 0.6 at 37°. The incubator was cooled to 16°C, and expression was induced by addition of 500 μM IPTG. Following overnight expression, the cells were centrifuged and resuspended in lysis buffer prior to lysis via three passes through a French pressure cell. Lysates were clarified through centrifugation and incubated with chitin resin for 30 min at 4°C. The resin was washed thoroughly before addition of 200 mM MESNA and a C-terminal peptide corresponding to residues 1653-1666 of p300. The expressed protein ligation reaction was allowed to proceed for 16 h followed by purification over a MonoS 5/50 GL strong cation exchange column (GE Healthcare) using linear gradients of NaCl (50 to 1000 mM). Purified semisynthetic p300 was concentrated and dialyzed against 20 mM HEPES, pH 7.9, 50 mM NaCl, 1 mM DTT, and 10% glycerol (v/v) prior to flash-freezing in liquid N2 and storage at -80°C. Protein concentration was determined by gel and by Bradford assay using BSA as a standard.
Initial screen of VLS hits
The top 194 p300 HAT inhibitor candidates identified by VLS were screened using a coupled spectrophotometric assay. In this assay, CoASH produced by the p300 reaction is used by α-ketoglutarate dehydrogenase (α-KGDH) to produce NADH, which can be monitored spectrophotometrically at 340 nm (Kim et al., 2000). Reactions were performed at 30°C in 1 M HEPES, pH 7.9, and contained 200 μM H4-15, 200 μM TPP, 5 mM MgCl2, 1 mM DTT, 50 μg/mL BSA, 200 μM NAD, 2.4 mM α-ketoglutarate, 200 μM acetyl-CoA, 0.1 units α-KGDH, and 100 nM p300. DMSO was kept at a constant 3.3%, and candidate compounds were screened at 100 μM. Reaction mixtures were incubated at 30°C for 10 min prior to initiation. Reactions were initiated with addition of H4-15 and followed over the linear portion of the progress curve, which provides the initial velocity via linear regression. Percent inhibition was determined by comparison with velocity without candidate added. Compounds that exhibited over 40% inhibition were subjected to further screening steps. To ensure that compounds were not inhibiting α-KGDH instead of p300, compounds were assayed with 0.2 units of α-KGDH, two times the amount used in the initial screen. To ensure that compounds were not inhibiting through nonspecific aggregation, compounds were assayed in the presence of 0.01% Triton X-100. Compounds whose inhibition was greatly decreased either by raising the α-KGDH concentration or by preventing nonspecific aggregation were removed from further consideration.
Compounds that passed the iterative verification process in the initial screen were then tested in a direct radioactive assay. In this assay, production of 14C-labeled Ac-H4-15 is monitored electrophoretically (Thompson et al., 2001). Reactions were performed in 20 mM HEPES, pH 7.9, and contained 5 mM DTT, 80 μM EDTA, 40 μg/mL BSA, 100 μM H4-15, and 5 nM p300. DMSO was kept constant at 2.5%, and inhibitors were screened at 25 μM. Reactions were incubated at 30°C for 10 minutes, initiated with addition of a 1:1 mixture of 12C-acetyl-CoA and 14C-acetyl-CoA to a final concentration of 20 μM, and allowed to run for 10 min at 30°C. Reactions are then quenched with addition of 14% SDS (w/v). Turnover was kept below 10%. All compounds were screened in duplicate. Samples were then loaded onto a 16% Tris-Tricine gel along with a BSA standard and run at 140 V for 90 minutes. Gels were washed and dried, and exposed in a PhosphorImager cassette for ∼2 days. Bands corresponding to Ac-H4-15 were then quantified using ImageQuant. Compounds exhibiting over 40% inhibition compared to control were then kinetically characterized.
