

Abstract
The cytosine analogue decitabine alters hematopoietic differentiation. For example, decitabine treatment increases self-renewal of normal hematopoietic stem cells. The mechanisms underlying decitabine induced shifts in differentiation are poorly understood, but likely relate to the ability of decitabine to deplete the chromatin-modifying enzyme DNA methyl-transferase 1 (DNMT1) that plays a central role in transcription repression. HOXB4 is a transcription factor that promotes hematopoietic stem cell self-renewal. In hematopoietic precursors induced to differentiate by the lineage-specifying transcription factor Pu.1, or by the cytokine granulocyte-colony stimulating factor (G-CSF), there is rapid repression of HOXB4 and other stem cell genes. Depletion of DNMT1 using shRNA or decitabine prevents HOXB4 repression by Pu.1 or G-CSF, and maintains hematopoietic precursor self-renewal. In contrast, depletion of DNMT1 by decitabine six hours after the differentiation stimulus, that is, after repression of HOXB4 has occurred, augments differentiation. Therefore, DNMT1 is required for the early repression of stem cell genes that occurs in response to a differentiation stimulus, providing a mechanistic explanation for the observation that decitabine can maintain or increase hematopoietic stem cell self-renewal in the presence of a differentiation stimulus. Using decitabine to deplete DNMT1 after this early repression phase does not impair progressive differentiation.
Keywords: Decitabine, hematopoietic stem cells, self-renewal, differentiation, transcription repression
Introduction
The cytosine analogue 5-aza-2’-deoxycytidine (decitabine), approved by the United States Food and Drug Administration (FDA) as a treatment for myelodysplastic syndrome (MDS), alters hematopoietic differentiation. Therapeutically important differentiation altering effects of decitabine include terminal differentiation of leukemia cells 1–5(supporting manuscript), increased erythropoiesis and fetal hemoglobin to treat sickle cell disease and β-thalassemia 6,7, and increased self-renewal of hematopoietic stem cells which could have a role in ex vivo expansion of hematopoietic stem cells 8–10. The mechanisms underlying these decitabine induced shifts in differentiation are poorly understood, but likely relate to the ability of decitabine to deplete the chromatin-modifying enzyme DNA methyl-transferase 1 (DNMT1) 11. DNMT1 duplicates methylation marks onto the newly synthesized DNA strand during mitosis (maintenance methylation) 12. DNMT1 is also a component of histone methyl-transferase protein-complexes that create methylation marks of transcription repression (reviewed in13) and histone demethylase protein-complexes that remove methylation marks of transcription activation 14. Accordingly, decitabine treatment to deplete DNMT1 not only hypomethylates DNA but can extensively modify chromatin, producing decondensation of chromatin structure 15.
Drugs that relax chromatin by inhibiting histone deacetylase (HDAC), produce changes in hematopoietic differentiation similar to that produced by decitabine: terminal differentiation of leukemia cells 4,16,17, increased fetal hemoglobin expression (reviewed in 18) and increased normal hematopoietic stem cell self-renewal 8,19,20. Since a shared property of HDAC inhibitors and DNMT1 depletion by decitabine is antagonism of transcription repression, this suggests that antagonism of transcription repression could have a mechanistic role in producing the observed shifts in differentiation by decitabine and by HDAC inhibitors.
The transition from one differentiation level to the next likely involves repression of key genes that maintain the initial or starting differentiation level. Here we examined the possibility that decitabine antagonizes the repression of stem cell associated genes by a differentiation inducing stimulus. We used two in vitro models of hematopoiesis to examine this possibility, a model of lineage-specifying transcription factor (Pu.1) driven differentiation of murine hematopoietic cells, and a model of cytokine (granulocyte-colony stimulating factor, G-CSF) driven differentiation of human hematopoietic cells. The generated observations provide a mechanistic explanation for some of the differentiation altering effects of decitabine treatment, and can guide the application of this drug.
Materials and Methods
Generation of PUER cells with stable suppression of Dnmt1 expression
A lentiviral vector pLenti6-DEST (Invitrogen, Carlsbad, CA) was used to construct short hairpin (sh) RNA for Dnmt1. The specific 21-bp target sequences for mouse shDnmt1 (5′-GAACGGCATCAAGGTGAAC-3′) were synthesized in sense and antisense orientation by Advanced DNA Technology (ADT), the single-strand oligos were then annealed to form double-strand oligos, and subsequently ligated with pENTRY vector (Invitrogen, Carlsbad, CA) downstream of an RNA promoter. The ligated constructs were transformed into E. Coli TOPO10. Positive clones were verified by DNA sequencing. The verified clones were then recombined into pLenti6-DEST vector using Invitrogen’s ViralPack kit, resulting in pLenti6-Dnmt1. The pLenti6-Dnmt1 constructs were then transfected together with envelop encoding plasmid (VSVG) into 293FT packaging cell line to produce lentivirus. The supernatant containing lentivirus was harvested at 48 hours after transfection. Titers were determined on NIH3T3 cells as transducing units using serial dilutions of vector stocks with 8 µg/ml polybrene (Sigma Chemical, St. Louis, MO).
