

Abstract
Epithelial-to-mesenchymal transition (EMT) is an important developmental process, participates in tissue repair and occurs during pathological processes of tumor invasiveness, metastasis and tissue fibrosis. The molecular mechanisms leading to EMT are poorly understood. While it is well documented that transforming growth factor (TGF)-β plays a central role in the induction of EMT, the targets of TGF-β signaling are poorly defined. We have shown earlier that Na,K-ATPase β1-subunit levels are highly reduced in poorly differentiated kidney carcinoma cells in culture and in patients’ tumor samples. In this study, we provide evidence that Na,K-ATPase is a new target of TGF-β1-mediated EMT in renal epithelial cells, a model system used in studies of both cancer progression and fibrosis. We show that following treatment with TGF-β1 the surface expression of the β1-subunit of Na,K-ATPase is reduced, prior to well-characterized EMT markers and is associated with the acquisition of a mesenchymal phenotype. RNAi mediated knockdown confirmed the specific involvement of the Na,K-ATPase β1-subunit in the loss of the epithelial phenotype and exogenous over-expression of the Na,K-ATPase β1-subunit attenuated TGF-β1-mediated EMT. We further show that both Na,K-ATPase α- and β-subunit levels are highly reduced in renal fibrotic tissues. These findings for the first time reveal that Na,K-ATPase is a target of TGF-β1-mediated EMT and is associated with the progression of EMT in both cancer and fibrosis.
Keywords: Transforming growth factor (TGF)-β1; Na,K-ATPase; Epithelial-to-mesenchymal transition (EMT); cancer; fibrosis
Introduction
Epithelial-to-mesenchymal transition (EMT) is a process in which polarized epithelial cells undergo multiple biochemical changes to assume a mesenchymal phenotype (1). In general, epithelia form an effective barrier between underlying tissue and external media. In order to perform this function epithelial cells possess extensive junctional networks that physically separate the plasma membrane into apical and basolateral domains. The junctional network also promotes adhesion, facilitates intercellular communication, restricts motility, and thus preserves tissue integrity and permits epithelial cells to function as a cohesive unit (2). The phenotype of epithelial cells with apical-basal polarity and junctional complexes is referred to as well-differentiated phenotype. Carcinoma arising from epithelial tissue is by far the most prevalent form of cancer accounting for about 90% of human malignancies. During carcinoma progression to advanced disease, epithelial cells often lose their well-differentiated phenotype and adopt a fibroblast-like, mesenchymal phenotype in a sequence of events often referred to as EMT. This process involves loss of cell-cell adhesion and intercellular junctions, destruction of the basement membrane, reorganization of the actin cytoskeleton, enhanced migratory capacity and invasiveness, activation of transcription factors, and down-regulation of epithelial markers and expression of mesenchymal genes (3). Molecular and morphologic features of EMT often correlate with poor histologic differentiation, destruction of tissue integrity and in general is associated with advancement in disease and deterioration in tissue function (4). Although, it is recognized that EMT is an important event during cancer progression as well as in fibrotic disease, molecular mechanisms leading to EMT and target molecules affected during this process are poorly understood.
A key molecule implicated in EMT is transforming growth factor-β1 (TGF-β1) (5, 6). The TGF-β growth factor superfamily comprises TGF-βs, bone morphogenetic proteins (BMPs), activins and other related proteins. The functional complex of TGF-β family receptors at the cell surface consists of two type II and two type I transmembrane serine/threonine receptors (5, 6), which exist as homodimers in the absence of ligand. Ligand binding to the type II receptor recruits the type I receptor into a heteromeric complex resulting in the transphosphorylation of type I receptor by type II receptor. Following phosphorylation of Smad2 or Smad3 by the activated type I receptor, a heteromeric complex is formed with Smad4, resulting in the translocation of the complex to the nucleus to directly or indirectly regulate gene transcription. In addition to Smad-dependent pathways, TGF-β also activates Smad-independent pathways such as the Erk, JNK and p38 MAPK signaling pathways (6, 7). The mechanism by which TGF-β1 mediated signaling in epithelial cells triggers EMT and its consequences remain to be clarified.
Na,K-ATPase is an abundantly expressed protein in epithelial cells and plays a crucial role in kidney function. Localized to the basolateral plasma membrane, the oligomeric Na, K-ATPase catalyzes an ATP-dependent transport of three Na+ out and two K+ into the cell per pump cycle to maintain Na+ and K+ gradients across the plasma membrane. This Na+ and K+ homeostasis is necessary to regulate the functions of the various ion and solute transporters in epithelial cells. Na,K-ATPase is composed of two essential polypeptide subunits, the α-subunit (~112 kDa) (8) and the β-subunit (~55 kDa) (9) and an optional regulatory γ-subunit with tissue-specific expression (~7 kDa) (10). Of the four α- and three β-subunit isoforms known, α1 (NaK-α1) and β1 (NaK-β1) are predominantly expressed in kidney (11).
We have shown that in addition to its epithelial transport function, Na,K-ATPase plays a fundamental role in the formation and maintenance of a well-differentiated, polarized epithelial phenotype in mammalian cells (12–16). Inhibition of Na,K-ATPase activity prevents the polarization of epithelial cells and disrupts tight junction structure and function in polarized epithelial monolayers (13, 15). Recently, we and others have shown that the Na,K-ATPase β1-subunit (NaK-β1) functions as a cell-cell adhesion molecule (16–19), is localized to the apical junctional complex in polarized epithelial cells (13) and may also function as a tumor suppressor (20). Na,K-ATPase also regulates tight junction function and formation during mouse preimplantation and blastocyst development (21, 22). In Drosophila, homologues of both NaK-α1 and NaK-β1 are localized to septate junctions (which are functionally similar to tight junctions in epithelial cells) and are involved in regulating their permeability (23, 24). Subsequent studies demonstrated that the Drosophila Na,K-ATPase has an ion-pump independent role in junction formation (25) and that Na,K-ATPase belongs to a new group of polarity proteins (26). In Zebrafish, Na,K-ATPase is essential for the epithelial integrity during heart morphogenesis (27) and lumen formation in the gut (28). Together, these studies indicate that Na,K-ATPase has a conserved role in the regulation of the well-differentiated phenotype of epithelial cells (29).