Kinetic characterization of verified inhibitors and analogs
IC50 values for the putative p300 HAT inhibitors identified through the initial screen were determined using the direct radioactive assay described above. Reactions were performed in 20 mM HEPES, pH 7.9, and contained 5 mM DTT, 80 μM EDTA, 40 μg/mL BSA, 100 μM H4-15, and 5 nM p300. Putative inhibitors were added over a range of concentrations, with DMSO concentration kept constant (<5%). Reactions were incubated at 30° for 10 minutes, then initiated with addition of a 1:1 mixture of 12C-acetyl-CoA and 14C-acetyl-CoA to 20 μM. After 10 min at 30°C, reactions were quenched with 14% SDS (w/v). All concentrations were screened in duplicate. Gels were run, washed, dried, and exposed to a PhosphorImager plate as described above, and production of Ac-H4-15 quantified to obtain IC50s.
Patterns of inhibition of putative p300 HAT inhibitors were determined in a similar fashion. One substrate was held constant while the other was varied over a range of three inhibitor concentrations (0, 0.5xIC50, and IC50). AcCoA was varied from 5-120 μM while holding H4-15 constant at 100 μM, and H4-15 was varied from 25-500 μM while holding acetyl-CoA constant at 10 μM. Reactions were performed in duplicate as described above; enzyme concentration and reaction time were varied to keep turnover below 10%. Following quantification, data were globally fit to equations for competitive or noncompetitive inhibition to determine the optimal pattern of inhibition (Copeland, 2000).
Determining inhibitor specificity for p300 HAT
C646, C375, and C146 were screened spectrophotometrically against PCAF (p300/CBP-associated factor, a histone acetyltransferase) and AANAT (arylalkylamine N-acetyltransferase, a non-histone acetyltransferase) using a similar coupled assay as described above. PCAF reactions were performed in 100 mM HEPES, pH 7.9, and contained 200 μM TPP, 5 mM MgCl2, 1 mM DTT, 50 mM NaCl, 0.05 mg/mL BSA, 200 μM NAD, 2.4 mM α-KG, 30 μM acetyl-CoA, 0.037 units α-KGDH, 3.3% DMSO, and 10 μM inhibitor. Reactions took place at 30°C. Reactions were initiated by addition of 10 nM PCAF and were followed at 340 nm over the linear portion of the curve below 10% turnover. AANAT reactions were performed in 100 mM NH4OAc, pH 6.8, and contained 200 μM TPP, 5 mM MgCl2, 1 mM DTT, 50 mM NaCl, 0.05 mg/mL BSA, 200 μM NAD, 2.4 mM α-KG, 200 μM AcCoA, 0.1 units α-KGDH, 3.3% DMSO, and 10 μM inhibitor. Reactions took place at 25°. Reactions were initiated with addition of 65 nM AANAT, and followed at 340 nm, as above. Percent inhibition values were compared to those with p300, which were repeated using the protocol given above. C646 was further analyzed as a potential HAT inhibitor with yeast GCN5, the Sas2/4/5 complex, MOZ, and Rtt109. Yeast GCN5, MOZ, and the Rtt109/Vps75 complex, were purified as described elsewhere (Poux et al., 2002; Tang et al., 2008; Holbert et al., 2007). The SAS complex was expressed and purified in E. coli. as detailed elsewhere (Shia et al., 2005; Sutton et al., 2003). Briefly, the SAS2, SAS4 and SAS5 proteins were co-expressed using the Duet system (Novagen) in BL21-CodonPlus(DE3)-RIL cells (Strategene). The complex was purified using a combination of nickel affinity, ion exchange (HisTrap SP) and gel filtration (Superdex 200) chromatography.
HAT assays with yeast GCN5, SAS complex, MOZ, and Rtt109/Vps75 complex used the direct radioactive assay described above. Reactions were carried out at 30 °C for times varying from 2 to 4 min under the following reaction conditions: 50 mM HEPES, pH 7.9, 50 mM NaCl, 0.05 mg/ml BSA, 5 mM DTT, 0.05 mM EDTA, 0.25% DMSO, 10 μM of X. laevis histone H3, and varying concentrations of C646 (0, 3, 10 μM). The reactions contained either 70 nanograms of Rtt109/Vps75, 15 nanograms of yGcn5, 300 nanograms of the SAS complex or 1 microgram of hMOZ. The amount of enzyme used in each assay was estimated by comparing Coomassie Blue staining of samples with bovine serum albumin standards, analyzed by SDS-PAGE. The mixture was allowed to equilibrate at 30°C for 10 min before the reaction was initialed with addition of a 1:1 mixture of 12C-AcCoA and 14C-AcCoA to a final concentration of 20 μM. After the appropriate time the reaction was quenched with 6 X Tris-Tricine gel loading buffer which contained 0.2 M Tris-Cl pH 6.8, 40% v/v glycerol, 14% w/v SDS, 0.3 M DTT, and 0.06% w/v Coomassie Blue. The 14C-labeled histone substrates were separated from reactants on a 16.5% Tris-Tricine SDS-PAGE gel. The rate of 14C-incorporation into histone H3 was quantified by autoradiography. We performed all assays in duplicate, and these generally agreed within 20%.
