

Abstract
A general methodology to design label-free fluorescent functional DNA sensors using unmodified DNA via a vacant site approach is described. By extending one end of DNA with a loop, a vacant site that binds an extrinsic fluorophore, 2-amino-5,6,7-trimethyl-1,8-naphthyridine (ATMND) could be created at a selected position in the DNA duplex region of DNAzymes or aptamers. When the vacant site binds ATMND, ATMND's fluorescence is quenched. This fluorescence can be recovered when one strand of the duplex DNA is released through either metal ion-dependent cleavage by DNAzymes or analyte-dependent structural-switching by aptamers. Through this design, label-free fluorescent sensors for Pb2+, UO22+, Hg2+, and adenosine have been successfully developed. These sensors have high selectivity and sensitivity; detection limits as low as 3 nM, 8 nM, 30 nM, and 6 μM have been achieved for UO22+, Pb2+, Hg2+ and adenosine, respectively. Control experiments using vacant-site-free DNA duplexes and inactive variants of the functional DNAs indicate that the presence of the vacant site and the activity of the functional DNAs are essential for the performance of the proposed sensors. The vacant site approach demonstrated here can be used to design many other label-free fluorescent sensors to detect a wide range of analytes.
Introduction
Driven by their impact on human health, the detection and quantification of metal ions and organic molecules in biological and environmental systems has attracted intense attention recently.1,2 Although instrumental analysis is the routine method of probing these systems, the cost and complicated operation of the required instruments limit their usefulness in carrying out the on-site and real-time detection that is crucial for these systems. To overcome these limitations, a number of highly sensitive and selective sensors have been developed that are portable and offer rapid quantification.3-14 While these results are promising, a more general platform must be developed so that one strategy can be used to select a sensor for any of a wide range of analytes.
One general sensor platform is based on functional DNAs. These functional DNA molecules are selected through a combinatorial method known as in vitro selection15 or Systematic Evolution of Ligands by Exponential Enrichment (SELEX).16 The DNAzymes (also called catalytic DNAs, deoxyribozymes, or DNA enzymes) and aptamers selected through these methods are reported to exhibit catalytic activity or binding affinity in the presence of a diverse number of targets, which range from metal ions and small organic molecules to macromolecules, and even viruses and cells.15-29 Unlike other molecules used for sensor design, functional DNAs have predictable secondary structures that can be easily functionalized them with fluorophores, chromophores or electrochemical tags, making it possible to transform the specific interactions between functional DNAs and their targets into detectable signals.26,28-47 Therefore, numerous functional DNA sensors, such as fluorescent,39,46-51 colorimetric,52-59 and electrochemical32,38,60 sensors based on this platform, have been developed. Among them, fluorescent sensors are particularly interesting because of their high sensitivity, simple instrumentation, and reproducible quantification.
Most fluorescent functional DNA sensors require covalent coupling of a fluorophore or a quencher to either the end, or the internal site of a DNA strand. The interaction between a functional DNA and its target induces the separation of the fluorophore and the quencher, causing an observable increase in fluorescence.48-50 However, DNA labeling can be complicated, expensive, and intrusive. The label might interfere with a functional DNA as it interacts with its targets.61,62 In addition, the label can make it difficult to introduce a labeled DNA into a biological system. To overcome these limitations, label-free fluorescent functional DNA sensors have been developed using intercalating dyes,63-65 malachite green,66,67 and abasic sites.68-70 We have previously reported70 that a dSpacer-DNA that was originally developed by Teramae's group71-73 as sensors for nucleobase recognition can be converted into a general platform for designing label-free functional DNA sensors. These sensors are highly sensitive and feature a controllable fluorophore-binding site. Teramae and coworkers have also demonstrated that a label-free approach could be achieved by incorporating Spacer C3 into adenosine aptamers.68,69 Nevertheless, these designs still require that the DNA be modified with dSpacer or Spacer C3, which is not only expensive to make, but also difficult to introduce into biological system through encoding. Developing label-free functional DNA sensors that use unmodified DNA to achieve a controllable fluorophore-binding site, and extending this method to both DNAzymes and aptamers remain as unmet challenges.
Here, we report a general method for designing label-free fluorescent functional DNA sensors using unmodified DNA containing a vacant site that strongly binds the extrinsic fluorophore 2-amino-5,6,7-trimethyl-1,8-naphthyridine (ATMND), and application of this approach to rational design of fluorescent sensors. To demonstrate the generality of this approach, we choose two targets detectable by DNAzymes (Pb2+ and UO22+) and two targets detectable by aptamers (adenosine and Hg2+). We show that these sensors exhibit high sensitivity and selectivity toward their targets.
Materials and Methods
The fluorophore 2-amino-5,6,7-trimethyl-1,8-naphthyridine (ATMND) was purchased from Ryan Scientific Inc. (Mt. Pleasant, SC) and was used as received. Metal ion salts, nucleotides, human serum, and other chemicals for buffers were purchased from Sigma-Aldrich Inc. (St. Louis, MO).The drinking water used is filtered tap water from the UIUC campus. The following oligonucleotides were purchased from Integrated DNA Technologies, Inc. (Coralville, IA):
Pb2+-dependent DNAzyme and substrate (with a single RNA nucleotide linkage rA):
| 17Eva | 5′-ACAGACATCTCTTCTCCGAGCCGGTCGAAATAGTG-3′ |
| 17Sva | 5′- GACGATGAAACATCGTCCCACTATrAGGAAGAGATGTCTGT-3′ |
| 17SvaG | 5′- GGACGATGAAACATCGTCCCACTATrAGGAAGAGATGTCTGT-3′ |
| 17EvaMut | 5′-ACAGACATCTCTTCCCCGAGCCGGTCGAAATAGTG-3′ |
Adenosine aptamer:
| AdAPva | 5′- GATCACAAAGTGATCCCACCTGGGGGAGTATTGCGGAGGAAGGT-3′ |
| AdAPvaM1 | 5′- GATCACAAAGTGATCCCACCTGGGGGTGTATTGCGGAGGAAGGT-3′ |
| AdAPvaM2 | 5′- GATCACAAAGTGATCCCACCTGGGGGAGTATTGCGGAGGTAGGT-3′ |
ssDNAs for hybridization with adenosine aptamer:
| AdL1va | 5′- CCAGGTG-3′ |
| AdL2va | 5′- CCCAGGTG-3′ |
| AdL3va | 5′- CCCCAGGTG-3′ |
| AdL2G | 5′- CCCAGGTGG-3′ |
UO22+-dependent DNAzyme and substrate (with a single RNA nucleotide linkage rA):
| 39Eva | 5′-CACGTCCATCTCTGCAGTCGGGTAGTTAAACCGACCTTCAGACATAGTG-3′ |
| 39Sva | 5′-GACGATGAAACATCGTCCCACTATrAGGAAGAGATGGACGTG-3′ |
Hg2+ aptamer:
| HgAPva | 5′- GACGATGAAACATCGTCCCTGTTTGTTTGTTGGCCCCCCTTCTTTCTTAC-3′ |
ssDNAs for hybridization with Hg2+ aptamer:
| HgLva | 5′- CAAACAG-3′ |
Fluorescence experiments
For a typical Pb2+ fluorescent sensing experiment, 480 μL buffer A (25 mM HEPES pH 7.0 and 100 mM NaCl), 5 μL ATMND stock solution (75 μM), 5 μL substrate 17Sva (100 μM), and 10 μL DNAzyme 17Eva (100 μM) were added sequentially into a 1.5 mL microcentrifuge tube. Upon vortexing, the tube was allowed to stand at room temperature for 2 min. The solution was then transferred to a cuvette and kept under a constant temperature control at 5 °C. After 6 min to allow the temperature to reach equilibrium, 5 μL of Pb2+ stock solution (0∼200 μM) in 1 mM HNO3 was added to the cuvette, which was then vortexed. A time-dependent fluorescent measurement at ex/em = 358/405 nm was immediately started.