In this study, using LLC-PK1 cells, a well-differentiated kidney epithelial cell line that undergoes TGF-β mediated EMT (30), we show that Na,K-ATPase is a target molecule of the TGF-β1 signaling pathways. We provide evidence that reduced Na,K-ATPase expression and function are accompanied by increased intracellular sodium levels and activation of MAPK signaling and might be one of the events associated with the loss of the polarized epithelial phenotype during TGF-β1 induced EMT in kidney epithelial cells.
Materials and Methods
Cell culture, siRNA and transfections
LLC-PK1 cells (ATCC, Rockville, MD) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum, 2 mM L-glutamine, 25 U/ml penicillin, and 25 μg/ml streptomycin. TGF-β1 (R&D, Minneapolis, MN) was added to subconfluent cultures at 4 ng/ml in serum-free DMEM for the time indicated after serum starvation for 4–6 hours. Control cells were serum-starved in parallel.
SiRNA against NaK-β1 (5′-AAAAGUGAUGCUGCUCACCAUCA-3′) and a scrambled control oligoribonucleotide (5′-AACUGCCGUACUAAGUUAAGACA-3′) were from Dharmacon (Lafayette, CO). The uniqueness of the sequences was confirmed using the GenBank™/EBI database. Cells were transfected with 150 nM NaK-β1 or control oligonucleotide using Cytofectene (BioRad, Hercules, CA) and harvested 72 h post-transfection. For stable knockdown of NaK-β1 in Caki-1 cells the NaK-β1 siRNA sequence was cloned into pSilencer 5.1 (Ambion, Austin, TX). Caki-1 cells (ATCC) were transfected with the cDNA using the calcium phosphate method and stable clones of NaK-β1knockdown Caki-1 cells were selected with 10 μg/ml puromycin.
Flag-tagged Smad7 cDNA was kindly provided by Dr. H.L. Moses (Vanderbilt University, Nashville, TN). Transfections were performed with FuGene6 reagent (Roche, Indianapolis, IN).
Antibodies
Antibodies against ZO-1 and occludin were obtained from Invitrogen Corporation (Carlsbad, CA), E-cadherin from BD Biosciences (San Jose, CA), Flag-epitope (DYKDDDK), p44/42-MAPK (Erk1/2) and phospho-p44/42-MAPK (Erk1/2) from Cell Signaling (Danvers, MA), TGF-β RII from Abcam Inc. (Cambridge, MA), and fibronectin, β-actin, and smooth muscle actin (SMA) from Sigma Chemical Co. (St. Louis, MO). Antibodies against NaK-α1 (M7-PB-E9) and NaK-β1 (M17-P5-F11) were kindly provided by Dr. William Ball Jr. (University of Cincinnati, Cincinnati, OH). Horseradish peroxidase (HRP)-conjugated secondary antibodies were from Cell Signaling, FITC- or CY3 conjugated secondary antibodies from Jackson ImmunoResearch Laboratories (West Grove, PA).
Immunoblotting
Cell lysates for NaK-α1, NaK-β1, fibronectin, SMA, and E-cadherin were prepared as described earlier (13, 16) in a buffer containing 95 mM NaCl, 25 mM Tris, pH 7.4, 0.5 mM EDTA, 2% SDS and protease inhibitors (1 mM phenylmethylsulfonyl fluoride, 5 μg/ml each of antipain, leupeptin, and pepstatin) or in a buffer of 20 mM Tris-HCl, pH 7.4, 100 mM NaCl, 1% Triton X–100, 1 mM EDTA, 1 mM EGTA, 1 mM sodium glycerolphosphate, 1 mM sodium orthovanadate, and protease inhibitors (phospho-Erk1/2, Erk1/2). Equal amounts of protein were separated by SDS-PAGE and immunoblotted as described (13, 16).
Immunofluorescence
Immunofluorescence was done as described previously (15, 16, 31). Epifluorescence pictures were captured with a SPOT CCD camera and SPOT imaging software, version 4.0.4 (Diagnostic Instruments Inc., MI) attached to an Olympus AX70 microscope.
TGF-β responsive promoter assay
LLC-PK1 cells transiently transfected with SBE-luc (Smad4 Binding Element luciferase) alone, Smad7-Flag alone or SBE-luc and Smad7-FLAG were treated with TGF-β1 48 hours after transfection. Luciferase activity was measured after 24 hours of TGF-β1 treatment using the Dual-Luciferase Reporter Assay System (Promega) as described (32). The firefly luciferase activity was normalized to the activity of the internal control (Renilla luciferase, pRL vector) that was co-transfected along with the reporter vector.
Real-time quantitative PCR
Primer sets used were: NaK-β1 forward, 5′-TTACCCTTACTACGGCAAGCTCCT-3 ′, reverse, 5′-TTCAGTGTCCATGGTGAGGTTGGT-3′; Snail forward, 5′-TCACCGGCTCCTTCGTCCTTC-3′; reverse, 5′-TCCTTGTTGCAGTATTTGCAGTT-3′; GAPDH forward, 5′-GTGAAGGTCGGAGTCAACGG-3′; reverse, 5′-TGATGACAAGCTTCCCGTTCTC-3′. Reactions were run in triplicate on an iCycler iQ with iQ SYBR Green supermix detection (both Bio-Rad). Variation in cDNA loading was normalized to GAPDH simultaneously amplified in each well.
Rubidium transport assay and atomic emission spectroscopy
Na, K-ATPase activity was determined as the ouabain-sensitive 86Rb+ transport as described (16). The samples were normalized to the protein content.
Intracellular ion concentrations were measured with an inductively coupled plasma atomic emission spectrometer (Vista Axial 730; Varian, Walnut Creek, CA) as described (16). The concentrations for Na+ (588.995 nm), K+ (766.941 nm), and Mg2+ (285.213 nm) were determined and Na+ and K+ concentrations were normalized to the total Mg2+ content (internalcontrol).
