

Abstract
The metal-dependent deacetylase UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase (LpxC) catalyzes the first committed step in lipid A biosynthesis, the hydrolysis of UDP-3-O-myristoyl-N-acetyl-glucosamine to form UDP-3-O-myristoyl-glucosamine and acetate. Consequently, LpxC is a target for the development of antibiotics, virtually all of which coordinate the active site metal ion. Here we examine the ability of Fe2+ to serve as a cofactor for wild-type E. coli LpxC and a mutant enzyme (EcC63A), wherein one of the ligands for the inhibitory metal binding site has been removed. LpxC exhibits higher activity (6- to 8-fold) with a single bound Fe2+ as the cofactor compared to Zn2+-LpxC; both metalloenzymes have a bell-shaped dependence on pH with similar pKa values, indicating that at least two ionizations are important for maximal activity. X-ray absorption spectroscopy experiments suggest that the catalytic metal ion bound to Fe2+-EcLpxC is 5-coordinate, suggesting that catalytic activity may correlate with coordination number. Furthermore, the ligand affinity of Fe2+-LpxC compared to the Zn2+ enzyme alters by up to 6-fold. In contrast to Zn2+-LpxC, the activity of Fe2+-LpxC is redox sensitive and a time-dependent decrease in activity is observed under aerobic conditions. The LpxC activity of crude E. coli cell lysates is also aerobically sensitive, consistent with the presence of Fe2+-LpxC. These data indicate that EcLpxC can use either Fe2+ or Zn2+ to activate catalysis in vitro and possibly in vivo, which may allow LpxC to function in E. coli grown under different environmental conditions.
Keywords: LpxC, metal-dependent deacetylase, regulation, iron, zinc, cambialistic enzyme
Treatment of infections caused by Gram-negative bacteria is often difficult, due in part to the antibiotic resistance associated with these organisms. Furthermore, Gram-negative organisms (e.g. Yersinia pestis, Francisella tularensis) have recently been identified as potential bioterror agents (1). For these reasons, new antibiotics that are effective in the treatment of Gram-negative bacterial infections are needed, including drugs that act on new targets. Possible targets in Gram-negative bacteria include the enzymes involved in the biosynthesis of lipopolysaccharides1 (LPS) that make up the outer membranes of these organisms (2-4). The lipid A portion of LPS is responsible for anchoring LPS to the cell surface; consequently, the lipid A biosynthetic pathway is a potential source of targets for antibiotic development (2). UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase (LpxC) catalyzes the committed, and second overall, step in the biosynthesis of lipid A – the deacetylation of UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine to form UDP-3-O-(R-3-hydroxymyristoyl)-glucosamine and acetate (Figure 1A) (5). Consequently, LpxC is an appealing target for antimicrobial development.
Figure 1.

(A) LpxC-catalyzed reaction. (B) Active site of LpxC from A. aeolicus containing two zinc ions: ZnA (catalytic) and ZnB (inhibitory). Figure was made from PDB 1P42. (C) Structure of L-161,240. (D) Structure of BODIPY-fatty acid.
LpxC was originally classified as a “Zn2+-dependent” deacetylase based on the findings that LpxC activity is reversibly inhibited by incubation with metal chelators (e.g. ethylene diamine tetraacetic acid, EDTA; dipicolinic acid, DPA) and LpxC co-purifies with Zn2+ under aerobic conditions (6). However, these previous studies did not evaluate the ability of Fe2+ to activate LpxC. Recently, there have been several examples of “Zn2+-dependent” enzymes that have been reclassified as “Fe2+-dependent” enzymes, including peptide deformylase (PDF) (7-9), LuxS (10), and possibly histone deacetylase 8 (HDAC8) (11). The misidentification of the native metal cofactor as Zn2+ is attributed to the aerobic purification of these enzymes, with the oxidation of Fe2+ to Fe3+ and substitution of Zn2+ at the active site.
Herein, we demonstrate that replacement of the Zn2+ cofactor with Fe2+ in E. coli LpxC both enhances the catalytic activity and alters the affinity of this enzyme for ligands. In fact, Fe2+-LpxC functions as a mononuclear metal-dependent deacetylase with a catalytic activity that is ~6-fold higher than Zn2+-LpxC. This enhancement in catalytic activity is due primarily to an increase in the value of the parameter kcat, not KM. X-ray absorption spectroscopy (XAS) experiments indicate that the metal ion in Fe2+-LpxC is 5 coordinate, suggesting that higher coordination numbers correlate with enhanced catalytic activity. In contrast to Zn2+-LpxC, the activity of the Fe2+-bound enzyme is sensitive to oxygen. Furthermore, the activity of native LpxC in crude E. coli cell lysates is aerobically sensitive, consistent with the presence of Fe2+-LpxC. These findings suggest that under normal growth conditions the native metal bound to LpxC in E. coli is Fe2+. However, it is possible that the active site metal bound to LpxC in vivo could switch depending on the metal ion availability, thus allowing LpxC to function using different metal ion cofactors under different environmental conditions.
Material and Methods
General procedures
E. coli (WT and C63A variant) and A. aeolicus LpxC (AaLpxC) were over-expressed and purified according to published procedures using DEAE-sepharose and Reactive Red-120 affinity dye columns at 4° C and room temperature, respectively (5, 6, 12, 13). The apo- and single metal bound forms of LpxC were prepared by treatment with metal chelators (DPA/EDTA) followed by reconstitution with Zn2+ or Fe2+, as previously described (6, 14). All solutions (except for enzyme) were degassed with Ar prior to use. For Fe2+ experiments, a 400 mM FeCl2 stock was prepared in 10 mM dithionite and diluted to 100 μM with 1X assay buffer prior to incubation with apo-LpxC. Similarly, a 100 mM ZnSO4 solution was prepared in 25 mM bis-tris propane, 1.5 mM triscarboxyethylphosphine (TCEP) pH 7.5-8.7 and diluted to 100 μM with 1X assay buffer prior to incubation with apo-LpxC. The concentrations of metal stocks were verified by ICP-MS. To maintain anaerobic conditions, experiments were carried out either in an anaerobic chamber (Coy Laboratory Products, Grass Lake, MI) or using assay buffers containing 10 mM TCEP and were completed in < 2 hr to ensure that LpxC was maximally active during the course of the assays. All ICP-MS analysis was done at the University of Michigan, Department of Geology by Dr. Ted Huston.
LpxC deacetylase Assay
[14C]-UDP-3-O-(3-hydroxymyristoyl)-N-acetyl-glucosamine was prepared, and the deacetylase activity was measured as previously described (6, 14, 15). To examine the metal ion stoichiometry of the Zn2+-LpxC catalyzed reactions, apo-LpxC was incubated (5 μM WT or C63A in 20 mM bis-tris propane, pH 7.5) with 0-2 equivalents of ZnSO4 for 1-2 hours at room temperature to allow for holoenzyme formation, and was then diluted to 100-200 nM in the assay buffer just prior to activity measurement. For Fe2+ stoichiometry, apo-LpxC (10 μM WT or C63A in 20 mM bis-tris propane, 2-10 mM TCEP, pH 7.5) was incubated with FeCl2 (1 to 500 μM) on ice for 5 to 60 minutes in an anaerobic chamber (if TCEP < 10 mM) and then diluted with assay buffer prior to measuring activity. Assay mixtures containing 20 mM bis-tris propane, pH 7.5, 10 mM TCEP (for Fe2+ measurements), bovine serum albumin (BSA, fatty acid free, 1 mg/mL); and [14C]-UDP-3-O-(3-hydroxymyristoyl)-N-acetyl-glucosamine (0.05 to 4 μM) were pre-equilibrated at 30 °C and the reactions were initiated by the addition of enzyme (0.5 to 5 nM). ICP-MS results indicate these samples contain ≤ 20 nM zinc. After incubation for various times, the reactions were quenched by the addition of sodium hydroxide, which also cleaves the myristate substituent from substrate and product for ease of separation. Substrate and product were separated on PEI-cellulose TLC plates (0.1 M guanidinium HCl), quantified by scintillation counting, and the initial rates of product formation (< 20% reaction) were determined from these data. To evaluate the steady-state kinetic parameters, activity was measured at seven to nine different concentrations of myrUDP-GlcNAc (50 nM to 4 μM). The steady-state parameters kcat, KM and kcat/KM were obtained by fitting the Michaelis-Menten equation to the initial linear velocities measured at the various substrate concentrations using the curve-fitting program Kaleidagraph (Synergy Software), which also calculates the asymptotic standard errors. For pH studies of C63A LpxC, 20 mM bis-tris (pH 5.7 – 6.5) or bis-tris propane (pH 7 - 10) with 10 mM TCEP was used and pKa values were obtained by fitting Eq. (1) to these data. Control experiments demonstrate that incubation of LpxC at various pH values for up to 30 min. does not lead to an irreversible decrease in activity.