The procedure for the UO22+ fluorescent sensing experiment is the same as that of the Pb2+ fluorescent sensing experiment, except that the following reagents were used: 470 μL buffer B (50 mM MES pH 5.5 and 300 mM NaCl), 5 μL ATMND stock solution (100 μM), 10 μL substrate 39Sva (100 μM), and 15 μL DNAzyme 39Eva (100 μM).
In the kinetic study of the above DNAzyme-based sensors, a fluorescence decrease was observed in the first 2 min, especially at low Pb2+ or UO22+ concentrations (Figure 1b and 3). This decrease is due to the temperature effect of adding Pb2+ or UO22+ stock solution at room temperature to the sensor solution equilibrated at 5 °C. As a result, a brief temperature increase occurred at the beginning. Since the fluorescence of the ATMND-DNA complex tends to decrease with increasing temperature71, the fluorescence decrease was then observed in the initial 2 min before the temperature equilibrated. Because of this issue, the data between 6∼8 min were acquired for quantification to avoid the disturbance from the temperature change. For samples with high concentrations of Pb2+ and UO22+ (more than 250 nM), this temperature effect is negligible because the fluorescence signal increase is much faster. In this case, the initial rate for the first 30 s of fluorescence change was recorded to avoid the effect of substrate DNA depletion on the signal changes.
Figure 1.
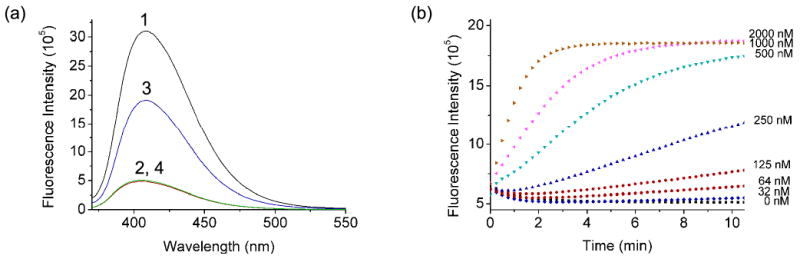
(a) Fluorescence spectra of (1) ATMND, (2) ATMND/17Sva/17Eva, (3) ATMND/17Sva/17Eva/Pb2+, (4) ATMND/17Sva/17Eva/Pb2+/EDTA. (b) Kinetics of fluorescence enhancement of ATMND/17Sva/17Eva in the presence of different amounts of Pb2+. Condition: 25 mM HEPES pH 7.0 and 100 mM NaCl at 5 °C. λex/λem = 358/405 nm.
Figure 3.
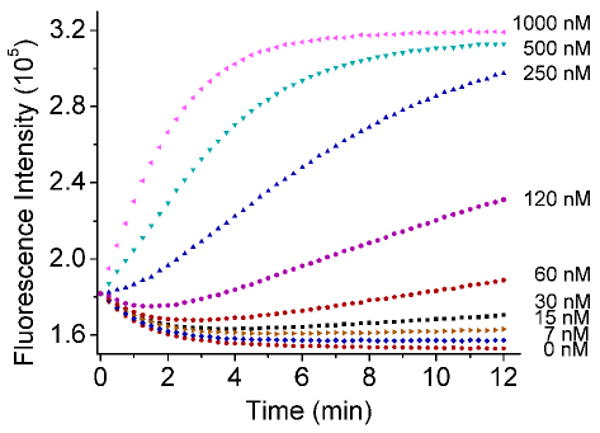
Kinetics of fluorescence enhancement of ATMND/39Sva/39Eva in the presence of different amounts of UO22+. Condition: 50 mM MES pH 5.5 and 300 mM NaCl at 5 °C. λex/λem = 358/405 nm.
In a typical adenosine sensing experiment, 480 μL buffer C (10 mM HEPES pH 7.0, 100 mM NaCl and 1mM EDTA), 5 μL aptamer AdAPva (100 μM), 10 μL ssDNA AdL1va∼AdL3va (100 μM), and 5 μL ATMND stock solution (50 μM) were added sequentially into a 1.5 mL microcentrifuge tube. After vortexing, the tube was allowed to stand at room temperature for 1 min. A 5 μL of adenosine stock solution (0∼1 mM) in buffer C was added to the above mixture, the solution was vortexed, and then it was allowed to stand at room temperature for 1 min. The solution was then transferred to a cuvette and kept under a constant temperature control at 5 °C. After 10 min, the fluorescence intensity at ex/em = 358/405 nm was recorded. The sample showed a stable intensity signal from 8∼30 min.
The procedure for Hg2+ fluorescent sensing experiment is the same as that of the adenosine sensing experiment, except that the following reagents were used: 480 μL buffer D (10 mM MOPS pH 7.2, 100 mM NaNO3), 5 μL aptamer HgAPva (30 μM), 5 μL ssDNA HgLva (35 μM), and 5 μL ATMND stock solution (10 μM).
Pb2+ detection in drinking water
Concentrated stock solutions of HEPES (500 mM) and NaCl (2 M) was added to drinking water containing different amounts of Pb2+ to achieve final concentration of HEPES and NaCl as 25 mM and 75 mM, respectively. Then, 480 μL of the sample, 5 μL ATMND stock solution (75 μM), 5 μL substrate 17Sva (100 μM), and 10 μL DNAzyme 17Eva (100 μM) were added sequentially into a 1.5 mL microcentrifuge tube. Upon vortexing, the tube was allowed to stand at room temperature for 2 min and then transferred to a cuvette and kept at 5 °C. After 25 min, the fluorescence intensity at ex/em = 358/405 nm was recorded.
Adenosine detection in human serum
Human serum was diluted five-fold by buffer C to produce a 20% serum sample. The detection of adenosine in this diluted serum sample is the same as that in buffer C as shown above. The concentration of adenosine detected in the diluted serum can be converted to the concentration in the original serum by multiplying the results by five.