Cell volume measurements
The procedure described by Kletzien et al (33) was modified for measuring the cell volume of polarized epithelia. Cells grown on 25 mm Millipore Isopore 0.8 micron pore membranes coated with bovine tendon collagen previously cross linked in ammonia vapor were rinsed and incubated in glucose-free MOPS–buffered saline, pH 7.35, 290 mOsm, containing 0.1 mM 14C-3-O-methyl-D-glucose (Dupont, NEN) at an activity of 0.1 μCi/ml. After 50 min of incubation filters were rinsed in ice-cold saline containing 0.1 mM phloretin and lysed with 0.4 M perchloric acid. After centrifugation, supernatant samples were taken for liquid scintillation counting and precipitates were analyzed for DNA by the diphenylamine spectrophotometric procedure. Using steady state conditions as above allows for conversion of μmoles of 3-O-methyl-D-glucose/μg DNA to ml of intracellular water/μg DNA.
Immunohistochemistry
Following Institutional Review Board approval, sections from biopsies were obtained from normal kidney and from three different diabetic patients diagnosed with mild, moderate or severe diabetic nephropathy. NaK-α1 and NaK-β1 were detected with mouse monoclonal antibodies M7-PB-E9 and M17-P5-F11, respectively, that have been used for immunohistochemical staining of human kidney previously (11). Standard VECTASTAIN ABC immunohistochemical staining was employed (Vector Laboratories, Burlingame, CA) and diaminobenzidine (DAB) was used as the chromogen. The sections were counterstained with Gill’s Hematoxylin plus bluing reagent.
Normal sections were obtained from kidneys of non-induced control rats and diabetic kidney sections from streptozotocin (STZ)-induced rats (12 weeks). NaK-α1 and NaK-β1 were detected with NaK-α1 (M7-PB-E9) antibody and a polyclonal antibody against NaK-β1 (generous gift of Dr. William Ball, University of Cincinnati) that both recognize rat Na,K-ATPase subunits. The sections were counterstained with Gill’s Hematoxylin. For all stainings complementary slides processed in the same manner minus primary antibody served as negative controls.
Statistics
Results are expressed as means ± SE. The statistical significance of differences was calculated using Student’s test.
Results
Na,K-ATPase is a target of TGF-β1 signaling in proximal tubule kidney epithelial cells
LLC-PK1 cells, a porcine proximal tubule-like kidney cell line, are well-established as a cell culture model of TGF-β1 induced EMT (30). Consistent with previous studies, treatment of LLC-PK1 cells with TGF-β1 induced a fibroblastic phenotype (Fig. 1A) with expression of fibronectin (FN) and α-smooth muscle actin (SMA), and reduced levels of the cell adhesion molecule E-cadherin (Fig. 1B). In addition, TGF-β1-induced EMT was accompanied by drastically reduced NaK-β1 (48 h: 48 ± 15%; 96 h: 27 ± 10% of control) and NaK-α1 (48 h: 81 ± 12%; 96 h: 32 ± 11% of control) protein levels.
Figure 1.

LLC-PK1 cells undergo TGF-β1-mediated EMT. A, Phase contrast microscopy of control (0h) and TGF-β1 treated (48, 96h) cells. B, Representative immunoblots for EMT markers fibronectin (FN) and α-smooth muscle actin (SMA), E-cadherin (E-cad), Na,K-ATPase α-subunit (NaK-α1) and β-subunit (NaK-β1). Actin immunoblot confirms equal loading. C, Immunostaining for E-cadherin, NaK-β1 and NaK-α1. NaK-β1 redistributed from the plasma membrane in control cells to intracellular vesicles in TGF-β1 treated cells as early as 18 hours after TGF-β1 treatment.
As expected, immunofluorescence analysis showed NaK-β1 and NaK-α1 predominantly localized to sites of cell-cell contact at the plasma membrane in control cells. In contrast, these subunits were localized to distinct intracellular vesicles surrounding the nucleus in TGF-β1-treated cells (Fig. 1C). More importantly, the redistribution of both Na,K-ATPase subunits from the plasma membrane to intracellular vesicles was observed early on, after 18 hours of TGF-β1 treatment, suggesting that altered Na,K-ATPase localization is an early event in TGF-β1 mediated induction of EMT. Consistent with the intracellular localization of the Na,K-ATPase subunits, the regulation of the NaK-β1 protein level seemed to occur at the post-translational level since quantitative real-time PCR did not show significant differences in NaK-β1 mRNA levels between control and TGF-β1 treated cells (Supplementary Fig. 1A). We have previously shown that the transcription factor Snail represses NaK-β1 in cancer cells (34). We did not find any significant differences in Snail mRNA levels in control and TGF-β1 treated cells (Supplementary Fig. 1B) further supporting that reduced NaK-β1 levels in TGF-β1 treated cells are due to post-translational regulation.
To test whether the relocation of NaK-β1 from the plasma membrane is a Smad-dependent or independent process we transiently expressed Smad7 in LLC-PK1 cells. Smad7 inhibits TGF-β dependent signaling by preventing Smad 4 to enter the nucleus following TGF-β1 treatment and has been reported to inhibit Smad-dependent TGF-β1 signaling in other kidney epithelial cells (35, 36). Similarly, TGF-β1 responsive promoter activity as assessed by SMAD binding element activity was inhibited in LLC-PK1 cells transiently expressing Smad7 (Fig. 2A). Immunofluoresence of Smad7-expressing LLC-PK1 cells treated with TGF-β1 revealed NaK-β1 still localized to the plasma membrane (Fig. 2B, arrow), whereas untransfected cells showed the previously observed vesicular staining pattern confirming that NaK-β1 expression and distribution are targets of Smad-dependent TGF-β1 signaling.
Figure 2.