For experiments with the hydroxamate inhibitor L-161,240 (13, 16, 17), a generous gift from Dr. Michael Pirrung, Zn2+- or Fe2+-LpxC (0.5 nM) was preincubated with inhibitor (0.5 to 500 nM, DMSO) or DMSO for 15 to 25 min at 30 °C in 20 mM bis-tris propane, 10 mM TCEP pH 7.5; 1 mg/mL BSA prior to initiation of the reaction by the addition of [14C]-UDP-3-O-(3-hydroxymyristoyl)-N-acetyl-glucosamine (200 nM). Initial rates were processed as described above, and the IC50 values were obtained by fitting Eq. (2) to these data.
| Eq. (1) |
| Eq. (2) |
Ultrafiltration Binding Assay
Dissociation constants (KD) of [14C]-UDP-3-O-(3-hydroxymyristoyl)-glucosamine from LpxC·product complexes were measured using ultrafiltration, as previously described (18). Briefly, the concentration of product was held constant (50 - 60 nM) and the concentration of wild-type or C63A LpxC was varied (0 to 24 μM). Enzyme and substrate were incubated at 30 °C for 15 – 30 min prior to the assay to allow for product formation and ligand equilibration. Assay mixtures were then transferred into ultrafiltration devices (Microcon MWCO 30K), and the free and bound products were separated by centrifuging the samples at 3000 rpm for 2.5 minutes. Equal volumes of the filtrate and retentate were removed, and the amounts of unbound (filtrate) and total product (retentate) were quantified using scintillation counting. The ratio of EP/Ptotal was determined as a function of [E]total and the KD values were obtained by fitting Eq. (4) to these data.
| Eq. (4) |
KDfatty acid determination using fluorescence anisotropy measurements
The KD value of 5-butyl-4,4-difluoro-4-bora-3a,4a-diaza-s-indacene-3-nonanoic acid (BODIPY® 500/510 C4, C9; Invitrogen) fatty acid for EcLpxC was determined using fluorescence anisotropy as previously described (18, 19). The concentration of BODIPY® fatty acid was held constant (0.1 μM; 25 mM HEPES, 10 mM TCEP pH 7.5, 100 μM FeCl2) and increasing concentrations of enzyme (0 to 100 μM) were titrated into the solution at 30 °C. The fluorescence anisotropy (Ex 480 nm, Em 516 nm) was measured ~ 3 minutes after each addition of LpxC, and KD values were determined by fitting Eq. (5) to these data.
| Eq. (5) |
X-ray Absorption Spectroscopy
XAS samples were prepared and analyzed as previously described (12). Briefly, a stoichiometric amount of apo-EcLpxC or apo-AaLpxC was added to a solution of 24 μM FeCl2 (anaerobic chamber), or ZnSO4 in 25 mM HEPES, 1.5 mM TCEP pH 7.5 and incubated on ice for 45 min (AaLpxC, room temperature for 25 min). The enzyme was concentrated using an Amicon Ultra-15 centrifugal filter unit (MWCO 10K) and unbound metal ions were removed by washing with buffer (25 mM HEPES, 1.5 mM TCEP pH 7.5) prior to concentration. Glycerol (20 μL) was mixed with the enzyme (60 μL) as a cryoprotectant, samples were transferred into Lucite cuvettes (3 × 2 × 25 mm) with 40 μm Kapton windows and frozen in N2(l). The concentration of Fe, Co and Zn were determined using ICP-MS, and all analyzed samples had a metal/enzyme stoichiometry of < 1 (range 0.14-0.63). XAS data were collected at the Stanford Synchotron Radiation Laboratory (beam line 9-3) under dedicated conditions as fluorescence excitation spectra, using a solid-state Ge detector array equipped with a filter and Soller slits focused on the sample. All channels of each scan were examined for glitches, and the good channels were averaged for each sample (two independent samples for each protein) to give the final spectrum.
The program EXAFSPAK (20) was used to extract and analyze EXAFS data as previously described (12), using ab inito phase and amplitude parameters calculated using FEFF 7.02 (21, 22). XANES data were normalized to tabulated absorption coefficients (23) using the program MBACK (24). The area of the 1s→3d transition in the XANES region was calculated by fitting the pre-edge region (7107-7118 eV) using the sum of a Gaussian and an arctan function; for comparison with previously published data, the fitted Gaussian area was normalized to the K-edge jump for Fe (3.556 × 102 cm2/g).
Native LpxC deacetylase activity
LB media was inoculated with BL21(DE3) or BL21(DE3)pLysS cells and grown in a shaker (250 rpm) overnight at 37 °C. The cells were harvested by centrifugation and the cell pellets resuspended in 20 mM bis-tris propane, 0 - 10 mM TCEP pH 7.5. The cells were washed with 5 mM CaCl2 (1 × 2 mL) followed by buffer (2 × 2 mL), resuspended in buffer, and stored at −80 °C. Control experiments demonstrate that Ca2+ (≤ 10 mM) has no significant effect on Zn2+-LpxC activity. Prior to assaying activity, cells were thawed, incubated with lysozyme (0.2 – 1.2 mg/mL) in assay buffer for 15 min at room temperature and the cell lysate was cleared by centrifugation (14000 rpm, 15 min). A sample of the cleared lysate was analyzed for metal content by ICP-MS; the concentration of total Fe in these lysates is ~2-fold greater than Zn (Fe/Zn = 1.4 to 3.0). The cleared lysate was diluted 10-fold in assay buffer prior to measurement of activity as described above ([S] = 200 nM). To demonstrate that the observed activity was catalyzed by LpxC, the activity was also measured in the presence of the inhibitor L-161,240. In vitro control experiments indicate that exchange of Zn2+ for Fe2+ is facile in vitro but the opposite metal exchange is not readily observed and is likely thermodynamically unfavorable (25). Therefore, the experiments in cell lysates should err on the side of underestimating the concentration of Fe2+-LpxC.
Results
The activity of Fe2+-LpxC is higher than Zn2+-LpxC
Activation of apo-LpxC by various divalent metal ions was previously demonstrated with Co2+ > Zn2+ > Ni2+ > Mn2+, while no increase in activity was observed following the addition of Mg2+, Ca2+, Cd2+ or Cu2+ to apo-LpxC (6). Stoichiometric addition of Zn2+ and Co2+ fully activate apo-LpxC, while excess concentration of Ni2+ (~3:1 Ni2+:LpxC) is required suggesting that LpxC has weaker affinity for Ni2+ (6). Based on these findings, and the fact that aerobically purified LpxC contains 1-3 Zn2+/enzyme, LpxC was characterized as a “zinc-dependent” deacetylase (6). Here we probe whether Fe2+ can also activate apo-LpxC.
Since Fe2+ is a redox sensitive metal ion, activation of LpxC by Fe2+ was measured under anaerobic conditions (glove box or 10 mM TCEP). Under these conditions, addition of Fe2+ (1 – 3 equivalents) activates apo-LpxC. In fact, the initial velocity for deacetylation of myrUDP-GlcNAc (0.2 μM) under standard assay conditions (pH 7.5) is 6- to 9-fold higher with Fe2+- compared to Zn2+-LpxC, indicating that Fe2+, like Co2+, is better suited than Zn2+ for enhancing the catalytic activity of LpxC. Furthermore, comparable activity is observed for LpxC in the presence of both 1 and 3 Fe2+ equivalents, suggesting that the enzyme is saturated with Fe2+ in the pre-incubation conditions.