Results and Discussion
General design
The success of fluorophore-labeled DNAzymes and aptamers as fluorescent sensors for Pb2+,21,51,74-76 UO22+,27,77 adenosine,55,78-80 and Hg2+ 81-85 with high sensitivity and selectivity encouraged us to transform these labeled sensors into label-free sensors. We hypothesized that a vacant site86 could serve as the binding site for a fluorophore in functional DNA sensors (Scheme 1). This site's affinity for fluorophores such as ATMND could be enhanced via hydrogen bonds, π-π stacking, and electrostatic interactions by positioning a cytosine opposite to the vacant site and two flanking guanines in DNA duplex. In the absence of targets, the functional DNA sensors could be designed to stabilize the vacant site to bind ATMND strongly, allowing the duplex DNA to quench the fluorescence of ATMND. A titration experiment carried out by adding different amounts of DNA-duplex containing a vacant site to ATMND suggested that they bound with high affinity (Ka > 106 M−1) and the resulting complex was stable in solution for at least one week (Figure S1, Supporting Information).The presence of a target, on the other hand, can cause either the catalytic cleavage of substrate by a DNAzyme or the structure-switching of an aptamer, resulting in perturbation of the vacant site, which lowers its affinity for ATMND. When ATMND is released, its fluorescence intensity increases. By monitoring this fluorescence change, the concentrations of target analytes can be measured. To demonstrate the generality of this vacant-site approach to functional DNA sensors, we chose a Pb2+-dependent DNAzyme, a UO22+-dependent DNAzyme, adenosine aptamer, and Hg2+-binding DNA with T-T mismatches (which may be considered to be an aptamer for Hg2+) as models,
Scheme 1.
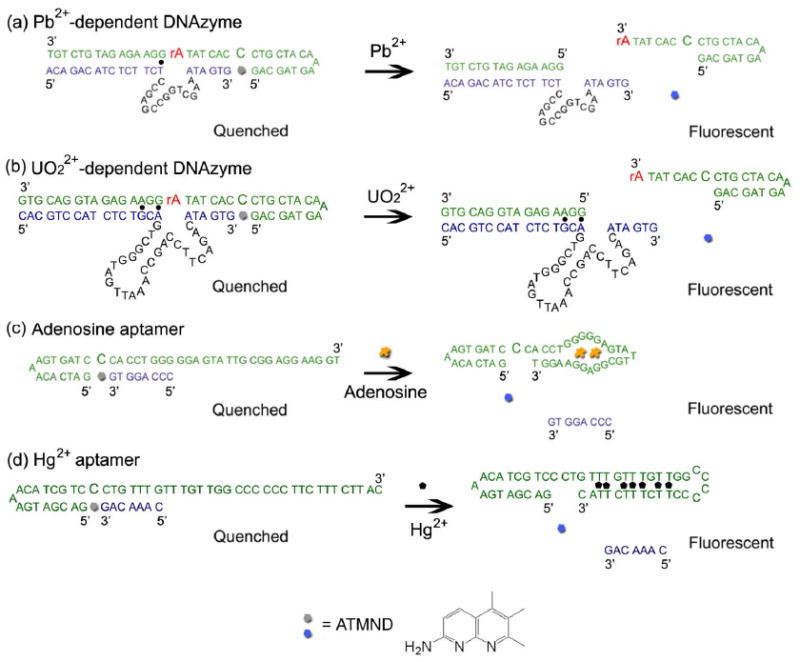
Fluorescence enhancement response of the functional DNA sensors specific to (a) Pb2+, (b) UO22+, (c) adenosine, and (d) Hg2+ using unmodified DNA via a vacant site approach.
Performance of the label-free functional DNA sensors with unmodified DNA
(1) Pb2+ sensor based on 8-17 DNAzyme
The design of label-free fluorescent sensor based on Pb2+-dependent 8-17 DNAzyme21 is shown in Scheme 1a. A loop was added to the 5′-end of the substrate strand (named 17Sva) of the DNAzyme (named 17Eva) to form a vacant site in the 17Sva/17Eva duplex. As illustrated in Figure 1a, the addition of 0.75 μM of ATMND into 1 μM 17Sva and 2 μM 17Eva resulted in ∼85% quenching of fluorescence signal of ATMND at 405 nm (compare Curves 1 and 2). This result suggested the strong binding of the ATMND to the vacant site as designed. Interestingly, the addition of 1 μM Pb2+ to the above system produced 275% increase of fluorescent signal within 6 min, (Curve 3), probably due to the Pb2+-induced cleavage of 17Sva and the release of ATMND from DNA duplex. To confirm the essential role of Pb2+ in the catalytic reaction, the above experiment was repeated in the presence of 1 mM EDTA, a metal ion chelator, and no fluorescent enhancement was observed (Curve 4). The kinetics of fluorescence enhancement were dependent on the concentration of Pb2+ (Figure 1b), displaying an increasing rate in the presence of higher concentrations of Pb2+. Instead of fluorescent intensity, the ratio of fluorescence enhancement rate over the fluorescence of a blank (ΔF/F0) was recorded, because the ratio is independent of fluorescence intensity and thus much less vulnerable to fluctuations in the background fluorescence. The blank fluorescence (F0) was that of ATMND/17Sva/17Eva free of Pb2+. The ΔF/F0 exhibited an approximately linear relationship with the concentration of Pb2+ between 0∼2 μM, and a calibration equation of ΔF/F0 (min−1) = 0.494×CPb2+ (μM) − 3.05 was derived from 7 data points within 0∼100 nM Pb2+ (Figure 2a). A detection limit of 8 nM was obtained based on 3σb/slope (σb, standard deviation of the blank samples) under the optimized condition. This detection limit is well below the maximum contamination level in drinking water (72 nM) defined by the US Environmental Protection Agency (EPA), and is either comparable or better than those Pb2+ sensors reported previously.21,51,74-76 This detection limit is very similar to those of fluorophore-labeled and dSpacer label-free methods using a Pb2+-dependent DNAzyme,20,21,70 suggesting that the detection limit is dependent on the functional DNA used and is not affected by the vacant sites incorporated into the DNA. Besides the high sensitivity, this vacant site approach did not sacrifice selectivity, as the selectivity toward Pb2+ over other divalent metal ions is similar to that reported before (Figure 2b).
Figure 2.
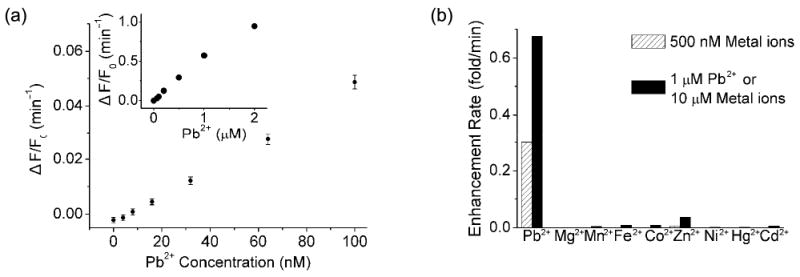
(a) Fluorescence enhancement rate as a function of Pb2+ concentration (0∼100 nM) for ATMND/17Sva/17Eva. Inset: 0∼2 μM Pb2+ range. (b) Selectivity of ATMND/17Sva/17Eva toward Pb2+ over other divalent metal ions. Condition: 25 mM HEPES pH 7.0 and 100 mM NaCl at 5 °C. λex/λem = 358/405 nm.