Smad-dependence and effects of altered Na,K-ATPase in TGF-β1-treated cells. A, TGF-β responsive SBE-Luc reporter activity. LLC-PK1 cells transiently transfected with SBE-luc alone (control, TGF-β1) or SBE luc and Smad7-FLAG (Smad 7, Smad7 + TGF-β1) and treated with TGF-β1 for 24 hours (TGF-β1, Smad7 + TGF-β1) or vehicle (Control, Smad7). The ratio of the Firefly luciferase activity over the Renilla luciferase is expressed as relative activity compared to control cells. Data are means ± SD from three independent determinations. B, Transiently expressed SMAD7 in TGF-β1-treated cells (72 hrs) was detected with anti-Flag antibody. Note the intense staining of NaK-β1 on the plasma membrane in Smad7 expressing cells (arrow). Representative image from two independent transfections is shown. C, Na,K-ATPase activity in TGF-β1-treated cells. Ouabain-sensitive 86Rb+ uptake in control (0h) and TGF-β1-treated cells (48, 96 hours) was determined as described in Materials and Methods. D, TGF-β1 treatment results in increased intracellular sodium. [Na+]i in control and TGF-β1-treated cells was determined by atomic emission spectrometry. Data are means ± SEM from four (C) or two (D) independent determinations done in triplicates.
TGF-β1 induced EMT is accompanied by increased intracellular sodium
Earlier, we observed that both Na,K-ATPase subunit levels (16) and enzyme activity (13–15) are essential for a polarized epithelial phenotype. Although we did not find a significant difference in Na,K-ATPase activity between control and TGF-β1 treated cells in ouabain-sensitive 86Rb+ uptake experiments (Fig. 2C) or in total cellular 86Rb+ uptake (data not shown), atomic emission spectrometry studies revealed increased intracellular sodium concentrations ([Na+]i) in TGF-β1 treated cells with a 1.85 ± 0.05 and 2.59 ± 0.03-fold increase in [Na+]i after 48 and 96 hours of TGF-β1 treatment, respectively (Fig. 2D). Intracellular potassium levels were not altered (data not shown). Increased [Na+]i seems to be a common feature of cells undergoing EMT. We found that poorly differentiated bladder, colon, and pancreatic cancer cells that express low levels of NaK-β1 (34) have higher [Na+]i levels when compared to well-differentiated cells of similar origin (Supplementary Fig. 2).
Consistent with the increase in [Na+]i, the water content of TGF-β1-treated cells was 1.36-fold higher (P<0.001) compared to control cells. While these data seem contradictory at first, it is well known that high [Na+]i will drive the Na,K-ATPase activity, resulting in a higher turnover rate per molecule which is similar to our earlier findings in MSV-transformed MDCK cells. MSV-MDCK cells that express low NaK-α1 and NaK-β1 levels did not display major differences in 86Rb+ uptake although [Na+]i was 2.8-fold higher as compared to MDCK cells (16). Together, these data show that TGF-β1 mediated EMT in LLC-PK1 cells is associated with increased intracellular sodium levels and accompanied change in cell volume.
NaK-β1 expression is essential to maintain a well-differentiated phenotype
Since we observed reduced NaK-β1 expression during the loss of the epithelial phenotype in TGF-β1 treated cells, we tested whether specific knockdown of this protein alters the phenotype of LLC-PK1 cells. Indeed, reducing NaK-β1 protein levels by 63 ± 8% in LLC-PK1 cells using an RNAi oligomer approach (Fig. 3A) resulted in the loss of the epithelial phenotype. Although E-cadherin levels remained unaffected (Fig. 3A), NaK-β1 siRNA cells displayed a fibroblastic morphology, while the parental cell line and scrambled oligomer-transfected control cells showed the cobblestone like morphology typical of well-differentiated epithelial cells (Fig. 3B). Since NaK-β1 is essential for the transport and stability of the NaK-α1, the reduced NaK-β1 level was accompanied by a reduced NaK-α1 level (Fig. 3A) which was also observed in TGF-β1 treated cells (Fig. 1B). Furthermore, stable knockdown of NaK-β1 in the human renal clear-cell carcinoma cell line, Caki-1 induced a fibroblastic morphology, accompanied by the expression of the EMT markers fibronectin and α-smooth muscle actin (Supplementary Fig. 3). Thus, the loss of NaK-β1 expression is sufficient to induce a fibroblastic phenotype.
Figure 3.

Effect of NaK-β1 knockdown and overexpression in LLC-PK1 cells. A and B, RNAi-mediated knockdown of NaK-β1. A, Immunoblot analysis for NaK-β1, NaK-α1 and E-cadherin in parental cells, control cells transfected with scrambled oligo (Scrambled) and NaK-β1 knockdown cells (NaK-β1 siRNA). B, NaK-β1 siRNA cells are fibroblastic (36 h post-transfection) and do not form characteristic cell domes in confluent monolayers (72 h). Bars, 20 μm (36 h); 100 μm (72 h). C–E, Ectopic expression of NaK-β1 delays the onset of TGF-β1 mediated EMT in LLC-PK1 cells. Immunoblots for NaK-β1 (C) and EMT markers fibronectin (FN) and α-smooth muscle actin (SMA) (D) and morphology (E) of control and NaK-β1 expressing cells upon TGF-β1 treatment. Bar, 20 μm.
Using a complementary approach (overexpression of NaK-β1), we further confirmed that high NaK-β1 expression levels delay the onset of phenotypic changes and induction of EMT markers. TGF-β1 treatment of LLC-PK1 cells expressing human NaK-β1 (LLC-PK1-NaK-β1) (Fig. 3C) showed minimal change in morphology at 48 and 96 hours, whereas control cells showed a typical mesenchymal phenotype (Fig. 3E). Although, NaK-β1 levels were reduced in control and LLC-PK1-NaK-β1 cells following TGF-β1 treatment (Fig. 3C), LLC-PK1-NaK-β1 cells had twice as much of NaK-β1 compared to control cells at 48 and 96 hours of treatment (Fig. 3C). FN and SMA levels inversely correlated with NaK-β1 levels. Fibronectin (FN) and α-smooth muscle actin (SMA) levels in TGF-β1-treated LLC-PK1-NaK-β1 cells were only 53% and 56%, respectively, of the levels of control cells (Fig. 3D). Taken together, these data demonstrate that reduction of NaK-β1 levels beyond a certain threshold is a critical step for the induction of EMT in kidney tubule cells.