Metal ion stoichiometry
Crystal structures of AaLpxC reveal that the LpxC active site contains two metal ion binding sites (Figure 1B, (26)): one catalytic site, ZnA, and one inhibitory site, ZnB. EcLpxC is activated by stoichiometric addition of either Zn2+ or Co2+, while the addition of excess Zn2+ inhibits enzyme activity (6). Similarly, the activity of apo-EcLpxC is activated by the addition of Fe2+. For WT LpxC, an initial increase in LpxC activity is observed as the concentration of metal is increased from 0 to 1 metal ions/enzyme for both Fe2+ and Zn2+, followed by a decrease in enzyme activity as the concentration of metal ion is increased further (Figure 2), although the inhibition by zinc is significantly more severe. These results are consistent with metal ion binding tightly at the catalytic site to enhance catalytic activity, followed by binding of a weaker, second metal ion at the inhibitory site that decreases the observed catalytic activity. Comparison of the maximal activities achieved with Fe2+ and Zn2+ suggests that LpxC is 6-9-fold more active with bound Fe2+ than Zn2+; however, direct comparison of these activities in the WT enzyme are complicated by metal ion binding at the inhibitory site.
Figure 2.

Activation of apo-LpxC by Zn2+ or Fe2+. Deacetylase activity as a function of either Zn2+ (closed circles) or Fe2+ (open circles) metal ion stoichiometry was measured for WT LpxC (A) and C63A LpxC (B). C63A LpxC activity was assayed with up to 50-fold excess Fe2+ (not shown). The deacetylase activity for the substrate myr-UDPGlcNAc (0.2 μM) was measured at 30 °C after incubation with varying equivalents of M2+, as described under “Materials and Methods”.
C63A mutation in LpxC decreases metal ion inhibition
To circumvent complications arising from metal ion inhibition, we proposed to preferentially decrease the metal affinity of the inhibitory site by selectively removing a ligand from this site. Structural data for AaLpxC indicate that the ligands for the inhibitory Zn2+ ion are the side chains of E78 and H265, a bound palmitate, and the catalytic water molecule (Figure 1B (26)), while EXAFS experiments indicate that the inhibitory Zn2+ in EcLpxC has at least one S/Cl ligand (12). Sequence alignment of EcLpxC with AaLpxC and mapping of the Cys side chains onto the AaLpxC structure indicated that the side chain of Cys63 (equivalent to Ser59 in AaLpxC, Figure 1B) is the only S atom within 10 Å of the inhibitory Zn2+ ion, implying that Cys63 is the third protein ligand for the inhibitory metal ion site in EcLpxC (18, 19). Since the side chains of E78 and H265 play important roles in the catalytic mechanism of LpxC (14, 27), we chose to prepare the EcC63A mutant to further compare the properties of Zn2+- and Fe2+-LpxC.
Stoichiometric addition of either Fe2+ or Zn2+ to C63A LpxC increases the catalytic activity to ≥80% of the maximal value observed at higher metal concentrations, indicating that C63A LpxC is activated by a single Fe2+ or Zn2+ ion. Furthermore, inhibition of C63A LpxC by metal ions is significantly reduced, consistent with the Cys63 side chain serving as a ligand for the inhibitory metal ion binding site. This decrease in metal ion inhibition facilitates a more accurate comparison of the maximal LpxC activity with the Fe2+ and Zn2+ cofactors.
Fe2+ activates steady-state turnover
Steady-state turnover was measured for LpxC (WT and C63A) reconstituted with stoichiometric Fe2+ or Zn2+ (Table 1) demonstrating that the value of kcat/KM for LpxC reconstituted with Fe2+ compared to Zn2+ is enhanced by 8-fold. The C63A mutation also increases the value of kcat/KM for LpxC reconstituted with Zn2+ and Fe2+ by 5-fold and 3.5-fold, respectively, relative to the wild-type enzyme, at least partly due to decreased metal inhibition. Since the data obtained for WT LpxC is complicated by differences in metal ion inhibition, we measured the relative effects of Zn2+ and Fe2+ on the steady-state parameters for the C63A mutant. These results (Table 1, Figure 3) show that switching the metal ion cofactor in C63A LpxC from Zn2+ to Fe2+ increases the values of both kcat (4-fold) and kcat/KM (6-fold) with little effect on the value of KM.
Table 1.
Steady-state activity parameters for EcLpxC
| Enzymea |
kcat (min−1)b |
KM (μM)b |
kcat/KM (min−1μM−1)b |
pKa1c | pKa2c |
kcat/KM (min−1μM−1) at pH maximumc |
|---|---|---|---|---|---|---|
| Zn2+-LpxCd | ND | ND | 34 ± 8d | 6.2 ± 0.2e | 9.2 ± 0.2e | ND |
| Fe2+-LpxC | 90 ± 3 | 0.32 ± 0.04 | 281 ± 24 | ND | ND | ND |
| Zn2+-C63A | 126 ± 12 | 0.7 ± 0.2 | 170 ± 34 | 6.6 ± 0.1 | 8.9 ± 0.1 | 260 ± 20 |
| Fe2+-C63A | 530 ± 30 | 0.5 ± 0.1 | 990 ± 130 | 6.8 ± 0.1 | 9.2 ± 0.1 | 760 ± 20 |
LpxC was reconstituted with stoichiometric (1:1) metal in a pre-incubation step, as described in the “Materials and Methods”. The measured values for kcat/KM catalyzed by apo-LpxC and apo-C63A LpxC are 15 and 11 min−1 μM−1, respectively, likely due to metal contamination in the assay.
The initial rates of LpxC deacetylase activity were determined at 30 °C (20 mM bis-tris propane, 10 mM TCEP pH 7.5, 1 mg/mL BSA) with myrUDP-GlcNAc as the substrate as described in the “Materials and Methods”. The kinetic parameters were obtained by fitting the Michaelis-Menten equation to the initial velocities.
Deacetylation of myrUDPGlcNAc catalyzed by LpxC (Fe2+-WT and C63A) was determined at 30 °C as a function of pH using substrate concentrations below the KM (< 0.2 μM). The pKa values were obtained from the pH dependence of the values using Eq. 1 as described in the “Materials and Methods”.
kcat/KM for WT ZnLpxC is reported as mean and standard deviation of 5 replicates performed at a single subsaturating substrate concentration (200 nM) with LpxC incubated with stoichiometric Zn2+.
Taken from reference (14)
Figure 3.
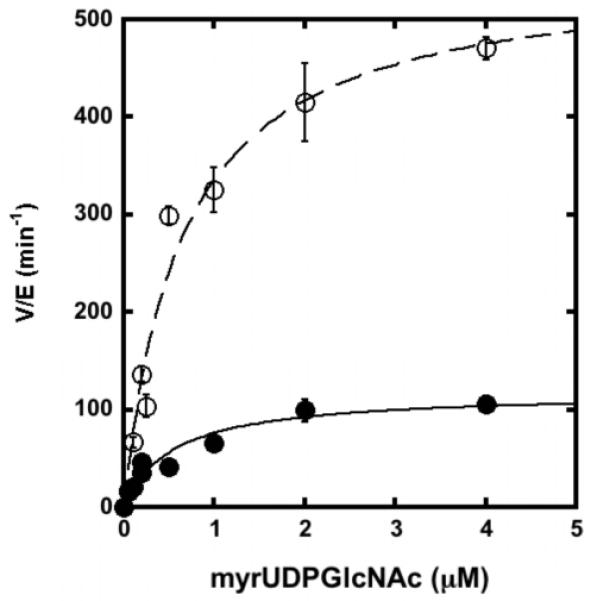
Steady-state turnover catalyzed by EcC63A substituted with Zn2+ (filled circle) or Fe2+ (open circle). The initial rates for deacetylation of myr-UDPGlcNAc (0.05 – 4 μM) were measured at 30 °C in 20 mM bis-tris propane, 10 mM TCEP pH 7.5, as described under “Materials and Methods” using apo-enzyme reconstituted with stoichiometric metal ion. The parameters kcat, KM and kcat/KM (Table 1) were obtained by fitting the Michaelis-Menten equation to these data.