Time course studies
Time courses of p300 HAT with C646 were determined using the radioactive assay described above. Reactions were performed using the conditions detailed above with 1.5 μM C646. Reactions were quenched at particular time intervals, then run on a 16% Tris-Tricine gel and quantified as described above. Similar studies were performed varying the time of p300 HAT pre-incubation with C646. Assays contained the conditions detailed above, with 1.5 μM C646 added at various times prior to initiation with 10 μM acetyl-CoA. Reactions were quenched after 5 min, then run on a 16% Tris-Tricine gel and quantified as described above. All time points were screened in duplicate.
Inhibition with p300 mutants
Sites for mutation were chosen by examination of the C646 binding model generated during VLS. T1411A, W1466F, Y1467F, and R1410A mutations were installed using QuikChange protocols. p300 variants were expressed in E. coli BL21(RIL)-DE3 cells and purified using expressed protein ligation as described above. IC50 values for C646 with all mutants were obtained using the methods described above. All assays contained 10 μM of a 1:1 mixture of 12C-acetyl-CoA and 14C-acetyl-CoA and 400 μM H4-15. Reaction time was varied between 5 and 10 min to keep turnover below 10%. Enzyme concentrations were altered for each mutant, as active site mutations affected enzyme activity. In a similar fashion, kinetic parameters for each mutant vs. AcCoA were determined. [H4-15] was 400 μM, and DMSO was held constant at 2.5%. Reactions proceeded for 6 minutes before being quenched and run on a 16% Tris-Tricine gel as described above. Data were quantified and fit to the Michaelis-Menten equation.
NMR studies
The 2D 1H-1H correlation spectra were acquired at 30 °C on an 11.7 T Varian INOVA spectrometer using a pentaprobe equipped with z-axis pulse-field gradient coils. Data were processed and analyzed using NMRPipe [61]. The sample contained 600 μL of 10 mM C646 DMSO-d6 solution.
Histone acetylation assays in mouse cells
C3H 10T1/2 mouse fibroblasts were grown in DMEM with 10 % FCS at 37 °C with 6 % CO2. Confluent cultures were rendered quiescent in DMEM with 0.5 % FCS for 18-20 h prior to treatment. Cells were treated with the following compounds: TSA (10 ng/ml [33 nM]; Sigma), C646 (25 μM), C37 (25 μM). Antibodies were used at the following concentrations: total H3 (1:10000; ab7834; Abcam); H4K12ac (1:2500; 06-761; Upstate). Rabbit anti-H3K9ac (1:10000) antibodies were generated in-house (Edmunds et al., 2008). Histones were isolated from cells by acid extraction, separated by SDS and acid-urea polyacrylamide gel electrophoresis and analyzed by Western blotting as described previously (Thomson et al., 1999; Clayton et al., 2000).
Cancer cell studies
Melanoma cell lines WM35, WM983A, and 1205Lu were generous gifts from Meenhard Herlyn's lab at the Wistar Institute (Philadelphia, PA). Non-small cell lung cancer (NSCLC) cell lines H23 and H838 were obtained from Dr. Charles Rudin's lab, and H1395 from Dr. Craig Peacock's lab, at Johns Hopkins. Melanoma cells were maintained in Dulbecco's Modified Eagle Medium. NSCLCs were maintained in RPMI Medium 1640. Both types of media were supplemented with 10% fetal bovine serum (FBS), penicillin-streptomycin, and L-glutamine. Media, pen-strep, and L-glutamine were purchased from Invitrogen. FBS was purchased from Gemini Bio-Products (#100106).