NaK-β1 knockdown activates Erk1/2 signaling
Activation of the MAPK signaling is a prerequisite for TGF-β1-induced EMT (7). While Smad signaling pathways have been well described, much less is known about the activation of Erk1/2 signaling upon TGF-β1 treatment. In LLC-PK1 cells TGF-β1 induced a transient activation of Erk1/2 that peaked at 10 min and diminished after 30 min of treatment (Fig. 4A). However, after 48 and 96 hours of TGF-β1-treatment Erk1/2 activation was increased when compared to untreated LLC-PK1 control cells (Fig. 4B). Interestingly, sustained activation of Erk1/2 correlated with reduced NaK-β1 expression (Fig. 1B) suggesting that reduced NaK-β1 levels might be associated with Erk1/2 activation. Indeed, in NaK-β1 siRNA cells, activated Erk1/2 levels were 3- and 4-fold higher compared to parental or scrambled oligo-transfected LLC-PK1 cells, respectively (Fig. 4C), suggesting that NaK-β1 levels are at least in part involved in sustained activation of Erk1/2 in TGF-β1 mediated EMT.
Figure 4.
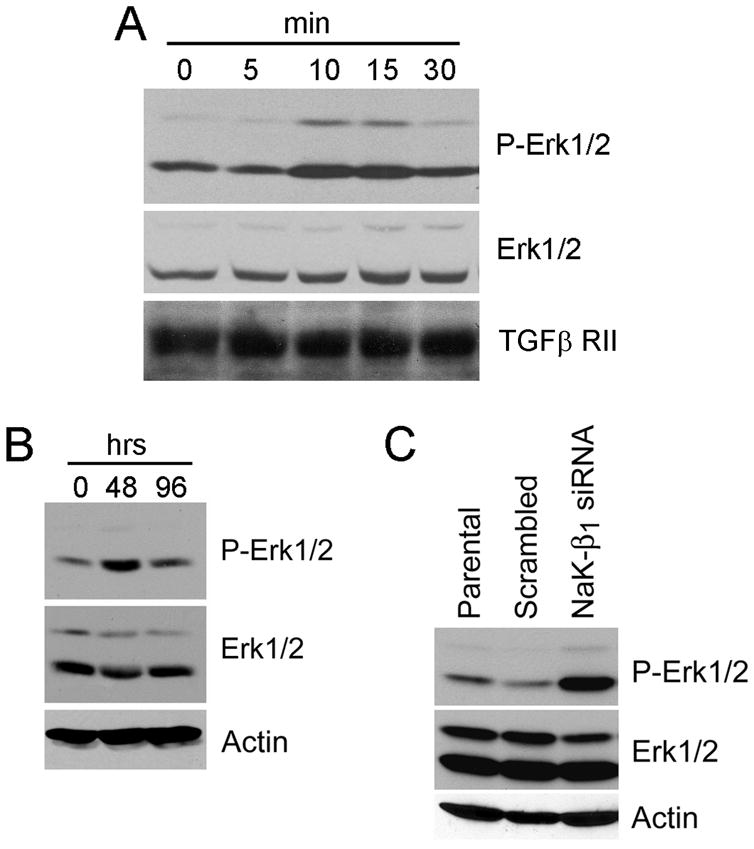
Activation of Erk1/2 signaling in TGF-β1-treated and NaK-β1 knockdown cells. Immunoblot analysis for activated and total Erk1/2 in TGF-β1-treated LLC-PK1 cells immediately after treatment (A) and after long-term treatment (B). Duration of TGF-β1 treatment is indicated in minutes (A) or hours (B). TGF-β RII (A) and actin (B) immunoblots are included to confirm equal amounts of protein used in analysis. C, Activation levels of Erk1/2 in NaK-β1 siRNA cells and corresponding parental and scrambled oligo control cells. Actin immunoblot confirms equal loading.
Na,K-ATPase subunit levels are reduced in different types of EMT
EMT is a process not only associated with cancer progression but also with fibrosis. In fact, LLC-PK1 cells have been used as a model to study the transdifferentiation of epithelial cells into interstitial fibrotic cells observed in fibrotic tissue (30). Since Na,K-ATPase subunits were affected in this cell culture system, we examined the NaK-β1 and NaK-α1 levels in well-defined fibrotic tissues by immunohistochemical analysis of kidney tissue sections from diabetic patients. Diabetic nephropathy is the most common contributor to end-stage renal disease and emerging evidence suggests that EMT of renal tubular epithelial cells to myofibroblasts is an important event in renal tubulointerstitial fibrosis (37–39). Renal biopsy samples were obtained from patients diagnosed with mild, moderate, and severe diabetic nephropathy and compared to normal kidney tissue sections. NaK-β1 and NaK-α1 were clearly reduced in fibrotic tissues compared to normal tissues (Fig. 5). In addition, immunohistochemistry for NaK-α1 and NaK-β1 on tissue sections obtained from STZ-induced diabetic rats revealed considerably reduced staining intensity in nephropathic tissues compared to normal tissues of non-induced rats (Fig. 6). Thus, reduced Na,K-ATPase subunit expression seems to be associated with fibrotic as well as cancerous EMT.
Figure 5.

Immunohistochemistry for NaK-α1 and NaK-β1 in kidneys of patients diagnosed with mild, moderate or severe diabetic nephropathy (Patients 1, 2, and 3, respectively). NaK-α1 and NaK-β1 show intense staining in tubules of normal kidney. Note the fibroblastic phenotype of epithelial cells in tubules undergoing EMT in patients with diabetic nephropathy (arrows). These tubules show no or little NaK-β1 and NaK-α1 staining. Insert in Patient 3/NaK-β1 is negative control.
Figure 6.

Immunohistochemistry for NaK-α1 and NaK-β1 of diabetic kidneys of STZ-induced rats. Staining shows reduced NaK-β1 and NaK-α1 staining in kidney tubules of STZ-induced rats compared to normal kidney. Arrows indicate glycogen-granule accumulation in tubular epithelium, a morphological change often associated with sustained hyperglycaemia.