The WT LpxC-catalyzed reaction exhibits a bell-shaped dependence on pH, wherein pKa1 and pKa2 are proposed to represent ionization of Glu78 and another group located near the metal ion, such as H265, respectively (14, 27). The pH-dependence of kcat/KM catalyzed by C63A LpxC activated by Zn2+ or Fe2+ (Figure 4) shows a bell-shaped pH profile, indicating that at least two ionizations are important for maximal activity, consistent with pH profiles previously measured for WT LpxC (14, 27). However, at high pH the decrease in activity for C63A LpxC has a squared dependence on the proton concentration (Figure 4), suggesting that two deprotonations affect the activity at high pH. Therefore, the C63A mutant is best described by three ionizations: one at low pH and two at high pH. These data are fit using an equation that assumes that the two high pH ionizations have comparable pKa values. The reactions catalyzed by EcC63A LpxC reconstituted with either stoichiometric Zn2+ or Fe2+ have nearly identical pH-profiles, indicating that the Fe-enzyme has higher catalytic activity regardless of the pH. Furthermore, the fitted values for pKa1 and pKa2 for the C63A mutant are slightly higher and comparable to, respectively, the values determined for wild-type LpxC, suggesting that the same ionizations are observed for both WT and the C63A mutant. The additional ionization at high pH observed for the C63A mutant could be due to several factors. For example, several ionizable LpxC residues not directly implicated in catalysis, such as H19 or K143 (18), could be perturbed in the C63A mutant and ionization of these residues could become observable in the steady-state kinetics. Alternatively, replacing the cysteine thiol with an alanine may lower the metal-water pKa such that it is observable at high pH. Ionization of these groups could directly affect the stability of the transition state or could reversibly destabilize the active structure of LpxC.
Figure 4.
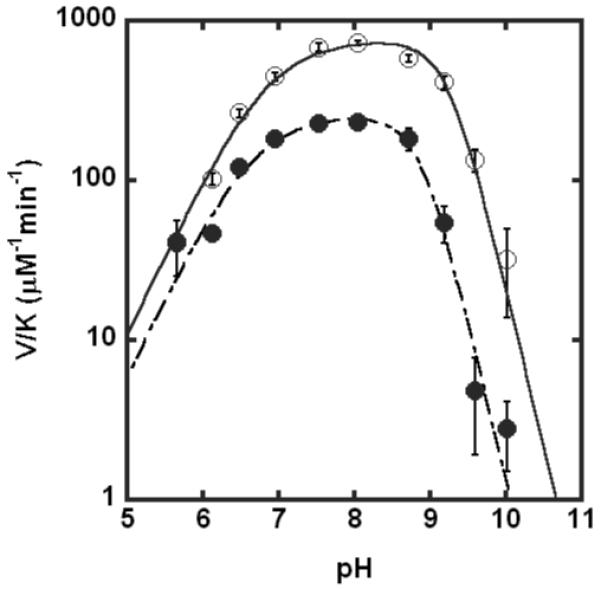
pH-dependence of LpxC-catalyzed deacetylase activity for Zn2+-C63A (●) or Fe2+ - C63A (○) LpxC. The values of V/K were measured at 30 °C using subsaturating concentrations of myr-UDPGlcNAc (≤ 0.2 μM) and enzyme reconstituted with stoichiometric metal ion, as described under “Materials and Methods”. The pKa values (see Table 1) were determined by fitting Eq. 1 to the data.
XAS analysis of LpxC
The change in LpxC activity observed with Fe2+ and Zn2+ cofactors may correlate with an alteration in the preferred coordination numbers and/or geometries of the respective metal ions. For peptide deformylase (PDF), the higher coordination number of Fe2+-PDF compared to Zn2+-PDF is proposed to contribute to the higher activity that is observed with the Fe2+ cofactor (28). In biological zinc sites a coordination number of 4 (tetrahedral geometry) is observed most frequently, although 5 and 6 ligands are also observed, while Fe2+ prefers higher coordination numbers (5 or 6) (29-31). Previous XAS studies of AaLpxC and Pseudomonas aeruginosa LpxC (PaLpxC) with a single bound Zn2+ (12) demonstrated that the catalytic zinc ion has four N/O ligands. Similarly, crystal structures of the zinc-inhibited and the cacodylate complex of AaLpxC indicate that the catalytic zinc ion is tetrahedral (26, 32). The best fits to the EXAFS data for EcLpxC with Zn2+ bound to both the catalytic and inhibitory sites is 3 N/O and 1 S/Cl while the fit for EcLpxC with Co2+ at the catalytic site is 5 N/O. These previous data suggest that the increased activity of the Co2+ EcLpxC (17) correlates with the higher coordination number.
To probe determinants of the enhanced activity of Fe2+-LpxC, we examined the coordination environments of EcLpxC and AaLpxC with stoichiometric Fe2+ bound at the catalytic site. The XANES and EXAFS data for these samples are shown in Figure 5. The XANES region of the XAS spectra (1s → 3d transitions) provides valuable information about metal ion coordination number and oxidation state. The 1s → 3d transitions (pre-edge areas) observed for 4-, 5- and 6-coordinate Fe are 19.8 - 25 × 10−2 eV, 12.4 - 18.8 × 10−2 eV, and 3.1 - 9.9 × 10−2 eV, respectively (33-35). The Fe pre-edge areas calculated for EcLpxC (12.1 × 10−2 eV) and AaLpxC (11.2 - 14.9 × 10−2 eV) are both consistent with a 5-coordinate Fe species (Table 3), and in particular, are significantly smaller than the values observed for tetrahedral Fe. This conclusion is supported by the EXAFS data for both WT Fe2+-EcLpxC and Fe2+-AaLpxC (Table 4, Figure 5). Consistent with our observations for Zn-AaLpxC and Zn-PaLpxC (12), the EXAFS data for Fe2+-EcLpxC and Fe2+-AaLpxC show significant disorder. Fits using 4, 5, or 6 low-Z ligands all give similar quality fits, with Debye-Waller factors that increase as the fitted coordination number increases (Table 4). Although it was possible in most cases to fit the data using a mixed ligation mode of 3-5 (O/N) + 1 S, in no case did these fits result in more than a modest (i.e., < 10%) improvement in fit quality, improvement that can be accounted for by the doubling in the number of adjustable parameters. Further evidence that the EXAFS data do not support the presence of mixed (N/O)+S ligation comes from the fact that in most cases the apparent Fe-S parameters were chemically unreasonable (e.g., apparent Fe-S distances < 2 Å, apparent Fe-S Debye-Waller factors σ2 > 0.01 Å2). In all cases, both (N/O) only and (N/O)+S fits, the apparent Fe-(N/O) distance was ~2.1 Å, significantly longer than that found for the 4-coordinate Zn2+ that is observed in many XAS and crystallographic studies of AaLpxC, PaLpxC and EcLpxC (12), and consistent with the distances found for 5-coordinate Fe2+. This conclusion is supported by the fact that the 5-coordinate Fe-(N/O) fits consistently give the bond-valence sum values (36, 37) closest to 2.
Figure 5.
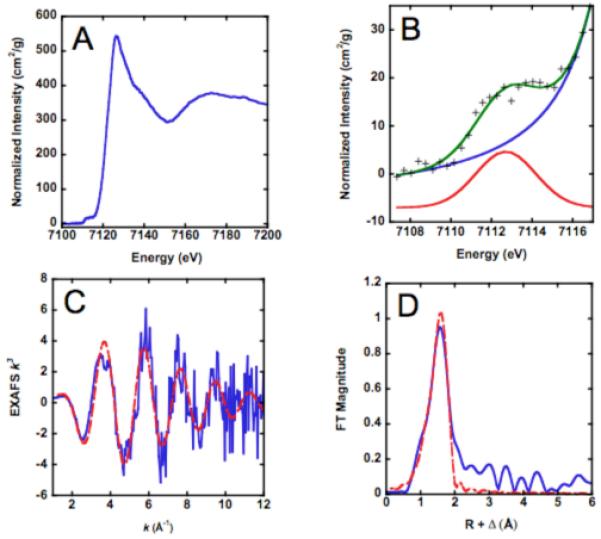
XAS of Fe2+-EcLpxC. (A) XANES region. (B) Expansion of XANES showing 1s->3d transition with calculated background (blue) and best fit (green). Gaussian fit to 1s->3d transition is shown offset vertically for clarity. (C) k3 weighted EXAFS data (blue) together with best fit using 5 oxygen ligands (red, dashed). (D) Fourier transform of EXAFS data: experimental (solid line) and fit (dashed line) to 5 N/O ligands.
Table 3.
XANES pre-edge areas
| Sample | 1s->3d (sample 1) |
1s->3d (sample 2) |
1s->3d average |
|---|---|---|---|
| Fe2+-AaLpxC | 11.2 × 10−2 eV | 14.9 × 10−2 eV | 13.1 × 10−2 eV |
| Fe2+-EcLpxC | 12.1 × 10−2 eV | 12.1 × 10−2 eV | 12.1 × 10−2 eV |
Table 4.