Before treating cells with p300 inhibitors (C646, Lys-CoA-Tat [Zheng et al., 2005; Guidez et al., 2005]) or control compounds (C37, Ac-Asp-Asp-Asp-Asp-Tat [also known as Ac-DDDD-Tat]), cells were plated at sub-confluent concentration (∼60%) and incubated at 37°C until attached to the plating surface. Compound stocks (10-20 mM in anhydrous DMSO) were directly added to culture media at desired concentrations. DMSO concentration was kept constant at 0.2% between different treatment conditions. Cells were seeded in 96-well plates at ∼5000 cells per well on average, depending on each cell line's doubling time. After attachment, cells were treated with p300 inhibitors, control compounds, or DMSO for 24 h. After treatment, 3H-thymidine (1 mCi/ml stock) was added to media at 10 μCi/ml final concentration. Cells were incubated for an additional 5 h, then trypsinized and collected onto a filter mat using a Cell Harvester (PerkinElmer). Radioactivity was measured with a MicroBeta plate reader (PerkinElmer). Each sample was tested in triplicate.
Cell Cycle Analysis
Cells treated with C646 or DMSO were stained with propidium iodide (PI) according to a published protocol (Robinson, 2009). Briefly, equal numbers of cells (greater than 106) in 0.5 ml PBS were fixed in 70% ethanol for at least 2 h at 4°C. A stock staining solution containing 10 ml of 0.1% Triton X-100 in PBS, 400 μl of RNase cocktail (equivalent of ∼200 units of RNase A) (Ambion), and 200 μl of 1 mg/ml PI was prepared. Fixed cells were spun at 200 g for 5 min (Beckman Coulter Allegra® X-12R, with an SX4750A rotor), re-hydrated in 5 ml PBS, and spun again to remove all traces of ethanol. Cells were stained with 1 ml staining solution for 20 min at 37°C, then immediately analyzed on a FACSCalibur flow cytometer (BD Biosciences) at the Johns Hopkins Flow Cytometry Core Facility. Data acquisition and analysis were performed with the CellQuest software (BD). WinMDI 2.9 (http://facs.scripps.edu.software.html) was also used for data presentation.
Supplementary Material
Scheme 1.

Acknowledgments
We thank the NIH, FAMRI Foundation, and Henry and Elaine Kaufman Foundation for support. EMB is grateful for predoctoral support from the NSF.
Footnotes
- --Virtual ligand screening was employed to identify novel inhibitors of p300 HAT activity.
- --C646 is a relatively potent, selective, cell-permeable small molecule p300 inhibitor.
- --C646 slows cancer cell growth and impedes intracellular histone acetylation.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abagyan R, Totrov M, Kuznetsov D. ICM-a new method for protein modeling and design: applications to docking and structure prediction from the distorted native conformation. J Comput Chem. 1994;15:488–506. [Google Scholar]
- Abagyan R, Totrov M, Kuznetsov D. ICM-a new method for protein modeling and design: applications to docking and structure prediction from the distorted native conformation. J Comput Chem. 1994;15:488–506. [Google Scholar]
- Arif M, Pradhan SK, Thanuja GR, Vedamurthy BM, Agrawal S, Dasgupta D, Kundu TK. Mechanism of p300 specific histone acetyltransferase inhibition by small molecules. J Med Chem. 2009;52:267–277. doi: 10.1021/jm800657z. [DOI] [PubMed] [Google Scholar]
- Balasubramanyam K, Swaminathan V, Ranganathan A, Kundu TK. Small molecule modulators of histone acetyltransferase p300. J Biol Chem. 2003;278:19134–19140. doi: 10.1074/jbc.M301580200. [DOI] [PubMed] [Google Scholar]