Discussion
In this study, we provide the first evidence that Na,K-ATPase is a target of TGF-β1-signaling during the induction of EMT. We show that following the treatment of renal proximal tubule cells with TGF-β1, NaK-β1 surface expression is reduced prior to well-characterized EMT markers, such as E-cadherin, and the induction of fibronectin and α-smooth muscle actin. We validated the significance of NaK-β1 in the induction of EMT by two complementary approaches. RNAi-mediated specific knockdown of NaK-β1 resulted in the loss of the epithelial phenotype, while ectopic expression of NaK-β1 delayed the induction of a fibroblastic phenotype and reduced the levels of fibronectin and α-smooth muscle actin following TGF-β1 treatment. We further show that NaK-β1 reduction is a common event in EMT irrespective of the type of EMT.
RNAi-mediated specific knockdown of NaK-β1 is sufficient to induce a fibroblastic phenotype of LLC-PK1 cells. We have shown earlier that NaK-β1 expression is reduced in a wide variety of poorly differentiated cell lines (34) and that NaK-β1 repletion induces a well-differentiated phenotype in kidney epithelial cells (16). Collectively, these data demonstrate that NaK-β1 subunit function is important to maintain the well-differentiated phenotype of epithelial cells. We and others have shown that NaK-β1 functions as a cell-cell adhesion molecule (16–19) and loss of its cell adhesion function upon its reduced plasma membrane expression might be associated with the EMT in TGF-β1 treated cells. In addition, we have shown earlier that E-cadherin is less stably associated with the actin cytoskeleton when the NaK-β1 levels are low (16). Since NaK-β1’s translocation from the plasma membrane occurs prior to E-cadherin, we suggest that reduced surface expression of NaK-β1 might lead to the destabilization of E-cadherin’s interaction with the actin cytoskeleton. The loss of E-cadherin’s cell-cell adhesion function, which is dependent on its stable association with the actin cytoskeleton (40) might then further contribute to the phenotypic changes associated with EMT.
The transcription factor Snail, that suppresses E-cadherin transcription plays an important role in TGF-β1 mediated EMT (41, 42). We have shown that Snail also suppresses NaK-β1 transcription (34). Interestingly, in TGF-β1-treated LLC-PK-1 cells NaK-β1 mRNA levels were not affected. Our data also indicate that Snail levels were not increased in TGF-β1-treated cells (Supplementary Fig. 1), suggesting that the reduced NaK-β1 levels are primarily due to post-translational mechanisms. This notion is further supported by our observations in LLC-PK1 cells ectopically expressing NaK-β1. Although NaK-β1 expression is regulated by a CMV promoter in these cells the protein level was ultimately reduced following TGF-β1 treatment (Fig. 3C). Further experiments will be necessary to understand the post-translational mechanisms that lead to reduced NaK-β1 levels during TGF-β1 induced EMT in kidney epithelial cells.
In this study we report for the first time that TGF-β1-induced EMT is associated with increased [Na+]i levels. We consistently observed that TGF-β1 treated cells appear larger than control cells, a phenomenon also observed by others (43). Increased intracellular sodium could explain such an increase in cell size due to osmotic regulation, a well documented phenomenon, and is consistent with our observation of increased water content in TGF-β1 treated cells.
TGF-β1 treatment activates Erk1/2 transiently during early time points and after 30 minutes the phospho Erk1/2 level is drastically reduced. At this time point, we did not detect any change in NaK-β1 levels or localization. Interestingly, knockdown of NaK-β1 constitutively activated Erk1/2 in LLC-PK1 cells as well as in other epithelial cell lines (data not shown). We also showed that reduced NaK-β1 is associated with increased Erk1/2 in vivo in tumor xenograft models (20). Thus, we propose that in general loss of NaK-β1 is associated with the activation of Erk1/2. In the context of TGF-β1-induced EMT, reduced NaK-β1 might be involved in the sustained activation of Erk1/2 that was observed after 48 and 96 hours of treatment and is known to be involved in events leading to cancer progression.
Recently, three subtypes of EMT have been suggested that include mesenchymal EMT during gastrulation or neural crest formation (Type 1), EMT in the context of inflammation and fibrosis (Type 2) and EMT of cancer with invasion and metastasis (Type 3) (1, 3). Loss of Na,K-ATPase expression during EMT in cancer has been reported in numerous studies from our group (11, 16, 20, 44, 45) and others (46–49). We now provide evidence that NaK-β1 and NaK-α1 are reduced during EMT in fibrotic tissues in human as well as in animals. This data is consistent with the idea that Na,K-ATPase is a potential marker for both type 2 and type 3 EMT. Although EMT type 2 and EMT type 3 have in common the loss of the epithelial phenotype, they have distinct pathological consequences. During fibrosis epithelial cells transdifferentiate into myofibroblasts that secrete an extensive amount of extracellular matrix. As these cells are embedded in this matrix, tissue architecture and function are lost. On the other hand, EMT during cancer progression results in increased motility, invasion and metastastic potential of cancer cells. Type 2 and type 3 EMT are mediated at least in part by the same growth factors and can derive from the same cells (e.g. renal clear-cell carcinoma and renal fibrosis are though to originate from renal proximal tubule cells). Our results indicate that loss of NaK-β1 is associated with EMT in both cancer and fibrosis and occurs early on (Fig. 1B, C). Thus loss of NaK-β1 expression might have important significance in the pathological consequences associated with loss of the epithelial phenotype and EMT. However, the molecular factors that contribute to one type of EMT over the other remain to be determined.
Supplementary Material
Acknowledgments
We are grateful to Drs. William James Ball Jr. for Na,K-ATPase antibodies and Harold Moses for the Flag-tagged Smad7 cDNA.
Financial support: NIH grant RO1-DK56216 (AKR); The Nemours Foundation (SAR and AKR).