(2) UO22+ sensor based on 39E DNAzyme
After demonstrating the label-free Pb2+ sensor using 8-17 DNAzyme via the vacant site approach, we wondered if this approach could be generally applied to other DNAzyme sensors, such as the UO22+-dependent 39E DNAzyme.27,77 The sensor was designed so that the 5′-end of the substrate (named 39Sva) was extended to form a vacant site in the DNA duplex between the DNAzyme (named 39Eva) and 39Sva (Scheme 1b). Similar to the Pb2+ sensor described above, a UO22+-dependent fluorescence enhancement (Figure 3) was observed when different amounts of UO22+ were added to the solution containing 1 μM ATMND, 2 μM 39Sva and 3 μM 39Eva. The rate of enhancement over blank increased with the concentration of UO22+. The increase of fluorescence is attributable to the UO22+-dependent cleavage of substrate 39Sva by enzyme 39Eva that released ATMND from the vacant site and recovered the quenched fluorescence of ATMND. A detection limit (defined as 3σb/slope, σb, standard deviation of the blank samples) of 3 nM and an approximately linear range at least within 0∼1 μM UO22+ as ΔF/F0 (min-1) = 0.302×CUO22+ (μM) – 1.9 (calibrated from the data within 0∼60 nM range) were obtained under the optimal condition (Figure 4a). Although this detection limit of 3 nM is higher than the 45 pM detection limit of the original fluorophore-labeled design,27 because of the higher concentration of DNA used to stabilize the vacant-site to enable efficient label-free fluorophore binding, the sensitivity is high enough to monitor uranium in real-world samples such as drinking water, as the detection limit is still above the US Environmental Protection Agency (EPA) maximum contamination level of 126 nM in drinking water). Similar to the original labeled design,27,77, the label-free sensor reported here maintained the excellent selectivity toward UO22+ over other metal ions (Figure 4b).
Figure 4.
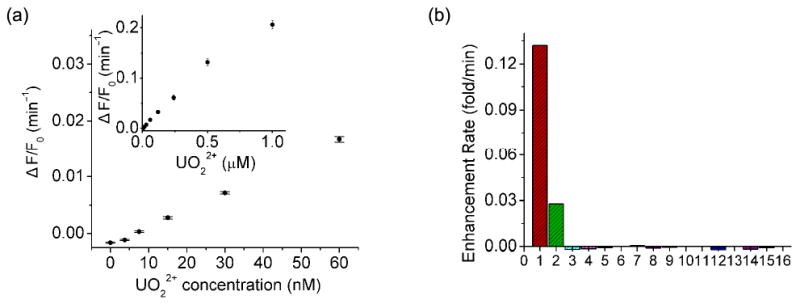
(a) Fluorescence enhancement rate as a function of UO22+ concentration (0∼60 nM) for ATMND/39Sva/39Eva. Inset: 0∼1 μM UO22+ range. (b) Selectivity of ATMND/39Sva/39Eva toward UO22+ over other metal ions. 1∼16: 500 nM UO22+, 125 nM UO22+, 2 μM Mg2+/Ca2+/Sr2+/Ba2+, Mn2+, Fe2+, VO2+, Th4+, Tb3+, Eu3+, Co2+, Ni2+, Cu2+, Zn2+, Pb2+, Cd2+, Hg2+. Condition: 50 mM MES pH 5.5 and 300 mM NaCl at 5 °C. λex/λem = 358/405 nm.
(3) Adenosine sensor based on adenosine aptamer
In addition to the DNAzymes, we further investigated the application of this vacant site approach to the design of aptamer sensors. To accomplish the goal, the 5′-end sequence of the adenosine aptamer55,78-80 was extended to form a loop. In doing so, a vacant site could also be formed in the DNA duplex between the adenosine aptamer (named AdAPva) and its partially complementary single strand DNA (named AdL2va) (Scheme 1c). Similar to what was observed in the DNAzyme sensors described above, the addition of 0.5 μM ATMND to a solution containing 1 μM AdAPva and 2 μM of AdL2va resulted in ∼80% quenching of the ATMND fluorescence (Figure 5a, Curves 1 and 2). Upon the addition of 200 μM adenosine to the above solution, the fluorescence was increased by 200% (Curve 3), suggesting that the structure-switching48,78,87 of the aptamer caused the release of both AdL2va and ATMND and recovered the fluorescence of ATMND. In contrast, cytidine could not recover any of the fluorescence (Curve 4), indicating the essential role of adenosine as a specific target to the aptamer AdAPva.
Figure 5.
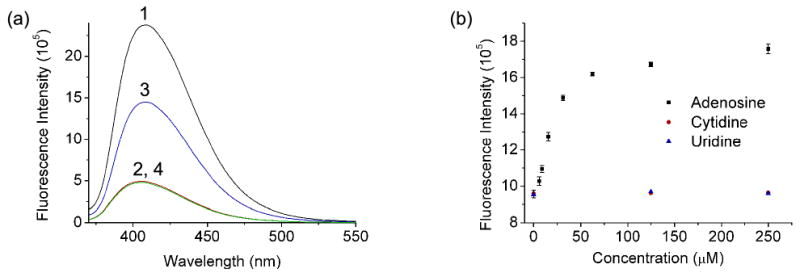
(a) Fluorescence spectra of (1) ATMND, (2) ATMND/AdAPva/AdL2va, (3) ATMND/AdAPva/AdL2va/adenosoine, (4) ATMND/AdAPva/AdL2va/cytidine. (b) Adenosine-dependent fluorescence enhancement of ATMND/AdAPva/AdL1va. Condition: 10 mM HEPES pH 7.0, 100 mM NaCl and 1 mM EDTA at 5 °C. λex/λem = 358/405 nm.
The fluorescence enhancement of ATMND was indeed dependent on the concentration of adenosine in the solution containing ATMND/AdAPva/AdL1va (Figure 5b). A 100% increase of fluorescence intensity was observed when the first 60 μM adenosine was added, while the fluorescence approached its maximum upon subsequent addition of adenosine up to 250 μM. A detection limit of 6 μM was obtained by the definition of 3σb/slope (σb, standard deviation of the blank samples) when using ATMND/AdAPva/AdL1va for adenosine quantification under the optimized condition. This detection limit is similar to that of the original labeled aptamer sensor,78 suggesting that the activity of the aptamer was not affected by the incorporation of a vacant site. The selectivity of the aptamer to adenosine over cytidine and uridine was also well preserved in this vacant-site-based sensor, because only adenosine could produce a fluorescence enhancement response among the three nucleotides in Figure 5b (guanosine was not tested here due to a solubility issue that hindered the preparation of a stock solution). More interestingly, by using ssDNAs (AdL1va∼AdL3va) that were complementary to AdAPva with different number of base pairs, the dynamic range of the sensor system could be tuned to 6∼60 μM, 12∼200 μM, or 32∼1000 μM, respectively (Figure 6).52,58,70,88 This tunable dynamic range makes it possible to apply this sensor system to samples containing different levels of analyte with a sensitive response.