- Balasubramanyam K, Varier RA, Altaf M, Swaminathan V, Siddappa NB, Ranga U, Kundu TK. Curcumin, a novel p300/CREB-binding protein-specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase-dependent chromatin transcription. J Biol Chem. 2004;279:51163–51171. doi: 10.1074/jbc.M409024200. [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Kouzarides T. The CBP co-activator is a histone acetyltransferase. Nature. 1996;384:641–643. doi: 10.1038/384641a0. [DOI] [PubMed] [Google Scholar]
- Bisson WH, Koch DC, O'Donnell EF, Khalil SM, Kerkvliet NI, Tanguay RL, Abagyan R, Kolluri SK. Modeling of the aryl hydrocarbon receptor (AhR) ligand binding domain and its utility in virtual ligand screening to predict new AhR ligands. J Med Chem. 2009;52:5635–5641. doi: 10.1021/jm900199u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavasotto CN, Orry AJ, Murgolo NJ, Czarniecki MF, Kocsi SA, Hawes BE, O'Neill KA, Hine H, Burton MS, Voigt JH, Abagyan RA, Bayne ML, Monsma FJ., Jr Discovery of novel chemotypes to a G-protein-coupled receptor through ligand-steered homology modeling and structure-based virtual screening. J Med Chem. 2008;51:581–588. doi: 10.1021/jm070759m. [DOI] [PubMed] [Google Scholar]
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- Clayton AL, Rose S, Barratt MJ, Mahadevan LC. Phosphoacetylation of histone H3 on c-fos- and c-jun-associated nucleosomes upon gene activation. EMBO J. 2000;19:3714–3726. doi: 10.1093/emboj/19.14.3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole PA. Chemical probes for histone-modifying enzymes. Nat Chem Biol. 2008;4:590–597. doi: 10.1038/nchembio.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland RA. Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis. 2nd. New York: Wiley-VCH; 2000. [Google Scholar]
- Dekker FJ, Haisma HJ. Histone acetyl transferases as emerging drug targets. Drug Discov Today. 2009;14:942–948. doi: 10.1016/j.drudis.2009.06.008. [DOI] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Edmunds JW, Mahadevan LC, Clayton AL. Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. EMBO J. 2008;27:406–420. doi: 10.1038/sj.emboj.7601967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng BY, Shelat A, Doman TN, Guy RK, Shoichet BK. High-throughput assays for promiscuous inhibitors. Nat Chem Biol. 2005;1:146–148. doi: 10.1038/nchembio718. [DOI] [PubMed] [Google Scholar]
- Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000;14:1553–1577. [PubMed] [Google Scholar]
- Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- Guidez F, Howell L, Isalan M, Cebrat M, Alani RM, Ivins S, Hormaeche I, McConnell MJ, Pierce S, Cole PA, Licht J, Zelent A. Histone acetyltransferase activity of p300 is required for transcriptional repression by the promyelocytic leukemia zinc finger protein. Mol Cell Biol. 2005;25:5552–5566. doi: 10.1128/MCB.25.13.5552-5566.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halgren TA, Nachbar RB. Merck molecular force field. IV. Conformational energies and geometries for MMFF94. J Comput Chem. 1996;17:587–615. [Google Scholar]
- Haval KP, Argade NP. General strategy for the synthesis of natural and unnatural dialkylmaleic anhydrides. J Org Chem. 2008;73:6936–6938. doi: 10.1021/jo801284r. [DOI] [PubMed] [Google Scholar]
- Hodawadekar SC, Marmorstein R. Chemistry of acetyl transfer by histone modifying enzymes: structure, mechanism and implications for effector design. Oncogene. 2007;26:5528–5540. doi: 10.1038/sj.onc.1210619. [DOI] [PubMed] [Google Scholar]