Fitting results for Fe-LpxC
| Na | R ( )b )b
|
σ2×103 ( 2×103) 2×103) |
Fc | BVSd | |
|---|---|---|---|---|---|
| Fe2+-AaLpxC | |||||
| Sample 1 | 4 | 2.100 | 5.7 | 0.66 | 1.5 |
| 5 | 2.100 | 7.7 | 0.67 | 1.9 | |
| 6 | 2.100 | 9.7 | 0.70 | 2.2 | |
| Sample 2 | 4 | 2.100 | 6.0 | 0.40 | 1.5 |
| 5 | 2.096 | 8.2 | 0.42 | 1.9 | |
| 6 | 2.097 | 10.5 | 0.44 | 2.3 | |
| Fe2+-EcLpxC | |||||
| Sample 1 | 4 | 2.100 | 6.4 | 0.30 | 1.5 |
| 5 | 2.105 | 8.2 | 0.30 | 1.8 | |
| 6 | 2.106 | 10.1 | 0.32 | 2.2 | |
| Sample 2 | 4 | 2.090 | 6.8 | 0.33 | 1.5 |
| 5 | 2.090 | 8.8 | 0.33 | 1.9 | |
| 6 | 2.090 | 10.9 | 0.35 | 2.3 |
EXAFS coordination number, fixed at integer values
Fe-O bond length. Although the experimental accuracy is estimated at ±0.02 Å, values are reported to 0.001 Å in order to show the precision of the data
Root-mean square deviation between data and fit
Bond-valence sum
Crystallographic analyses of AaLpxC and PaLpxC complexed with hydroxamate inhibitors or palmitate indicate that the catalytic zinc changes geometry upon ligand binding to square pyramidal (5 coordinate) (26, 38). These data indicate that the geometry of the metal site is flexible, consistent with our finding that the Fe2+ site appears to adopt a 5-coordinate structure even in the absence of added inhibitors. Since the Fe-LpxC coordination sphere is best fit to 5 (N/O) ligands, the added ligand is most likely a water molecule as observed in other proteins where metal substitution leads to a higher coordination number, such as carbonic anhydrase (39) and peptide deformylase (28), These observations are consistent with the suggestion that the higher activity and ligand affinity of Fe2+-EcLpxC correlates with the higher coordination number of Fe2+.
Effect of Fe2+ on molecular recognition
The metal ion status of LpxC was previously shown to influence the binding affinity of LpxC for small molecules, which has important implications for the development of LpxC inhibitors as potential antibiotics (18, 19). Here, we examine whether substitution of EcLpxC with Fe2+ alters the affinity of small molecules for LpxC compared to Zn2+ by measuring the affinities of myrUDPGlcNAc (KDProduct) and a fluorescent fatty acid (KDfatty acid) for LpxC (WT and C63A) using ultrafiltration and fluorescence anisotropy, respectively. The C63A mutation in LpxC causes a small enhancement in the binding affinity of these ligands, irregardless of the metal ion at the active site (Table 2); the value of KDProduct and KDfatty acid are lowered 3- to 4- and < 2-fold, respectively, compared to WT LpxC. The value of KDProduct is not significantly altered when comparing Fe2+- to Zn2+-EcLpxC, in either WT or C63A, suggesting that product binding in these enzymes is similar and/or that there is not a direct metal ion-product interaction in these complexes. In contrast, a ~6-fold increase in KDfatty acid is observed for LpxC (WT and C63A) substituted with Fe2+ compared to Zn2+, indicating that changing the metal ion significantly alters recognition of this small molecule, consistent with the direct metal ion-fatty acid interaction observed in crystal structures of the LpxC·palmitate complex (14, 32). Finally, a modest (2-fold) enhancement in inhibition (IC50 value) by the hydroxamate inhibitor L-161,240 is observed for LpxC with bound Fe2+ compared to Zn2+ (Table 2). This is comparable to the 2-fold decrease in Ki for inhibition of histone deacetylase 8 by suberoylanilide hydroxamic acid upon switching the active site metal ion from Zn2+ to Fe2+ (11).
Table 2.
LpxC molecular recognition parameters
| Enzymea |
KDProduct (μM)b |
KDFatty acid (μM)c |
IC50 (nM)d |
|---|---|---|---|
| Apo-LpxC | 1.5 ± 0.5 | 25 ± 1 | ND |
| Zn2+-LpxC | 1.6 ± 0.2e | 7 ± 0.6f | ND |
| Fe2+-LpxC | 1.9 ± 0.3 | 45 ± 3 | ND |
| Apo-C63A | 0.5 ± 0.1 | 17 ± 1 | ND |
| Zn2+-C63A | 0.40 ± 0.07 | 7 ± 1 | 4 ± 0.4 |
| Fe2+-C63A | 0.6 ± 0.1 | 44 ± 3 | 2 ± 0.2 |
Metal substituted LpxC was reconstituted with stoichiometric metal ion (1:1) as described in the “Materials and Methods”.
Product (myr-UDPGlcNH2) binding to LpxC was measured at 30 °C (20 mM bis-tris propane, 10 mM TCEP pH 7.5) using ultrafiltration as described in the “Materials and Methods”. The KD values were obtained by fitting Eq. 4 to the data.
BODIPY-fatty acid binding to LpxC was measured at 30 °C (20 mM bis-tris propane, 10 mM TCEP pH 7.5, 100 μM ZnSO4 or FeCl2) using fluorescence anisotropy as described in the “Materials and Methods”. The KD values were obtained by fitting Eq. 5 to the data.
Hydroxamate L161, 240 inhibition of LpxC activity was measured at 30 °C (20 mM bis-tris propane, 10 mM TCEP pH 7.5, 1 mg/mL BSA) and 200 nM myrUDPGlcNAc as described in the “Materials and Methods”. The IC50 values were obtained by fitting Eq. 2 to the data.
Data are adapted from ref (18).
Data are adapted from ref (19).
Oxygen sensitivity of Fe2+-EcLpxC activity
Since Fe2+ is a redox sensitive metal ion, it is susceptible to oxidation. The initial rate for deacetylation catalyzed by LpxC after reconstitution with stoichiometric Fe2+ is decreased ≥ 8-fold when assayed under aerobic conditions compared to anaerobic conditions (glove box). Furthermore, similar decreases were observed when the deacetylase activity of Fe2+-LpxC was measured in the presence of catalase (100 μg/mL), dithiothreitol (2 mM), dithionite (≤10 mM) or TCEP (≤ 2 mM) under aerobic conditions, suggesting that these conditions are not sufficient for maintaining Fe in the reduced form. However, when higher concentrations of TCEP (10 mM) are added to the assay, comparable deacetylase activity is observed for Fe2+-LpxC under both aerobic and anaerobic conditions over 2 hours (Figure 6). In contrast, there is no change in Zn2+-LpxC activity when the concentration of TCEP is varied under these conditions (Figure 6A). Note that the activity of Fe2+-EcLpxC does not go to zero, but to the level observed for Zn2+-EcLpxC activity suggesting that under these conditions the Fe2+ cofactor is replaced by nM concentrations of adventitious Zn2+ in the assay. In vitro control experiments demonstrate that Fe2+-LpxC inactivated by exposure to oxygen can be reactivated by addition of divalent metal ions. These results demonstrate that the activity of Fe2+-LpxC, and not Zn2+-LpxC, is sensitive to exposure to oxygen.
Figure 6.

Dependence of LpxC activity on reducing agents. (A) The activity of Fe2+- and Zn2+-EcLpxC was measured at 30 °C in buffer containing either 10 mM (black bars) or 0.5 mM (gray bars) TCEP as described in the “Materials and Methods” section. (B) Apo-EcLpxC was reconstituted with either Fe2+ (open circle) or Zn2+ (filled circle) and the resulting activity was measured at 30 °C (20 mM bis-tris propane, 10 mM TCEP, pH 7.5) as a function of time.