PUER cells 21 are murine hematopoietic precursor cells that have been retrovirally transduced to express Pu.1 fused to the estrogen receptor. To knock-down Dnmt1 in cells, murine PUER cells were grown in Iscove's modified Eagle's medium, without phenol-red, with 10% fetal bovine serum, 2.5ng/ml mouse IL-3, 1µg/ml Puromycin, 55µM beta-ME, 1% penicillin/streptomycin at 37°C in a humidified atmosphere with 5% CO2 in air. The lentivirus-containing supernatant was added to the cell culture at appropriate 4 particles/cell concentration with 8 µg/ml polybrene. Twenty-four hours after infection, 4ug/ml of blasticidin (bln) was added to the cell culture for positive clone selection. The Bln-resistant cells were analyzed for Dnmt1 expression by RQ-PCR and Western blot. Control cells were PUER transduced with empty vector (PUER Control).
Addition of tamoxifen (OHT) to PUER triggers their terminal differentiation into macrophages 21. Differentiation status was analyzed by: (i) presence of adherent cells by light microscopy, (ii) morphological changes in Giemsa stained cytospin preparations, and (iii) cKit (eBioscience, San Diego, CA, catalog number 11-1171-82) and F4/80 (eBioscience catalog number 15-4801-82) expression by flow cytometry. Cell analysis was performed on a Coulter Epics XL-MCL flow cytometer equipped with CXP software (Beckman-Coulter, Miami, FL).
Cell Fractionation and Protein Extraction
Approximately 5 million PUER Control and PUER shDnmt1 cells were used to prepare cell-lysates. After removal of the medium, cells were transferred to 15 ml conical tubes and washed twice with 5 mL ice cold 1x PBS. Cells were resuspended in 500 µL of 1× Hypotonic Buffer containing 10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 10 mM PMSF, and protease inhibitor cocktail (Sigma-Aldrich, A8340), and incubated for 10 min on ice. 20 µL of 10 % NP-40 was added to cell suspensions to break the cell membrane. After another 10 min incubation on ice, cell suspensions were centrifuged at 500g for 10 min. The supernatant was transferred to clean 1.5 mL Eppendorf tubes, and labeled as the cytoplasm fraction. Nuclear pellets were washed twice with ice cold water, and resuspended in 50 µL of 50 mM Tris-HCl pH 8.0, 1 mM MgCl2, 10 mM PMSF, protease inhibitor cocktail (Sigma-Aldrich, A8340) and DNASE 1 (Sigma-Aldrich, D5915, 50 unit final concentration). The nuclear suspensions were incubated on ice for 30 min with vigorous vortex every 5 min. At the end of incubation, 50 µL protein extraction buffer containing 4% SDS, 10 mM DTT, 20% Glycerol in 50 mM Tris-HCl was added and the mixture was sonicated on ice and centrifuged at 140,000 rpm for 5 min. The supernatant containing nuclear proteins was transferred to clean tubes and protein concentration was determined by BCA assay.
1D SDS-PAGE and Western Blotting
Approximately 50 µg of cytoplasmic and nuclear protein extracts from PUER Control and PUER shDnmt1 cells, together with molecular weight markers, were subjected to 1D SDS-PAGE on 4–12% gradient gels (Invitrogen). After electrophoresis per manufacturer’s manual (Invitrogen), proteins were transferred to PVDF membranes (Millipore, Billerica, MA) at 35 constant voltage for 1 hour using Invitrogen’s semidry blotting apparatus. Western analyses of PVDF membranes utilized established protocols and rabbit monoclonal anti-Pu.1 (Cell Signaling, Danvers, MA #2258) and anti-β-Actin peroxidase (Sigma-Aldrich, A3854).