- Hodgkiss RJ, Begg AC, Middleton RW, Parrick J, Stratford MR, Wardman P, Wilson GD. Fluorescent markers for hypoxic cells. A study of novel heterocyclic compounds that undergo bio-reductive binding. Biochem Pharmacol. 1991;41:533–541. doi: 10.1016/0006-2952(91)90625-f. [DOI] [PubMed] [Google Scholar]
- Holbert MA, Sikorski T, Carten J, Snowflack D, Hodawadekar S, Marmorstein R. The human monocytic leukemia zinc finger histone acetyltransferase domain contains DNA-binding activity implicated in chromatin targeting. J Biol Chem. 2007;282:36603–36613. doi: 10.1074/jbc.M705812200. [DOI] [PubMed] [Google Scholar]
- Hosoya T, Aoyama H, Ikemoto T, Kihara Y, Hiramatsu T, Endoc M, Suzuki M. Dantrolene analogues revisited: General synthesis and specific functions capable of discriminating two kinds of Ca2+ release from sarcoplasmic reticulum of mouse skeletal muscle. Bioorg Med Chem. 2003;11:663–673. doi: 10.1016/s0968-0896(02)00600-4. [DOI] [PubMed] [Google Scholar]
- Itoh Y, Suzuki T, Miyata N. Isoform-selective histone deacetylase inhibitors. Current Pharm Des. 2008;14:529–544. doi: 10.2174/138161208783885335. [DOI] [PubMed] [Google Scholar]
- Iyer NG, Xian J, Chin SF, Bannister AJ, Daigo Y, Aparicio S, Kouzarides T, Caldas C. p300 is required for orderly G1/S transition in human cancer cells. Oncogene. 2007;26:21–29. doi: 10.1038/sj.onc.1209771. [DOI] [PubMed] [Google Scholar]
- Kim JB, Lee YS. Peptide synthesis with polymer bound active ester. II. Synthesis of pyrazolone resin and its applications in acylation reaction. Bull Korean Chem Soc. 1991;12:376–379. [Google Scholar]
- Kim Y, Tanner KG, Denu JM. A continuous, nonradioactive assay for histone acetyltransferases. Anal Biochem. 2000;280:308–314. doi: 10.1006/abio.2000.4546. [DOI] [PubMed] [Google Scholar]
- Kitz R, Wilson IB. Esters of methanesulfonic acid as irreversible inhibitors of acetylcholinesterase. J Biol Chem. 1962;237:3245–3249. [PubMed] [Google Scholar]
- Langner M, Remy P, Bolm C. Highly modular synthesis of C1-symmetric aminosulfoximines and their use as ligands in copper-catalyzed asymmetric Mukaiyama-Aldol reactions. Chem Eur J. 2005;11:6254–6265. doi: 10.1002/chem.200500497. [DOI] [PubMed] [Google Scholar]
- Lau OD, Courtney AD, Vassilev A, Marzilli LA, Cotter RJ, Nakatani Y, Cole PA. p300/CBP-associated factor histone acetyltransferase processing of a peptide substrate. Kinetic analysis of the catalytic mechanism. J Biol Chem. 2000;275:1953–1959. doi: 10.1074/jbc.M003219200. [DOI] [PubMed] [Google Scholar]
- Lau OD, Kundu TK, Soccio RE, Ait-Si-Ali S, Khalil EM, Vassilev A, Wolffe AP, Nakatani Y, Roeder RG, Cole PA. HATs off: selective synthetic inhibitors of the histone acetyltransferases p300 and PCAF. Mol Cell. 2000;5:589–595. doi: 10.1016/s1097-2765(00)80452-9. [DOI] [PubMed] [Google Scholar]
- Liu X, Wang L, Zhao K, Thompson PR, Hwang Y, Marmorstein R, Cole PA. The structural basis of protein acetylation by the p300/CBP transcriptional coactivator. Nature. 2008;451:846–850. doi: 10.1038/nature06546. [DOI] [PubMed] [Google Scholar]
- Liu Y, Dentin R, Chen D, Hedrick S, Ravnskjaer K, Schenk S, Milne J, Meyers DJ, Cole P, Yates J, 3rd, Olefsky J, Guarente L, Montminy M. A fasting inducible switch modulates gluconeogenesis via activator/coactivator exchange. Nature. 2008;456:269–273. doi: 10.1038/nature07349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macek B, Mann M, Olsen JV. Global and site-specific quantitative phosphoproteomics: principles and applications. Annu Rev Pharmacol Toxicol. 2009;49:199–221. doi: 10.1146/annurev.pharmtox.011008.145606. [DOI] [PubMed] [Google Scholar]