Native LpxC activity
To analyze the metal ion cofactor bound to LpxC in vivo, we measured the oxygen sensitivity of natively expressed LpxC in E. coli cell lysates. E. coli cells (BL21(DE3) without the LpxC expression plasmid) were grown, lysed and the resulting LpxC activity was measured over time (15 min - 3 hr following lysis) in buffers containing either ≤ 1 mM or 10 mM TCEP. The total native EcLpxC activity in cell lysates is ~3-fold higher in assays containing 10 mM TCEP compared to 0.1 mM TCEP (Figure 7 – column A). Furthermore, over 3 hours the activity decreases ~5-fold (Figure 7, column B) to a residual activity that is stable in the cell lysate. If the residual activity measured in the presence of the LpxC hydroxamate inhibitor L-161,240 (Figure 7, column C), reflecting background deacetylation catalyzed by other enzymes, is subtracted from the oxygen sensitive activity, then the observed activity decreases 5- to 7-fold in the presence of oxygen. This decrease is consistent with the higher activity of Fe2+-LpxC compared to Zn2+-LpxC. These findings suggest that the majority of the native LpxC expressed in E. coli contains a bound Fe2+ cofactor.
Figure 7.
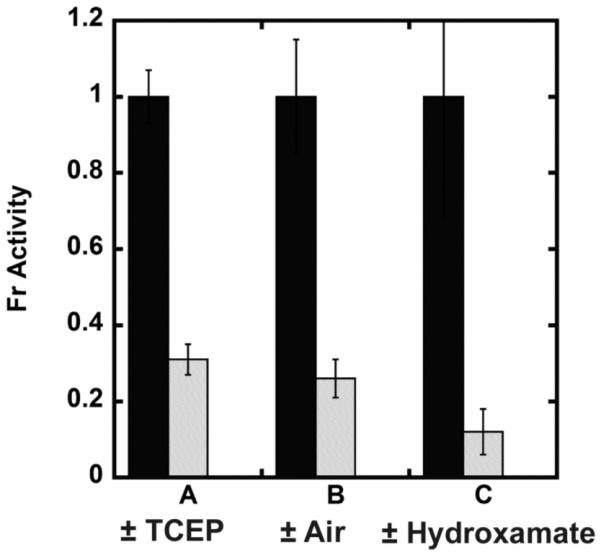
Native LpxC activity. The LpxC deacetylase activity of E. coli crude cell lysates was measured at 30 °C as described in “Materials and Methods”. (A) LpxC activity of E. coli cell lysate measured in 20 mM bis-tris propane pH 7.5 containing 10 mM (black bars) or 0.1 mM TCEP (gray bars). (B) E. coli cell lysate activity assayed in 10 mM TCEP immediately following lysis (black bars) or after incubation on ice for 3 hours under aerobic (benchtop) conditions post-lysis (gray bars). (C) The LpxC inhibitor L-161,240 inhibits 90% of the deacetylase activity in the E. coli cell lysate (20 mM bis-tris propane, 10 mM TCEP pH 7.5). The concentration of L-161,240 was 0 μM (black bars) or (1 μM) (gray bars).
Discussion
LpxC activity correlates with metal geometry
The kinetic experiments indicate that Fe2+ can serve as a cofactor for E. coli LpxC, a mononuclear metal-dependent deacetylase, and that the Fe2+ cofactor alters the functional properties of LpxC compared to Zn2+. Specifically, LpxC substituted with a single catalytic Fe2+ has 6- to 8-fold higher activity than Zn2+-LpxC. Comparison of the steady-state kinetic parameters for the C63A mutant indicate that switching from Zn2+ to Fe2+ leads to a 6-fold increase in the value of kcat/KM, with a nearly comparable effect on the value of kcat (4-fold). The single site mutation, C63A, eliminates much of the metal inhibition that complicates analysis of LpxC activity, likely by removing a ligand to the inhibitory metal.
Metal substitution in LpxC increases the value of both the steady-state parameters kcat/KM and kcat. The dependence of the value of kcat/KM for LpxC on the active site metal ion and on mutations (Table 1; (14, 18)) argue that substrate association is not the rate-limiting step under these conditions even though the value of kcat/KM (≤ 1.6 × 107 M−1s−1; Table 1) approaches that of diffusion-controlled rate constants measured for many enzymes (107 – 108 M−1s−1) (40). Additionally, the solvent kinetic isotope effect of 2 observed for kcat/KM catalyzed by LpxC (18) suggests that the chemical step is a rate contributing step for this kinetic parameter. Additionally, under conditions of saturating substrate and high enzyme concentration, a burst of product formation is not observed, suggesting that the rate-limiting step in kcat occurs at or before the chemical step (27). Therefore, it is reasonable to assume that the increases in kcat/KM and kcat upon substitution of Zn2+ with Fe2+ are attributable mainly to an enhancement in the chemical step.
The best fit of the models to the XAS data is 5 N/O ligands for both Fe2+-substituted EcLpxC and AaLpxC compared to the 4 N/O ligands observed for WT Zn2+-LpxC presumably due to an additional water ligand (12). These results are consistent with the hypothesis that the higher activity of the Fe2+-substituted EcLpxC correlates with the coordination number. Previously, the enhanced activity of Fe2+-peptide deformylase (PDF) has been proposed to be due, at least in part, to the higher coordination number of Fe2+-PDF compared to Zn2+-PDF (8, 28, 41-43). The XAS and activity data for Fe2+-EcLpxC, in concert with recent crystallography data demonstrating a square pyramidal zinc site in LpxC-complexed with hydroxamate inhibitors or palmitate (26, 38), suggest that the proposed mechanism for deacetylation should be modified to incorporate a 5-coordinate metal ion, in both the ground and transition states (Figure 8). In this mechanism, the metal ion serves both to lower the pKa of the metal-bound water and to coordinate the substrate (myrUDPGlcNAc). Coordination of the substrate to the metal ion can assist in polarization of the carbonyl group, enhancing the electrophilicity of the carbonyl carbon. However, enhancement of activity by the 5-coordinate Fe2+ cofactor could also be due to small differences in metal-ligand bond length and ligand geometry that optimize the positions of the metal-bound water, the substrate carbonyl carbon and active site side chains for catalyzing deacetylation Following substrate binding, the side chain of E78 serves as a general base catalyst to activate the metal-water for attack on the carbonyl carbon of the substrate (14, 27); the resulting oxyanion intermediate is stabilized by the side chain of T191 and the metal ion (18). The mechanism that is most consistent with the crystallographic, theoretical studies and mutagenesis data (14, 18, 44) is that the side chain of protonated H265 (rather than protonated E78 as proposed for most metalloproteinases (45)) facilitates breakdown of the tetrahedral intermediate by protonation of the leaving group.
Figure 8.

Proposed mechanism for EcLpxC.
Molecular recognition is affected by metal substitution
Previously it has been demonstrated that apo-LpxC, LpxC with a single bound zinc, and the zinc-inhibited form of LpxC (Zn2-LpxC) bind ligands with different affinity (19); similarly, switching the catalytic metal from Zn2+ to Fe2+ alters molecular recognition as well. The affinity of Fe2+-EcLpxC for a hydroxamate inhibitor increases by a small degree (~2-fold), consistent with crystal structures indicating that the metal geometry in LpxC-hydroxamate complexes is 5-coordinate (square pyramidal) (26, 38). A similar increase in hydroxamate inhibitor affinity has been observed upon switching the active site metal from Zn2+ to Fe2+ in histone deacetylase 8 (11). In addition, the affinity of Fe2+-EcLpxC for the BODIPY fatty acid inhibitor is decreased by 6-fold compared to the Zn-bound enzyme. Both changes in affinity are likely due to alteration of the geometry of the metal-ligand coordination leading to alterations in interactions with other active site groups. Since the majority of LpxC inhibitors currently being developed as antibiotics interact with the active site metal ion, it is clear that the identity of this metal ion will affect the inhibitor efficacy.
Fe2+-LpxC is redox-sensitive in vitro and present in E. coli
Differences in the in vitro behaviors of Fe2+-LpxC and Zn2+-LpxC may provide information that will enable identification of the metal ion cofactor used by LpxC in vivo. In particular, the higher activity and oxygen sensitivity of Fe2+-LpxC compared to Zn2+-LpxC provides a means to evaluate the metal cofactor bound to LpxC in E. coli lysates. A majority of the native LpxC activity in E. coli cell lysates is lost upon exposure to oxygen (Figure 7). This suggests that E. coli grown aerobically in rich media (LB) use Fe2+ as the main metal cofactor in LpxC and not Zn2+. However, both Fe2+ and Zn2+ can activate LpxC-catalyzed deacetylation and this metal-switching capability may allow the LpxC to function, and E. coli to grow, under different environmental conditions, including limiting iron concentrations. Thus, LpxC likely fits into the category of a “cambialistic” enzyme that can be activated by either Fe or Zn, like several other enzymes, including the cambialistic Mn or Fe superoxide dismutases (46), metallo-beta-lactamase L1 (47, 48), and the glyoxylases (49-51).