Figure 6.
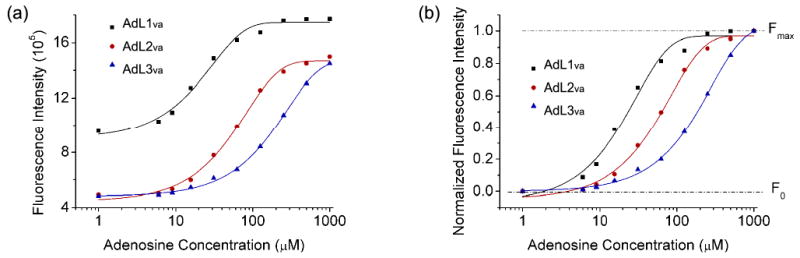
(a) Tuning the dynamic range of adenosine detection for ATMND/AdAPva/AdL1va∼AdL3va. (b) Figure with normalized fluorescence change. Condition: 10 mM HEPES pH 7.0, 100 mM NaCl and 1 mM EDTA at 5 °C. λex/λem = 358/405 nm.
(4) Hg2+ sensor based on T-T mismatch
Since the above sections show that the vacant site approach can be applied to both DNAzyme and aptamer sensor design, we wish to further expand the approach to Hg2+ sensing based on T-T mismatch. T-T mismatches have been reported to be a very efficient binding site for Hg2+ in aqueous solution.81-83,85 It has been the basis of several Hg2+ sensors.81-85 Here, a loop was linked to the 5′-end of the DNA containing T-T mismatches (named HgAPva) to build a vacant site with the single strand DNA (HgLva) that was partially complementary to HgAPva (Scheme 1d). Similar to the DNAzyme and aptamer systems described above, the fluorescence of 0.1 μM ATMND was quenched upon binding to 0.3 μM HgAPva and 0.35 μM HgLva. The subsequent addition of Hg2+ caused the fluorescence enhancement of ATMND, probably by releasing HgLva and ATMND from the DNA duplex via the Hg2+-induced structure-switching.48,78,84,87 The fluorescence intensity of the solution was found to be proportional to the concentration of Hg2+ (Figure 7a), with a detection limit of about 30 nM (defined as 3σb/slope, σb, standard deviation of the blank samples) and a linear range at least within 0∼1000 nM Hg2+. The moderate sensitivity of our sensor compared to labeled sensors84 was possibly due to the fact that the vacant site approach requires a larger amount of HgAPva loading (300 nM) to stabilize the vacant site and that each DNA strand might need several Hg2+ ions to induce structure-switching. Nevertheless, the selectivity of the sensor here toward Hg2+ over other divalent metal ions was as good as the reported Hg2+ sensors81,84 using the T-T mismatch design (Figure 7b).
Figure 7.
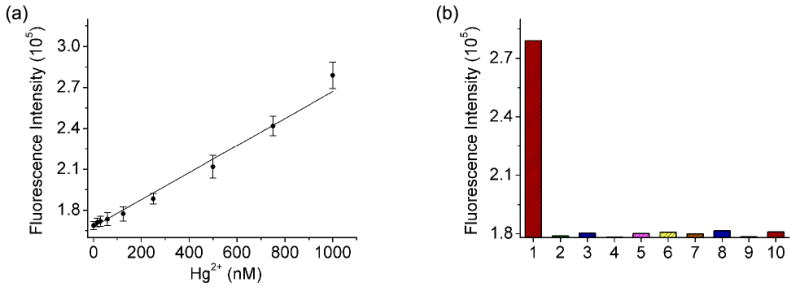
(a) Hg2+-dependent fluorescence enhancement of ATMND/HgAPva/HgLva. (b) Selectivity of ATMND/HgAPva/HgLva toward Hg2+ over other divalent metal ions. 1∼10: 1 μM Hg2+, 2 μM Mg2+, Ca2+, Cu2+, Zn2+, Mn2+, Co2+, Cd2+, Ni2+, Pb2+. Condition: 10 mM MOPS buffer pH 7.2 and 100 mM NaNO3 at 5 °C. λex/λem = 358/405 nm.
Effects of pH and salt concentration
The pH and salt concentration are important for the performance of the functional DNA sensors described in this work, because both the hybridization of DNA and the activity of the functional DNAs may be affected by the conditions. As shown in Figure S2 (Supporting Information), when the pH was maintained at 7.0, 5.5, 7.0 and 7.2 for Pb2+,21,75 UO22+,27,77 adenosine55,78 and Hg2+ sensors,84 respectively, as those conditions used in the published reports, 100 mM sodium salt was found to be the optimal salt concentration to yield the highest signal response. The performance of the UO22+ sensor is similar in the presence of either 100 or 300 mM NaCl. By fixing the salt concentration at 100 mM for the Pb2+, adenosine, and Hg2+sensors, and at 300 mM for the UO22+ sensor, the effect of pH was investigated (Figure S3, Supporting Information). For the Pb2+ and UO22+ sensors, the optimal pHs of 7.0 and 5.5, respectively, were found to be similar to the optimal pHs reported in the previous studies. 21,27,77 Even though the Pb2+ sensor exhibited a faster rate of fluorescence enhancement at pH 7.75 than at pH 7.0, a higher background fluorescence increase was also observed (Figure S2a). For the adenosine and Hg2+ sensors, a neutral pH was found to be optimal (Figure S3).
Effects of vacant site and DNA sequences
To further confirm that the observed fluorescence enhancement of ATMND was induced by the weakened binding of the vacant site to ATMND via the interaction between a functional DNA and its target, two control experiments were conducted using Pb2+-dependent DNAzyme and adenosine aptamer as models.
First, to confirm the role of the vacant site in the sensing, the vacant site in the loop of the 8-17 DNAzyme in Scheme 1a was “filled” by incorporating an additional G nucleotide to the 5′-end of substrate strand (named 17SvaG) to eliminate the binding site of fluorophore. In contrast to the vacant site-containing system's 200% increase of fluorescent signal in the presence of Pb2+ as shown in Scheme 1a (17Sva/17Eva), this vacant-site-free control showed little fluorescence change (Figure S4a, Supporting Information). Moreover, the background fluorescence in the absence of Pb2+ for the vacant-site-free design was much higher than that of the vacant site-containing design, probably because ATMND hardly bound to the DNA duplex without a vacant site (Figure S4a). Similar results were also observed when the vacant site in the loop of the adenosine aptamer (AdAPva) in Scheme 1c was filled by an incorporated G nucleotide (Figure S4b). These results suggested that the vacant site, which served as binding site of fluorophore, was essential for the performance of these label-free fluorescent functional DNA sensors composed of unmodified DNA.