Real time PCR (RQ-PCR)
mRNA levels were assayed using RQ-PCR. Briefly, total cellular RNA is isolated from 5×105 cells using RNeasy Plus (QIAGEN), according to the manufacturer's protocol. For cDNA synthesis, after a denaturation step of 5 min at 70°C, 1 µg of RNA were reverse transcribed to single-stranded cDNA using a mix of random hexamers and oligo dT primers and M-MLV reverse transcriptase for first strand synthesis (Promega, Madison, WI). Real-time PCR was performed with Real-time PCR Master Mix containing Sybrgreen I and hotstart Taq DNA polymerase (Takara, Madison, WI). GAPDH was amplified as control. Primer sequences available upon request. Real-time detection of the emission intensity of SYBR Green bound to double-stranded DNA will be detected using the iCycler instrument (Bio-Rad, Hercules, CA). Data is reported as relative expression value’ which was determined by raising 2 to the power of the negative value of delta-delta CT for each sample.
Treatment of cells with decitabine
Decitabine stock solution (5 mM) was generated by reconstituting lypholized decitabine in 100% methanol. Stock solution was stored at −20 °C for up to 3 weeks. Working solution was generated by diluting the stock solution 1:100 in PBS immediately before addition to the cells at a further dilution as per the intended final concentration. Similar amounts of methanol are added to untreated control cells. Cells were treated with decitabine (0.5 µM) with timings as designated per the text and figure legends.
Normal cord blood samples
Umbilical cord blood was collected during normal full-term deliveries following written informed consent of the mother on a CASE IRB approved protocol.
Isolation of CD34+ cells
CD34+ cells from umbilical cord blood were immunopositively purified using a magnetic cell sorting system a CD34 MicroBead Kit (catalog #: 130-046-702, Miltenyi Biotec) according to manufacturer instructions. The purity of the CD34+ population (ranged typically from 95 to 99%) was determined by immunolabeling with FITC-conjugated monoclonal antibodies against CD34 (Clone 581, Beckman Coulter) that reacted with an epitope other than the antibody used for separation.
Human hematopoietic cell culture and clonogenic progenitor assays
CD34+ selected normal human hematopoietic cells were cultured in IMDM supplemented with 10% fetal bovine serum and 10 ng/ml of the following human cytokines: stem cell factor (SCF), FLT3 ligand (FLT3), thrombopoietin (TPO), interleukin-3 (IL-3) and interleukin-6 (IL-6). Cells were treated once with decitabine 0.5 µM either concurrently with the addition of G-CSF 50 ng/ml or 6 hours after G-CSF 50 ng/ml. Cells were harvested from liquid culture on day 9 and plated into methyl-cellulose at a concentration of 20,000 cells per ml of semi-solid media (Methocult, Stem Cell Technologies, Vancouver, BC, Canada) supplemented with SCF (50 ng/mL), GM-CSF (10 ng/mL), IL-3 (10 ng/mL), and erythropoietin (3 U/mL). Colonies were counted/photographed Day 12 of semi-solid culture.
Results
Dnmt1 depletion by shRNA prevents Pu.1 induced HoxB4 repression and differentiation
Hematopoietic cell fate is driven by key transcription factors such as HOXB4, which promotes hematopoietic stem cell self-renewal 22, and the lineage-specifying factor PU.1, which is required for macrophage and B-cell lineage-commitment and differentiation (reviewed in 23). We wished to examine if Dnmt1 is required for the repression of HoxB4 and other stem cell associated genes by Pu.1. PUER cells are murine Pu.1−/− hematopoietic precursors which have been transduced with a retroviral vector to express Pu.1 fused to the estrogen-receptor (ER) 21. In cell-culture with murine interleukin-3 (mIL-3), PUER cells self-renew indefinitely. Addition of tamoxifen (OHT) to these cells (as an estrogen agonist) causes functional reintroduction of Pu.1 by translocation into the nucleus, and triggers terminal macrophage differentiation 21.
Although not normal hematopoietic stem cells, PUER self-renewing in mIL3 express high levels of the self-renewal promoting factor HoxB4, and other genes that are expressed at high levels in stem cells, Bmi-1 and c-Kit. After addition of OHT, there is repression of HoxB4, Bmi-1 and c-Kit, followed by activation of genes expressed at high levels in macrophages (macrophage colony stimulating factor receptor - Mcsfr, granulocyte monocyte colony stimulating factor - Gmcsfr, F4/80) (figure 1A). These expression changes were accompanied by acquisition of morphologic changes of macrophage differentiation (increase in cell size, decrease in nuclear-cytoplasmic ratio, clumping of nuclear chromatin, adherence to culture plates, cytoplasmic vacuolization) (figure 1A).
Figure 1. Dnmt1 depletion by shRNA prevented Pu.1 mediated repression of stem cell associated genes and allowed continued self-renewal in the presence of nuclear Pu.1.