- Mallya M, Phillips RL, Saldanha SA, Gooptu B, Brown SC, Termine DJ, Shirvani AM, Wu Y, Sifers RN, Abagyan R, Lomas DA. Small molecules block the polymerization of Z alpha1-antitrypsin and increase the clearance of intracellular aggregates. J Med Chem. 2007;50:5357–5363. doi: 10.1021/jm070687z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantelingu K, Reddy BA, Swaminathan V, Kishore AH, Siddappa NB, Kumar GV, Nagashankar G, Natesh N, Roy S, Sadhale PP, Ranga U, Narayana C, Kundu TK. Specific inhibition of p300-HAT alters global gene expression and represses HIV replication. Chem Biol. 2007;14:645–657. doi: 10.1016/j.chembiol.2007.04.011. [DOI] [PubMed] [Google Scholar]
- Moreau F, Desroy N, Genevard JM, Vongsouthi V, Gerusz V, Le Fralliec G, Oliveira C, Floquet S, Denis A, Escaich S, Wolf K, Busemann M, Aschenbrenner A. Discovery of new Gram-negative antivirulence drugs: structure and properties of novel E. coli WaaC inhibitors. Bioorg Med Chem Lett. 2008;18:4022–4026. doi: 10.1016/j.bmcl.2008.05.117. [DOI] [PubMed] [Google Scholar]
- Nemethy G, et al. Energy parameters in polypeptides. 10. Improved geometrical parameters and nonbonded interactions for use in the ECEPP/3 algorithm, with application to proline-containing peptides. J Phys Chem. 1992;96:6472–6484. [Google Scholar]
- Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y. Nuclear receptor coactivator ACTR is a novel histone acetyltransferase and forms a multimeric activation complex with P/CAF and CBP/p300. Cell. 1997;90:569–80. doi: 10.1016/s0092-8674(00)80516-4. [DOI] [PubMed] [Google Scholar]
- Poux AN, Cebrat M, Kim CM, Cole PA, Marmorstein R. Structure of the GCN5 histone acetyltransferase bound to a bisubstrate inhibitor. Proc Natl Acad Sci U S A. 2002;99:14065–14070. doi: 10.1073/pnas.222373899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao Y, Molina H, Pandey A, Zhang J, Cole PA. Chemical rescue of a mutant enzyme in living cells. Science. 2006;311:1293–1297. doi: 10.1126/science.1122224. [DOI] [PubMed] [Google Scholar]
- Ravindra KC, Selvi BR, Arif M, Reddy BA, Thanuja GR, Agrawal S, Pradhan SK, Nagashayana N, Dasgupta D, Kundu TK. Inhibition of lysine acetyltransferase KAT3B/p300 activity by a naturally occurring hydroxynaphthoquinone, plumbagin. J Biol Chem. 2009;284:24453–24464. doi: 10.1074/jbc.M109.023861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JP, editor. Current Protocols in Cytometry. Wiley Interscience; NY: 2009. [Google Scholar]
- Shechter D, Dormann HL, Allis CD, Hake SB. Extraction, purification and analysis of histones. Nat Protoc. 2007;2:1445–1457. doi: 10.1038/nprot.2007.202. [DOI] [PubMed] [Google Scholar]
- Shia WJ, Osada S, Florens L, Swanson SK, Washburn MP, Workman JL. Characterization of the yeast trimeric-SAS acetyltransferase complex. J Biol Chem. 2005;280:11987–11994. doi: 10.1074/jbc.M500276200. [DOI] [PubMed] [Google Scholar]
- Shogren-Knaak MA, Alaimo PJ, Shokat KM. Recent advances in chemical approaches to the study of biological systems. Annu Rev Cell Dev Biol. 2001;17:405–433. doi: 10.1146/annurev.cellbio.17.1.405. [DOI] [PubMed] [Google Scholar]
- Stimson L, Rowlands MG, Newbatt YM, Smith NF, Raynaud FI, Rogers P, Bavetsias V, Gorsuch S, Jarman M, Bannister A, Kouzarides T, McDonald E, Workman P, Aherne GW. Isothiazolones as inhibitors of PCAF and p300 histone acetyltransferase activity. Mol Cancer Ther. 2005;4:1521–1532. doi: 10.1158/1535-7163.MCT-05-0135. [DOI] [PubMed] [Google Scholar]