Next, to confirm the role of the functional DNA sequences in the sensing, an inactive mutant of the Pb2+-dependent 8-17 DNAzyme20,21,75 (named 17EvaMut) was investigated under the same conditions as a control of its active analogue (17Eva) in Scheme 1a. As shown in Figure S5a (Supporting Information), the inactive DNAzyme could quench the fluorescence of ATMND as efficiently as active 17Eva in the presence of 17Sva because a vacant site was formed within the DNA duplex in both cases. However, the addition of Pb2+ could result in the recovery of ATMND's fluorescence only for 17Eva but not for 17EvaMut, indicating that the fluorescence enhancement was indeed the result of catalytic cleavage of substrate by DNAzyme. Similarly, two inactive adenosine aptamers55,89 (named AdAPvaM1 and AdAPvaM2) could partially quench the fluorescence of ATMND when they formed vacant sites with AdL1va (Figure S5b) as AdAPva. However, no fluorescence change could be observed even when a high concentration of adenosine was present. These results suggested that the activity of a DNAzyme or the binding of an aptamer -- and the subsequent perturbation of the vacant site and release of ATMND -- was the origin of fluorescence enhancement.
The performance of the sensors in a complex sample medium and the application of the sensors in real-world samples
After demonstrating the good sensitivity and selectivity of the sensors in simple buffered solutions, we further investigated the sensors' performance in a more complex sample matrix. As shown in Figure S6 (Supporting Information), both the Pb2+ and UO22+ sensors displayed a specific response to the corresponding metal ion at low nanomolar concentrations in samples containing 1 μM Cd2+, Fe2+, Ni2+, Co2+, Ca2+, Mg2+, and the responses were similar to those obtained in the absence of these competing metal ions. The adenosine sensor was also tested in the presence of an additional 0.5 mM cytidine and uridine, with results consistent with those obtained in the absence of these competing nucleotides (Figure S6).
To explore their potential applications in real sample analysis, the Pb2+ sensor and adenosine sensors designed here were used to quantify Pb2+ in drinking water and adenosine in 20% human serum, respectively. First, calibration curves were obtained by using drinking water and serum containing different amounts of analyte (Figure S7, Supporting Information). Then, blind tests were conducted using the calibration curves to evaluate the reliability of the method. As shown in Table 1, the results suggest that the sensors were successful in detecting their respective targets in the real-world samples.
Table 1.
Detection of Pb2+ and adenosine in drinking water and human serum.
| Added | Found | Recovery (%) | SD (%), n = 4 | |
|---|---|---|---|---|
| Pb2+ in drinking water | 60.0 nM | 57.8 nM | 96.3 | 6.9 |
| 250 nM | 262 nM | 104.8 | 4.3 | |
| Adenosine in 20% human serum | 32.0 μM | 30.6 | 95.6 | 4.8 |
| 125 | 121 | 96.8 | 5.5 |
Conclusions
In summary, we have demonstrated a general vacant site approach that makes it possible to use unmodified DNA to design label-free fluorescent sensors based on functional DNA molecules such as DNAzymes and aptamers. It was accomplished by extending these DNA molecules with a loop to form a vacant site in the DNA duplex for the controllable binding of a fluorophore, resulting in functional DNA sensors for Pb2+, UO22+, Hg2+, and adenosine. These sensors are highly selective to their targets over other similar metal ions or nucleotides, and are very sensitive, with detection limits of 3nM 8 nM, 30 nM, and 6 μM for UO22+, Pb2+, Hg2+, and adenosine, respectively. Control experiments with functional DNAs free of vacant site or of mutated inactive sequences reveal that the vacant site and the activity of the DNAzyme or aptamer are crucial for the sensors to exhibit a fluorescence enhancement response in the presence of their targets. This vacant site approach could further facilitate the design of other label-free fluorescent sensors for a wide range of analytes.
Supplementary Material
Acknowledgments
We wish to thank Hannah Ihms for proof-reading the manuscript, the US Department of Energy (DE-FG02-08ER64568), National Institute of Health (ES16865) and the National Science Foundation (CTS-0120978) for financial support.
References
- 1.Nolan EM, Lippard SJ. Chem Rev. 2008;108:3443–3480. doi: 10.1021/cr068000q. [DOI] [PubMed] [Google Scholar]
- 2.Que EL, Domaille DW, Chang CJ. Chem Rev. 2008;108:1517–1549. doi: 10.1021/cr078203u. [DOI] [PubMed] [Google Scholar]
- 3.Burdette SC, Walkup GK, Spingler B, Tsien RY, Lippard SJ. J Am Chem Soc. 2001;123:7831–7841. doi: 10.1021/ja010059l. [DOI] [PubMed] [Google Scholar]
- 4.Chang CJ, Jaworski J, Nolan EM, Sheng M, Lippard SJ. Proc Nat Acad Sci USA. 2004;101:1129–1134. doi: 10.1073/pnas.0308079100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen P, He C. J Am Chem Soc. 2004;126:728–729. doi: 10.1021/ja0383975. [DOI] [PubMed] [Google Scholar]
- 6.Yoon S, Albers AE, Wong AP, Chang CJ. J Am Chem Soc. 2005;127:16030–16031. doi: 10.1021/ja0557987. [DOI] [PubMed] [Google Scholar]
- 7.Wu ZS, Guo MM, Zhang SB, Chen CR, Jiang JH, Shen GL, Yu RQ. Anal Chem. 2007;79:2933–2939. doi: 10.1021/ac0622936. [DOI] [PubMed] [Google Scholar]