Abbreviations List
- BMP
Bone morphogenetic protein
- DMEM
Dulbecco’s modified Eagle’s medium
- EMT
Epithelial-to-mesenchymal transition
- NaK-α1
Na, K-ATPase α1-subunit
- NaK-β1
Na, K-ATPase β1-subunit
- TGF-β1
Transforming growth factor-β1
References
- 1.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–8. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Farquhar MG, Palade GE. Junctional complexes in various epithelia. J Cell Biol. 1963;17:375–412. doi: 10.1083/jcb.17.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest. 2009;119:1429–37. doi: 10.1172/JCI36183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Christiansen JJ, Rajasekaran AK. Reassessing epithelial to mesenchymal transition as a prerequisite for carcinoma invasion and metastasis. Cancer Res. 2006;66:8319–26. doi: 10.1158/0008-5472.CAN-06-0410. [DOI] [PubMed] [Google Scholar]
- 5.Derynck R, Akhurst RJ, Balmain A. TGF-[beta] signaling in tumor suppression and cancer progression. Nat Genet. 2001;29:117–29. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- 6.Massague J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. 2000;103:295–309. doi: 10.1016/s0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- 7.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–84. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 8.Shull GE, Schwartz A, Lingrel JB. Amino-acid sequence of the catalytic subunit of the (Na+ + K+)ATPase deduced from a complementary DNA. Nature. 1985;316:691–5. doi: 10.1038/316691a0. [DOI] [PubMed] [Google Scholar]
- 9.Shull GE, Lane LK, Lingrel JB. Amino-acid sequence of the beta-subunit of the (Na+ + K+)ATPase deduced from a cDNA. Nature. 1986;321:429–31. doi: 10.1038/321429a0. [DOI] [PubMed] [Google Scholar]
- 10.Beguin P, Wang X, Firsov D, et al. The gamma subunit is a specific component of the Na,K-ATPase and modulates its transport function. EMBO J. 1997;16:4250–60. doi: 10.1093/emboj/16.14.4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rajasekaran SA, Ball WJ, Jr, Bander NH, Liu H, Pardee JD, Rajasekaran AK. Reduced expression of beta-subunit of Na,K-ATPase in human clear-cell renal cell carcinoma. J Urol. 1999;162:574–80. [PubMed] [Google Scholar]
- 12.Rajasekaran AK, Rajasekaran SA. Role of Na-K-ATPase in the assembly of tight junctions. Am J Physiol Renal Physiol. 2003;285:F388–96. doi: 10.1152/ajprenal.00439.2002. [DOI] [PubMed] [Google Scholar]
- 13.Rajasekaran SA, Barwe SP, Gopal J, Ryazantsev S, Schneeberger EE, Rajasekaran AK. Na-K-ATPase regulates tight junction permeability through occludin phosphorylation in pancreatic epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2007;292:G124–33. doi: 10.1152/ajpgi.00297.2006. [DOI] [PubMed] [Google Scholar]
- 14.Rajasekaran SA, Hu J, Gopal J, et al. Na,K-ATPase inhibition alters tight junction structure and permeability in human retinal pigment epithelial cells. Am J Physiol Cell Physiol. 2003;284:C1497–507. doi: 10.1152/ajpcell.00355.2002. [DOI] [PubMed] [Google Scholar]
- 15.Rajasekaran SA, Palmer LG, Moon SY, et al. Na,K-ATPase activity is required for formation of tight junctions, desmosomes, and induction of polarity in epithelial cells. Mol Biol Cell. 2001;12:3717–32. doi: 10.1091/mbc.12.12.3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rajasekaran SA, Palmer LG, Quan K, et al. Na,K-ATPase beta-subunit is required for epithelial polarization, suppression of invasion, and cell motility. Mol Biol Cell. 2001;12:279–95. doi: 10.1091/mbc.12.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barwe SP, Kim S, Rajasekaran SA, Bowie JU, Rajasekaran AK. Janus model of the Na,K-ATPase beta-subunit transmembrane domain: distinct faces mediate alpha/beta assembly and beta-beta homo-oligomerization. J Mol Biol. 2007;365:706–14. doi: 10.1016/j.jmb.2006.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kitamura N, Ikekita M, Sato T, et al. Mouse Na+/K+-ATPase beta1-subunit has a K+-dependent cell adhesion activity for beta-GlcNAc-terminating glycans. Proc Natl Acad Sci U S A. 2005;102:2796–801. doi: 10.1073/pnas.0409344102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shoshani L, Contreras RG, Roldan ML, et al. The polarized expression of Na+,K+-ATPase in epithelia depends on the association between beta-subunits located in neighboring cells. Mol Biol Cell. 2005;16:1071–81. doi: 10.1091/mbc.E04-03-0267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Inge LJ, Rajasekaran SA, Yoshimoto K, et al. Evidence for a potential tumor suppressor role for the Na,K-ATPase beta1-subunit. Histol Histopathol. 2008;23:459–67. doi: 10.14670/hh-23.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Madan P, Rose K, Watson AJ. Na/K-ATPase beta1 subunit expression is required for blastocyst formation and normal assembly of trophectoderm tight junction-associated proteins. J Biol Chem. 2007;282:12127–34. doi: 10.1074/jbc.M700696200. [DOI] [PubMed] [Google Scholar]
- 22.Violette MI, Madan P, Watson AJ. Na+/K+-ATPase regulates tight junction formation and function during mouse preimplantation development. Dev Biol. 2006;289:406–19. doi: 10.1016/j.ydbio.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 23.Genova JL, Fehon RG. Neuroglian, Gliotactin, and the Na+/K+ ATPase are essential for septate junction function in Drosophila. J Cell Biol. 2003;161:979–89. doi: 10.1083/jcb.200212054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paul SM, Ternet M, Salvaterra PM, Beitel GJ. The Na+/K+ ATPase is required for septate junction function and epithelial tube-size control in the Drosophila tracheal system. Development. 2003;130:4963–74. doi: 10.1242/dev.00691. [DOI] [PubMed] [Google Scholar]