Given the effects of the active site metal bound to LpxC on turnover and molecular recognition, and the ongoing efforts to identify small-molecular inhibitors for this enzyme, most of which feature moieties that bind the catalytic metal ion, further experiments to determine the behavior and specificity of this enzyme in vivo are needed.
ACKNOWLEDGMENT
We gratefully acknowledge Ted Huston for ICP analysis, Chris Walsh for the gift of acyl carrier protein, Chris Raetz for the LpxA plasmid, and Michael Pirrung for the inhibitor L-161,240. We also thank Rebekah Kelly for help with EXAFS analysis as well as Matthew Lattimer and the staff of SSRL beamline 9-3.
Footnotes
- LPS
- lipopolysaccharide
- LpxC
- UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase
- EcLpxC
- Escherichia coli LpxC
- AaLpxC
- Aquifex aeolicus LpxC
- EDTA
- ethylene diamine tetraacetic acid
- DPA
- dipicolinic acid
- UDP-GlcNAc
- uridine-5′-diphosphate-N-acetylglucosamine
- myrUDP-GlcNAc
- UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine
- myrUDP-GlcNH2
- UDP-3-O-(R-3-hydroxymyristoyl)-glucosamine
- peptide deformylase
- HDAC
- histone deacetylase
- XAS
- x-ray absorption spectroscopy
- EXAFS
- extended x-ray absorption fine structure
- XANES
- x-ray absorption near edge structure
- ICP-MS
- inductively coupled plasma emission spectroscopy-mass spectrometry
- WT
- wild type
- BSA
- bovine serum albumin
- TCEP
- triscarboxyethylphosphine
- DTT
- dithiothreitol
- DMSO
- dimethylsulfoxide
- MWCO
- molecular weight cutoff
- BODIPY® 500/510 C4, C9
- 5-butyl-4,4-difluoro-4-bora-3a,4a-diaza-s-indacene-3-nonanoic acid
REFERENCES
- (1).Roth LD, Khan AS, Lillibridge SR, Ostroff SM, Hughes JM. Public health assessment of potential biological terrorism agents. Emerging infectious diseases. 2002;8:225–230. doi: 10.3201/eid0802.010164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Raetz CRH, Whitfield C. Lipopolysaccharide endotoxins. Annual Review of Biochemistry. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Wyckoff TJO, Raetz CRH, Jackman JE. Antibacterial and anti-inflammatory agents that target endotoxin. Trends in Microbiology. 1998;6:154–159. doi: 10.1016/s0966-842x(98)01230-x. [DOI] [PubMed] [Google Scholar]
- (4).White RJ, Margolis PS, Trias J, Yuan ZY. Targeting metalloenzymes: a strategy that works. Current Opinion in Pharmacology. 2003;3:502–507. doi: 10.1016/s1471-4892(03)00115-2. [DOI] [PubMed] [Google Scholar]
- (5).Young K, Silver LL, Bramhill D, Cameron P, Eveland SS, Raetz CRH, Hyland SA, Anderson MS. The envA permeability cell division gene of Escherichia coli encodes the second enzyme of lipid A biosynthesis - UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase. Journal of Biological Chemistry. 1995;270:30384–30391. doi: 10.1074/jbc.270.51.30384. [DOI] [PubMed] [Google Scholar]
- (6).Jackman JE, Raetz CRH, Fierke CA. UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase of Escherichia coli is a zinc metalloenzyme. Biochemistry. 1999;38:1902–1911. doi: 10.1021/bi982339s. [DOI] [PubMed] [Google Scholar]
- (7).Groche D, Becker A, Schlichting I, Kabsch W, Schultz S, Wagner AFV. Isolation and crystallization of functionally competent Escherichia coli peptide deformylase forms containing either iron or nickel in the active site. Biochemical and Biophysical Research Communications. 1998;246:342–346. doi: 10.1006/bbrc.1998.8616. [DOI] [PubMed] [Google Scholar]
- (8).Rajagopalan PTR, Yu XC, Pei DH. Peptide deformylase: A new type of mononuclear iron protein. Journal of the American Chemical Society. 1997;119:12418–12419. [Google Scholar]
- (9).Becker A, Schlichting I, Kabsch W, Groche D, Schultz S, Wagner AFV. Iron center, substrate recognition and mechanism of peptide deformylase. Nature Structural Biology. 1998;5:1053–1058. doi: 10.1038/4162. [DOI] [PubMed] [Google Scholar]
- (10).Zhu JG, Dizin E, Hu XB, Wavreille AS, Park J, Pei DH. S-ribosylhomocysteinase (LuxS) is a mononuclear iron protein. Biochemistry. 2003;42:4717–4726. doi: 10.1021/bi034289j. [DOI] [PubMed] [Google Scholar]
- (11).Gantt SL, Gattis SG, Fierke CA. Catalytic activity and inhibition of human histone deacetylase 8 is dependent on the identity of the active site metal ion. Biochemistry. 2006;45:6170–6178. doi: 10.1021/bi060212u. [DOI] [PubMed] [Google Scholar]
- (12).McClure CP, Rusche KM, Peariso K, Jackman JE, Fierke CA, Penner-Hahn JE. EXAFS studies of the zinc sites of UDP-(3-O-acyl)-N- acetylglucosamine deacetylase (LpxC) Journal of Inorganic Biochemistry. 2003;94:78–85. doi: 10.1016/s0162-0134(02)00611-6. [DOI] [PubMed] [Google Scholar]
- (13).Jackman JE, Fierke CA, Tumey LN, Pirrung M, Uchiyama T, Tahir SH, Hindsgaul O, Raetz CRH. Antibacterial agents that target lipid A biosynthesis in Gram-negative bacteria - Inhibition of diverse UDP-3-O-(R-3- hydroxymyristoyl)-N-acetyglucosamine deacetylases by substrate analogs containing zinc binding motifs. Journal of Biological Chemistry. 2000;275:11002–11009. doi: 10.1074/jbc.275.15.11002. [DOI] [PubMed] [Google Scholar]
- (14).Hernick M, Gennadios HA, Whittington DA, Rusche KM, Christianson DW, Fierke CA. UDP-3-O-((R)-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase functions through a general acid-base catalyst pair mechanism. Journal of Biological Chemistry. 2005;280:16969–16978. doi: 10.1074/jbc.M413560200. [DOI] [PubMed] [Google Scholar]
- (15).Sorensen PG, Lutkenhaus J, Young K, Eveland SS, Anderson MS, Raetz CRH. Regulation of UDP-3-0- R-3-hydroxymyristoyl) -N- acetylglucosamine deacetylase in Escherichia coli - The second enzymatic step of lipid a biosynthesis. Journal of Biological Chemistry. 1996;271:25898–25905. doi: 10.1074/jbc.271.42.25898. [DOI] [PubMed] [Google Scholar]
- (16).Onishi HR, Pelak BA, Gerckens LS, Silver LL, Kahan FM, Chen MH, Patchett AA, Galloway SM, Hyland SA, Anderson MS, Raetz CRH. Antibacterial agents that inhibit lipid A biosynthesis. Science. 1996;274:980–982. doi: 10.1126/science.274.5289.980. [DOI] [PubMed] [Google Scholar]
- (17).Jackman JE, Raetz CR, Fierke CA. UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase of Escherichia coli is a zinc metalloenzyme. Biochemistry. 1999;38:1902–1911. doi: 10.1021/bi982339s. [DOI] [PubMed] [Google Scholar]
- (18).Hernick M, Fierke CA. Catalytic Mechanism and Molecular Recognition of E. coli UDP-3-O-(R-3-Hydroxymyristoyl)-N-acetylglucosamine Deacetylase Probed by Mutagenesis. Biochemistry. 2006;45:15240–15248. doi: 10.1021/bi061405k. [DOI] [PubMed] [Google Scholar]
- (19).Hernick M, Fierke CA. Molecular recognition by Escherichia coli UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase is modulated by bound metal ions. Biochemistry. 2006;45:14573–14581. doi: 10.1021/bi061625y. [DOI] [PubMed] [Google Scholar]
- (20).George GN. see http://www-ssrl.slac.stanford.edu/exafspak.html.