- 8.Zhang YL, Huang Y, Jiang JH, Shen GL, Yu RQ. J Am Chem Soc. 2007;129:15448–15449. doi: 10.1021/ja0773047. [DOI] [PubMed] [Google Scholar]
- 9.Chen PR, He C. Curr Opin Chem Biol. 2008;12:214–221. doi: 10.1016/j.cbpa.2007.12.010. [DOI] [PubMed] [Google Scholar]
- 10.Xue XJ, Wang F, Liu XG. J Am Chem Soc. 2008;130:3244–+. doi: 10.1021/ja076716c. [DOI] [PubMed] [Google Scholar]
- 11.Wong BA, Friedle S, Lippard SJ. J Am Chem Soc. 2009;131:7142–7152. doi: 10.1021/ja900980u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xue XJ, Xu W, Wang F, Liu XG. J Am Chem Soc. 2009;131:11668–+. doi: 10.1021/ja904728v. [DOI] [PubMed] [Google Scholar]
- 13.Xu W, Xue XJ, Li TH, Zeng HQ, Liu XG. Angew Chem, Int Ed. 2009;48:6849–6852. doi: 10.1002/anie.200901772. [DOI] [PubMed] [Google Scholar]
- 14.Wu Z, Zhen Z, Jiang JH, Shen GL, Yu RQ. J Am Chem Soc. 2009;131:12325–12332. doi: 10.1021/ja9038054. [DOI] [PubMed] [Google Scholar]
- 15.Breaker RR, Joyce GF. Chem Biol. 1994;1:223–229. doi: 10.1016/1074-5521(94)90014-0. [DOI] [PubMed] [Google Scholar]
- 16.Ellington AD, Szostak JW. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 17.Cuenoud B, Szostak JW. Nature. 1995;375:611–614. doi: 10.1038/375611a0. [DOI] [PubMed] [Google Scholar]
- 18.Carmi N, Shultz LA, Breaker RR. Chem Biol. 1996;3:1039–1046. doi: 10.1016/s1074-5521(96)90170-2. [DOI] [PubMed] [Google Scholar]
- 19.Santoro SW, Joyce GF, Sakthivel K, Gramatikova S, Barbas CF., III J Am Chem Soc. 2000;122:2433–2439. doi: 10.1021/ja993688s. [DOI] [PubMed] [Google Scholar]
- 20.Li J, Zheng WC, Kwon AH, Lu Y. Nucleic Acids Res. 2000;28:481–488. doi: 10.1093/nar/28.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li J, Lu Y. J Am Chem Soc. 2000;122:10466–10467. [Google Scholar]
- 22.Bruesehoff PJ, Li J, Augustine AJ, Lu Y. Comb Chem High Throughput Screening. 2002;5:327–335. doi: 10.2174/1386207023330264. [DOI] [PubMed] [Google Scholar]
- 23.Mei SHJ, Liu Z, Brennan JD, Li Y. J Am Chem Soc. 2003;125:412–420. doi: 10.1021/ja0281232. [DOI] [PubMed] [Google Scholar]
- 24.Wang Y, Silverman SK. J Am Chem Soc. 2003;125:6880–6881. doi: 10.1021/ja035150z. [DOI] [PubMed] [Google Scholar]
- 25.Lee JF, Hesselberth JR, Meyers LA, Ellington AD. Nucleic Acids Res. 2004;32:D95–D100. doi: 10.1093/nar/gkh094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Navani NK, Li Y. Curr Opin Chem Biol. 2006;10:272–281. doi: 10.1016/j.cbpa.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 27.Liu J, Brown AK, Meng X, Cropek DM, Istok JD, Watson DB, Lu Y. Proc Nat Acad Sci USA. 2007;104:2056–2061. doi: 10.1073/pnas.0607875104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Song S, Wang L, Li J, Fan C, Zhao J. TrAC, Trends Anal Chem. 2008;27:108–117. [Google Scholar]
- 29.Liu J, Cao Z, Lu Y. Chem Rev. 2009;109:1948–1998. doi: 10.1021/cr030183i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rajendran M, Ellington AD. Comb Chem High Throughput Screening. 2002;5:263–270. doi: 10.2174/1386207023330246. [DOI] [PubMed] [Google Scholar]
- 31.Lu Y. Chem Eur J. 2002;8:4588–4596. doi: 10.1002/1521-3765(20021018)8:20<4588::AID-CHEM4588>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 32.Willner I, Zayats M. Angew Chem, Int Ed. 2007;46:6408–6418. doi: 10.1002/anie.200604524. [DOI] [PubMed] [Google Scholar]
- 33.Willner I, Shlyahovsky B, Zayats M, Willner B. Chem Soc Rev. 2008;37:1153–1165. doi: 10.1039/b718428j. [DOI] [PubMed] [Google Scholar]
- 34.Li Y, Lu Y. Functional Nucleic Acids for Sensing and Other Analytical Applications. Springer; New York: 2009. [Google Scholar]
- 35.Xiao Y, Lubin AA, Heeger AJ, Plaxco KW. Angew Chem, Int Ed. 2005;44:5456–5459. doi: 10.1002/anie.200500989. [DOI] [PubMed] [Google Scholar]
- 36.Xiao Y, Piorek BD, Plaxco KW, Heeger AJ. J Am Chem Soc. 2005;127:17990–17991. doi: 10.1021/ja056555h. [DOI] [PubMed] [Google Scholar]
- 37.Baker BR, Lai RY, Wood MS, Doctor EH, Heeger AJ, Plaxco KW. J Am Chem Soc. 2006;128:3138–3139. doi: 10.1021/ja056957p. [DOI] [PubMed] [Google Scholar]
- 38.Zayats M, Huang Y, Gill R, Ma CA, Willner I. J Am Chem Soc. 2006;128:13666–13667. doi: 10.1021/ja0651456. [DOI] [PubMed] [Google Scholar]
- 39.Shlyahovsky B, Li D, Weizmann Y, Nowarski R, Kotler M, Willner I. J Am Chem Soc. 2007;129:3814–+. doi: 10.1021/ja069291n. [DOI] [PubMed] [Google Scholar]
- 40.Zuo XL, Song SP, Zhang J, Pan D, Wang LH, Fan CH. J Am Chem Soc. 2007;129:1042–1043. doi: 10.1021/ja067024b. [DOI] [PubMed] [Google Scholar]
- 41.He SJ, Li D, Zhu CF, Song SP, Wang LH, Long YT, Fan CH. Chem Commun. 2008:4885–4887. doi: 10.1039/b811528a. [DOI] [PubMed] [Google Scholar]
- 42.Zhang J, Wang LH, Pan D, Song SP, Boey FYC, Zhang H, Fan CH. Small. 2008;4:1196–1200. doi: 10.1002/smll.200800057. [DOI] [PubMed] [Google Scholar]
- 43.Schlosser K, Li YF. Chem Biol. 2009;16:311–322. doi: 10.1016/j.chembiol.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 44.Swensen JS, Xiao Y, Ferguson BS, Lubin AA, Lai RY, Heeger AJ, Plaxco KW, Soh HT. J Am Chem Soc. 2009;131:4262–4266. doi: 10.1021/ja806531z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zuo XL, Xiao Y, Plaxco KW. J Am Chem Soc. 2009;131:6944–+. doi: 10.1021/ja901315w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Freeman R, Finder T, Willner I. Angew Chem, Int Ed. 2009;48:7818–7821. doi: 10.1002/anie.200902395. [DOI] [PubMed] [Google Scholar]
- 47.Freeman R, Sharon E, Tel-Vered R, Willner I. J Am Chem Soc. 2009;131:5028–+. doi: 10.1021/ja809496n. [DOI] [PubMed] [Google Scholar]