- Sutton A, Shia WJ, Band D, Kaufman PD, Osada S, Workman JL, Sternglanz R. Sas4 and Sas5 are required for the histone acetyltransferase activity of Sas2 in the SAS complex. J Biol Chem. 2003;278:16887–16892. doi: 10.1074/jbc.M210709200. [DOI] [PubMed] [Google Scholar]
- Szewczuk LM, Saldanha SA, Ganguly S, Bowers EM, Javoroncov M, Karanam B, Culhane JC, Holbert MA, Klein DC, Abagyan R, Cole PA. De novo discovery of serotonin N-acetyltransferase inhibitors. J Med Chem. 2007;50:5330–5338. doi: 10.1021/jm0706463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Holbert MA, Wurtele H, Meeth K, Rocha W, Gharib M, Jiang E, Thibault P, Verreault A, Cole PA, Marmorstein R. Fungal Rtt109 histone acetyltransferase is an unexpected structural homolog of metazoan p300/CBP. Nat Struct Mol Biol. 2008;15:738–745. doi: 10.1038/nsmb.1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson S, Clayton AL, Hazzalin CA, Rose S, Barratt MJ, Mahadevan LC. The nucleosomal response associated with immediate-early gene induction is mediated via alternative MAP kinase cascades: MSK1 as a potential histone H3/HMG-14 kinase. EMBO J. 1999;18:4779–4793. doi: 10.1093/emboj/18.17.4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson PR, Kurooka H, Nakatani Y, Cole PA. Transcriptional coactivator protein p300. Kinetic characterization of its histone acetyltransferase activity. J Biol Chem. 2001;276:33721–33729. doi: 10.1074/jbc.M104736200. [DOI] [PubMed] [Google Scholar]
- Thompson PR, Wang D, Wang L, Fulco M, Pediconi N, Zhang D, An W, Ge Q, Roeder RG, Wong J, Levrero M, Sartorelli V, Cotter RJ, Cole PA. Regulation of the p300 HAT domain via a novel activation loop. Nat Struct Mol Biol. 2004;11:308–315. doi: 10.1038/nsmb740. [DOI] [PubMed] [Google Scholar]
- Totrov M, Abagyan R. Flexible protein–ligand docking by global energy optimization in internal coordinates. Proteins Suppl. 1997;1:215–220. doi: 10.1002/(sici)1097-0134(1997)1+<215::aid-prot29>3.3.co;2-i. [DOI] [PubMed] [Google Scholar]
- Totrov M, Abagyan R. Derivation of sensitive discrimination potential for virtual ligand screening. Proceedings of the third annual international conference on computational molecular biology; 1999. pp. 312–320. [Google Scholar]
- Vasyunkina TN, Bykova LM, Plotkin VN, Ramsh SM. Synthesis of 1,4-dihydropyrano[2,3-c]pyrazole derivatives. Russ J Org Chem. 2005;41:742–744. [Google Scholar]
- Walsh CT. Post-translational Modification of Proteins. Greenwood Village, CO: Roberts and Co.; 2006. [Google Scholar]
- Wang L, Tang Y, Cole PA, Marmorstein R. Structure and chemistry of the p300/CBP and Rtt109 histone acetyltransferases: implications for histone acetyltransferase evolution and function. Curr Opin Struct Biol. 2008;18:741–747. doi: 10.1016/j.sbi.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XJ, Seto E. Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol, Cell. 2008;31:449–461. doi: 10.1016/j.molcel.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M, Kijima M, Akita M, Beppu T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J Biol Chem. 1990;265:17174–17179. [PubMed] [Google Scholar]
- Yu M, Magalhães ML, Cook PF, Blanchard JS. Bisubstrate inhibition: Theory and application to N-acetyltransferases. Biochemistry. 2006;245:14788–14794. doi: 10.1021/bi061621t. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Balasubramanyam K, Cebrat M, Buck D, Guidez F, Zelent A, Alani RM, Cole PA. Synthesis and evaluation of a potent and selective cell-permeable p300 histone acetyltransferase inhibitor. J Am Chem Soc. 2005;127:17182–17183. doi: 10.1021/ja0558544. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.