- (21).Ankudinov AL, Rehr JJ. Relativistic calculations of spin-dependent x-ray-absorption spectra. Physical Review B. 1997;56:R1712–R1715. [Google Scholar]
- (22).Ankudinov AL, Ravel B, Rehr JJ, Conradson SD. Real-space multiple-scattering calculation and interpretation of x-ray-absorption near-edge structure. Physical Review B. 1998;58:7565–7576. [Google Scholar]
- (23).McMaster WH, Del Grande JH, Mallet NK, Hubbell JH. Compilation of X-Ray Cross Sections. US Department of Commerce; 1969. Lawrence Livermore National Laboratory Report UCRL-50174, Sec II, Rev. 1, NIST. [Google Scholar]
- (24).Weng TC, Waldo GS, Penner-Hahn JE. A method for normalization of X-ray absorption spectra. Journal of Synchrotron Radiation. 2005;12:506–510. doi: 10.1107/S0909049504034193. [DOI] [PubMed] [Google Scholar]
- (25).Irving H, Williams RJP. Order of Stability of Metal Complexes. Nature. 1948;162:746–747. [Google Scholar]
- (26).Whittington DA, Rusche KM, Shin H, Fierke CA, Christianson DW. Crystal structure of LpxC, a zinc-dependent deacetylase essential for endotoxin biosynthesis. Proc Natl Acad Sci USA. 2003;100:8146–8150. doi: 10.1073/pnas.1432990100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).McClerren AL, Zhou P, Guan Z, Raetz CRH, Rudolph J. Kinetic Analysis of the Zinc-Dependent Deacetylase in the Lipid A Biosynthetic Pathway. Biochemistry. 2005;44:1106–1113. doi: 10.1021/bi048001h. [DOI] [PubMed] [Google Scholar]
- (28).Jain RK, Hao B, Liu RP, Chan MK. Structures of E-coli peptide deformylase bound to formate: Insight into the preference for Fe2+ over Zn2+ as the active site metal. Journal of the American Chemical Society. 2005;127:4558–4559. doi: 10.1021/ja0503074. [DOI] [PubMed] [Google Scholar]
- (29).Lippard SJ, Berg JM. Principles of Bioinorganic Chemistry. University Science Books; Mill Valley, California: 1994. [Google Scholar]
- (30).Frausto da Silva JJR, Williams RJP. The Biological Chemistry of the Elements. Oxford University Press; New York: 2001. [Google Scholar]
- (31).Kaim W, Schwederski . Bioinorganic Chemistry: Inorganic Elements in the Chemistry of Life. John Wiley and Sons LTD; Chichester: 1994. [Google Scholar]
- (32).Gennadios HA, Christianson DW. Binding of uridine 5′-diphosphate in the “basic patch” of the zinc deacetylase LpxC and implications for substrate binding. Biochemistry. 2006;45:15216–15223. doi: 10.1021/bi0619021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Shu L, Chiou YM, Orville AM, Miller MA, Lipscomb JD, Que L., Jr. X-ray absorption spectroscopic studies of the Fe(II) active site of catechol 2,3-dioxygenase. Implications for the extradiol cleavage mechanism. Biochemistry. 1995;34:6649–6659. doi: 10.1021/bi00020a010. [DOI] [PubMed] [Google Scholar]
- (34).Westre TE, Kennepohl P, DeWitt JG, Hedman B, Hodgson KO, Solomon EI. A multiplet analysis of Fe K-edge 1s->3d pre-edge features of iron complexes. Journal of the American Chemical Society. 1997;119:6297–6314. [Google Scholar]
- (35).Roe AL, Schneider DJ, Mayer RJ, Pyrz JW, Widom J, Que L. X-Ray Absorption-Spectroscopy of Iron-Tyrosinate Proteins. Journal of the American Chemical Society. 1984;106:1676–1681. [Google Scholar]
- (36).Thorp HH. Bond Valence Sum Analysis of Metal-Ligand Bond Lengths in Metalloenzymes and Model Complexes. Inorganic Chemistry. 1992;31:1585–1588. [Google Scholar]
- (37).Brown ID, Altermatt D. Bond-Valence Parameters Obtained from a Systematic Analysis of the Inorganic Crystal-Structure Database. Acta Crystallographica Section B-Structural Science. 1985;41:244–247. [Google Scholar]
- (38).Mochalkin I, Knafels JD, Lightle S. Crystal structure of LpxC from Pseudomonas aeruginosa complexed with the potent BB-78485 inhibitor. Protein Sci. 2008;17:450–457. doi: 10.1110/ps.073324108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Hakansson K, Wehnert A, Liljas A. X-Ray-Analysis of Metal-Substituted Human Carbonic-Anhydrase-Ii Derivatives. Acta Crystallographica Section D-Biological Crystallography. 1994;50:93–100. doi: 10.1107/S0907444993008790. [DOI] [PubMed] [Google Scholar]
- (40).Fersht AR. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding. W. H. Freeman and Company; New York: 1999. [Google Scholar]
- (41).Rajagopalan PT, Datta A, Pei D. Purification, characterization, and inhibition of peptide deformylase from Escherichia coli. Biochemistry. 1997;36:13910–13918. doi: 10.1021/bi971155v. [DOI] [PubMed] [Google Scholar]
- (42).Rajagopalan PT, Pei D. Oxygen-mediated inactivation of peptide deformylase. J Biol Chem. 1998;273:22305–22310. doi: 10.1074/jbc.273.35.22305. [DOI] [PubMed] [Google Scholar]
- (43).Chan MK, Gong W, Rajagopalan PT, Hao B, Tsai CM, Pei D. Crystal structure of the Escherichia coli peptide deformylase. Biochemistry. 1997;36:13904–13909. doi: 10.1021/bi9711543. [DOI] [PubMed] [Google Scholar]
- (44).Robinet JJ, Gauld JW. DFT investigation on the mechanism of the deacetylation reaction catalyzed by LpxC. The journal of physical chemistry B. 2008;112:3462–9. doi: 10.1021/jp075415m. [DOI] [PubMed] [Google Scholar]
- (45).Christianson DW, Cox JD. Catalysis by metal-activated hydroxide in zinc and manganese metalloenzymes. Annual Review of Biochemistry. 1999;68:33–57. doi: 10.1146/annurev.biochem.68.1.33. [DOI] [PubMed] [Google Scholar]
- (46).Martin ME, Byers BR, Olson MOJ, Salin ML, Arceneaux JEL, Tolbert C. A Streptococcus-Mutans Superoxide-Dismutase That Is Active with Either Manganese or Iron as a Cofactor. Journal of Biological Chemistry. 1986;261:9361–9367. [PubMed] [Google Scholar]
- (47).Hu Z, Spadafora LJ, Hajdin CE, Bennett B, Crowder MW. Structure and Mechanism of Copper- and Nickel-Substituted Analogues of Metallo-beta-lactamase L1 (dagger) Biochemistry. 2009;48:2981–2989. doi: 10.1021/bi802295z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Hu ZX, Gunasekera TS, Spadafora L, Bennett B, Crowder MW. Metal content of metallo-beta-lactamase L1 is determined by the bioavailability of metal ions. Biochemistry. 2008;47:7947–7953. doi: 10.1021/bi8004768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Limphong P, McKinney RM, Adams NE, Makaroff CA, Bennett B, Crowder MW. The metal ion requirements of Arabidopsis thaliana Glx2-2 for catalytic activity. J Biol Inorg Chem. 2010;2:249–258. doi: 10.1007/s00775-009-0593-6. [DOI] [PubMed] [Google Scholar]
- (50).Limphong P, Nimako G, Thomas PW, Fast W, Makaroff CA, Crowder MW. Arabidopsis thaliana Mitochondrial Glyoxalase 2-1 Exhibits beta-Lactamase Activity. Biochemistry. 2009;48:8491–8493. doi: 10.1021/bi9010539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Limphong P, McKinney RM, Adams NE, Bennett B, Makaroff CA, Gunasekera T, Crowder MW. Human Glyoxalase II Contains an Fe(II)Zn(II) Center but Is Active as a Mononuclear Zn(II) Enzyme. Biochemistry. 2009;48:5426–5434. doi: 10.1021/bi9001375. [DOI] [PMC free article] [PubMed] [Google Scholar]