- 48.Nutiu R, Li Y. Chem Eur J. 2004;10:1868–1876. doi: 10.1002/chem.200305470. [DOI] [PubMed] [Google Scholar]
- 49.Cho EJ, Rajendran M, Ellington AD. Top Fluoresc Spectrosc. 2005;10:127–155. [Google Scholar]
- 50.Cao Z, Suljak SW, Tan W. Curr Proteomics. 2005;2:31–40. [Google Scholar]
- 51.Liu J, Lu Y. Methods Mol Biol. 2006;335:275–288. doi: 10.1385/1-59745-069-3:275. [DOI] [PubMed] [Google Scholar]
- 52.Liu J, Lu Y. J Am Chem Soc. 2003;125:6642–6643. doi: 10.1021/ja034775u. [DOI] [PubMed] [Google Scholar]
- 53.Pavlov V, Xiao Y, Shlyahovsky B, Willner I. J Am Chem Soc. 2004;126:11768–11769. doi: 10.1021/ja046970u. [DOI] [PubMed] [Google Scholar]
- 54.Huang CC, Huang YF, Cao Z, Tan W, Chang HT. Anal Chem. 2005;77:5735–5741. doi: 10.1021/ac050957q. [DOI] [PubMed] [Google Scholar]
- 55.Liu J, Lu Y. Angew Chem, Int Ed. 2006;45:90–94. [Google Scholar]
- 56.Lee JS, Han MS, Mirkin CA. Angew Chem, Int Ed. 2007;46:4093–4096. doi: 10.1002/anie.200700269. [DOI] [PubMed] [Google Scholar]
- 57.Lee JH, Wang Z, Liu J, Lu Y. J Am Chem Soc. 2008;130:14217–14226. doi: 10.1021/ja803607z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang Z, Lee JH, Lu Y. Adv Mater. 2008;20:3263–3267. [Google Scholar]
- 59.Zhao W, Lam JC, Chiuman W, Brook MA, Li Y. Small. 2008;4:810–816. doi: 10.1002/smll.200700757. [DOI] [PubMed] [Google Scholar]
- 60.Xiao Y, Rowe AA, Plaxco KW. J Am Chem Soc. 2007;129:262–263. doi: 10.1021/ja067278x. [DOI] [PubMed] [Google Scholar]
- 61.Jiang Y, Fang X, Bai C. Anal Chem. 2004;76:5230–5235. doi: 10.1021/ac049565u. [DOI] [PubMed] [Google Scholar]
- 62.Wang J, Jiang Y, Zhou C, Fang X. Anal Chem. 2005;77:3542–3546. doi: 10.1021/ac050165w. [DOI] [PubMed] [Google Scholar]
- 63.Joseph MJ, Taylor JC, McGown LB, Pitner B, Linn CP. Biospectroscopy. 1996;2:173–183. [Google Scholar]
- 64.Li B, Wei H, Dong S. Chem Commun. 2007:73–75. doi: 10.1039/b612080f. [DOI] [PubMed] [Google Scholar]
- 65.Wang Y, Liu B. Analyst. 2008;133:1593–1598. doi: 10.1039/b806908e. [DOI] [PubMed] [Google Scholar]
- 66.Babendure JR, Adams SR, Tsien RY. J Am Chem Soc. 2003;125:14716–14717. doi: 10.1021/ja037994o. [DOI] [PubMed] [Google Scholar]
- 67.Stojanovic MN, Kolpashchikov DM. J Am Chem Soc. 2004;126:9266–9270. doi: 10.1021/ja032013t. [DOI] [PubMed] [Google Scholar]
- 68.Xu Z, Morita K, Sato Y, Dai Q, Nishizawa S, Teramae N. Chem Commun. 2009:6445–6447. doi: 10.1039/b908345f. [DOI] [PubMed] [Google Scholar]
- 69.Xu Z, Sato Y, Nishizawa S, Teramae N. Chem Eur J. 2009;15:10375–10378. doi: 10.1002/chem.200901226. [DOI] [PubMed] [Google Scholar]
- 70.Xiang Y, Tong A, Lu Y. J Am Chem Soc. 2009;131:15352–15357. doi: 10.1021/ja905854a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yoshimoto K, Nishizawa S, Minagawa M, Teramae N. J Am Chem Soc. 2003;125:8982–8983. doi: 10.1021/ja029786m. [DOI] [PubMed] [Google Scholar]
- 72.Sankaran NB, Nishizawa S, Seino T, Yoshimoto K, Teramae N. Angew Chem, Int Ed. 2006;45:1563–1568. doi: 10.1002/anie.200502979. [DOI] [PubMed] [Google Scholar]
- 73.Ihara T, Uemura A, Futamura A, Shimizu M, Baba N, Nishizawa S, Teramae N, Jyo A. J Am Chem Soc. 2009;131:1386–1387. doi: 10.1021/ja809023n. [DOI] [PubMed] [Google Scholar]
- 74.Liu JW, Lu Y. J Am Chem Soc. 2002;124:15208–15216. doi: 10.1021/ja027647z. [DOI] [PubMed] [Google Scholar]
- 75.Brown AK, Li J, Pavot CMB, Lu Y. Biochemistry. 2003;42:7152–7161. doi: 10.1021/bi027332w. [DOI] [PubMed] [Google Scholar]
- 76.Kim HK, Rasnik I, Liu JW, Ha TJ, Lu Y. Nat Chem Biol. 2007;3:763–768. doi: 10.1038/nchembio.2007.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Brown AK, Liu JW, He Y, Lu Y. ChemBioChem. 2009;10:486–492. doi: 10.1002/cbic.200800632. [DOI] [PubMed] [Google Scholar]
- 78.Nutiu R, Li Y. J Am Chem Soc. 2003;125:4771–4778. doi: 10.1021/ja028962o. [DOI] [PubMed] [Google Scholar]
- 79.Liu J, Lu Y. Advanced Materials. 2006;18:1667–1671. [Google Scholar]
- 80.Liu J, Lee JH, Lu Y. Anal Chem. 2007;79:4120–4125. doi: 10.1021/ac070055k. [DOI] [PubMed] [Google Scholar]
- 81.Ono A, Togashi H. Angew Chem, Int Ed. 2004;43:4300–4302. doi: 10.1002/anie.200454172. [DOI] [PubMed] [Google Scholar]
- 82.Miyake Y, Togashi H, Tashiro M, Yamaguchi H, Oda S, Kudo M, Tanaka Y, Kondo Y, Sawa R, Fujimoto T, Machinami T, Ono A. J Am Chem Soc. 2006;128:2172–2173. doi: 10.1021/ja056354d. [DOI] [PubMed] [Google Scholar]
- 83.Tanaka Y, Oda S, Yamaguchi H, Kondo Y, Kojima C, Ono A. J Am Chem Soc. 2007;129:244–245. doi: 10.1021/ja065552h. [DOI] [PubMed] [Google Scholar]
- 84.Wang ZD, Lee JH, Lu Y. Chem Commun. 2008:6005–6007. doi: 10.1039/b812755g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Okamoto I, Iwamoto K, Watanabe Y, Miyake Y, Ono A. Angew Chem, Int Ed. 2009;48:1648–1651. doi: 10.1002/anie.200804952. [DOI] [PubMed] [Google Scholar]
- 86.Li N, Mei L, Xiang Y, Tong A, Nishizawa S, Teramae N. Anal Chim Acta. 2007;597:97–102. doi: 10.1016/j.aca.2007.06.031. [DOI] [PubMed] [Google Scholar]
- 87.Nutiu R, Li Y. Angew Chem, Int Ed. 2005;44:1061–1065. doi: 10.1002/anie.200461848. [DOI] [PubMed] [Google Scholar]
- 88.Vallee-Belisle A, Ricci F, Plaxco KW. Proc Nat Acad Sci USA. 2009;106:13802–13807. doi: 10.1073/pnas.0904005106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Huizenga DE, Szostak JW. Biochemistry. 1995;34:656–665. doi: 10.1021/bi00002a033. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.