- 25.Paul SM, Palladino MJ, Beitel GJ. A pump-independent function of the Na,K-ATPase is required for epithelial junction function and tracheal tube-size control. Development. 2007;134:147–55. doi: 10.1242/dev.02710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laprise P, Lau KM, Harris KP, et al. Yurt, Coracle, Neurexin IV and the Na+,K+-ATPase form a novel group of epithelial polarity proteins. Nature. 2009;459:1141–5. doi: 10.1038/nature08067. [DOI] [PubMed] [Google Scholar]
- 27.Cibrian-Uhalte E, Langenbacher A, Shu X, Chen JN, Abdelilah-Seyfried S. Involvement of zebrafish Na+,K+ ATPase in myocardial cell junction maintenance. J Cell Biol. 2007;176:223–30. doi: 10.1083/jcb.200606116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bagnat M, Cheung ID, Mostov KE, Stainier DY. Genetic control of single lumen formation in the zebrafish gut. Nat Cell Biol. 2007;9:954–60. doi: 10.1038/ncb1621. [DOI] [PubMed] [Google Scholar]
- 29.Rajasekaran SA, Rajasekaran AK. Na,K-ATPase and epithelial tight junctions. Front Biosci. 2009;14:2130–48. doi: 10.2741/3367. [DOI] [PubMed] [Google Scholar]
- 30.Masszi A, Di Ciano C, Sirokmany G, et al. Central role for Rho in TGF-beta1-induced alpha-smooth muscle actin expression during epithelial-mesenchymal transition. Am J Physiol Renal Physiol. 2003;284:F911–24. doi: 10.1152/ajprenal.00183.2002. [DOI] [PubMed] [Google Scholar]
- 31.Rajasekaran SA, Gopal J, Espineda C, Ryazantsev S, Schneeberger EE, Rajasekaran AK. HPAF-II, a cell culture model to study pancreatic epithelial cell structure and function. Pancreas. 2004;29:e77–83. doi: 10.1097/00006676-200410000-00016. [DOI] [PubMed] [Google Scholar]
- 32.Reeves A, Zagurovskaya M, Gupta S, Shareef MM, Mohiuddin M, Ahmed MM. Inhibition of Transforming Growth Factor-I2 Signaling in Normal Lung Epithelial Cells Confers Resistance to Ionizing Radiation. International journal of radiation oncology, biology, physics. 2007;68:187–95. doi: 10.1016/j.ijrobp.2006.12.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kletzien RF, Pariza MW, Becker JE, Potter VR. A method using 3-O-methyl-D-glucose and phloretin for the determination of intracellular water space of cells in monolayer culture. Anal Biochem. 1975;68:537–44. doi: 10.1016/0003-2697(75)90649-1. [DOI] [PubMed] [Google Scholar]
- 34.Espineda CE, Chang JH, Twiss J, Rajasekaran SA, Rajasekaran AK. Repression of Na, K-ATPase beta1-subunit by the transcription factor snail in carcinoma. Mol Biol Cell. 2004;15:1364–73. doi: 10.1091/mbc.E03-09-0646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dai C, Yang J, Liu Y. Transforming growth factor-beta1 potentiates renal tubular epithelial cell death by a mechanism independent of Smad signaling. J Biol Chem. 2003;278:12537–45. doi: 10.1074/jbc.M300777200. [DOI] [PubMed] [Google Scholar]
- 36.Nakagawa T, Lan HY, Zhu HJ, Kang DH, Schreiner GF, Johnson RJ. Differential regulation of VEGF by TGF-beta and hypoxia in rat proximal tubular cells. Am J Physiol Renal Physiol. 2004;287:F658–64. doi: 10.1152/ajprenal.00040.2004. [DOI] [PubMed] [Google Scholar]
- 37.Eddy AA. Molecular basis of renal fibrosis. Pediatr Nephrol. 2000;15:290–301. doi: 10.1007/s004670000461. [DOI] [PubMed] [Google Scholar]
- 38.Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112:1776–84. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lan HY. Tubular epithelial-myofibroblast transdifferentiation mechanisms in proximal tubule cells. Curr Opin Nephrol Hypertens. 2003;12:25–9. doi: 10.1097/00041552-200301000-00005. [DOI] [PubMed] [Google Scholar]
- 40.Gumbiner BM. Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell. 1996;84:345–57. doi: 10.1016/s0092-8674(00)81279-9. [DOI] [PubMed] [Google Scholar]
- 41.Medici D, Hay ED, Goodenough DA. Cooperation between snail and LEF-1 transcription factors is essential for TGF-beta1-induced epithelial-mesenchymal transition. Mol Biol Cell. 2006;17:1871–9. doi: 10.1091/mbc.E05-08-0767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peinado H, Quintanilla M, Cano A. Transforming growth factor beta-1 induces snail transcription factor in epithelial cell lines: mechanisms for epithelial mesenchymal transitions. J Biol Chem. 2003;278:21113–23. doi: 10.1074/jbc.M211304200. [DOI] [PubMed] [Google Scholar]
- 43.Lamouille S, Derynck R. Cell size and invasion in TGF-{beta} induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J Cell Biol. 2007;178:437–51. doi: 10.1083/jcb.200611146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Espineda C, Seligson DB, James Ball W, Jr, et al. Analysis of the Na,K-ATPase alpha- and beta-subunit expression profiles of bladder cancer using tissue microarrays. Cancer. 2003;97:1859–68. doi: 10.1002/cncr.11267. [DOI] [PubMed] [Google Scholar]
- 45.Seligson DB, Rajasekaran SA, Yu H, et al. Na,K-adenosine triphosphatase alpha1-subunit predicts survival of renal clear cell carcinoma. J Urol. 2008;179:338–45. doi: 10.1016/j.juro.2007.08.094. [DOI] [PubMed] [Google Scholar]
- 46.Blok LJ, Chang GT, Steenbeek-Slotboom M, et al. Regulation of expression of Na+, K+-ATPase in androgen-dependent and androgen-independent prostate cancer. Br J Cancer. 1999;81:28–36. doi: 10.1038/sj.bjc.6690647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mijatovic T, Roland I, Van Quaquebeke E, et al. The alpha1 subunit of the sodium pump could represent a novel target to combat non-small cell lung cancers. J Pathol. 2007;212:170–9. doi: 10.1002/path.2172. [DOI] [PubMed] [Google Scholar]
- 48.Mobasheri A, Fox R, Evans I, Cullingham F, Martin-Vasallo P, Foster CS. Epithelial Na, K-ATPase expression is down-regulated in canine prostate cancer; a possible consequence of metabolic transformation in the process of prostate malignancy. Cancer Cell Int. 2003;3:8. doi: 10.1186/1475-2867-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sakai H, Suzuki T, Maeda M, et al. Up-regulation of Na(+), K(+)-ATPase alpha 3-isoform and down-regulation of the alpha1-isoform in human colorectal cancer. FEBS Lett. 2004;563:151–4. doi: 10.1016/S0014-5793(04)00292-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.