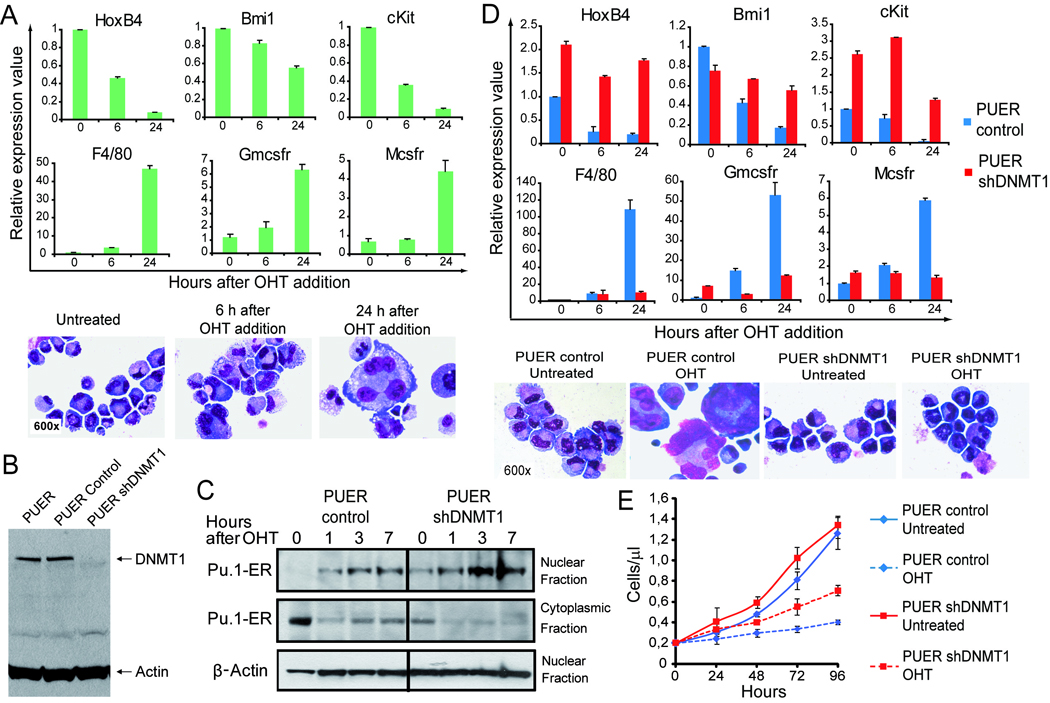
PUER are Pu.1 −/− murine hematopoietic precursors retrovirally transduced to express Pu.1 fused to the estrogen receptor. Tamoxifen (OHT) 100nM translocates Pu.1 to the nucleus and triggers macrophage differentiation. A) After addition of OHT, there is repression of stem cell associated genes HoxB4, Bmi-1 and c-Kit, followed by activation of genes associated with macrophage differentiation Mcsfr, Gmcsfr, F4/80. Giemsa-stained cytospin preparations 48h after addition of OHT. Gene expression measured by RQ-PCR, average of 6 independent experiments. B) Dnmt1 was knocked-down in PUER by lentiviral transduction of shRNA (PUER shDnmt1) with PUER transduced with empty vector as control (PUER Control). Dnmt1 measured by Western blot. C) Addition of OHT to shDnmt1 cells caused Pu.1-ER to be rapidly translocated from the cytoplasm into the nucleus at high levels. Pu.1-ER measured by Western blot in nuclear and cytoplasmic fractions. Actin in nuclear fraction. D) OHT treated shDnmt1 cells continued to express high levels of stem cell associated genes and low levels of differentiation genes. Gene expression by RQ-PCR, average of 3 independent experiments. Giemsa-stained cytospin preperations 48h after OHT addition. E) OHT treated shDnmt1 cells proliferated at a lower rate than shDnmt1 cells without OHT, however, the cells continued to exponentially expand at a significantly higher rate than OHT treated Control. Cell counts by automated cell counter, average of 3 independent experiments. Error bars = standard error.
Dnmt1 was knocked-down in PUER by lentiviral transduction of shRNA (PUER shDnmt1). Control cells were PUER transduced with empty vector (PUER Control). Knockdown of Dnmt1 by shDnmt1 was > 70% by Western Blot (figure 1B).
Addition of OHT to shDnmt1 cells caused Pu.1-ER to be rapidly translocated from the cytoplasm into the nucleus at high levels (figure 1C).
OHT treated control cells rapidly repressed expression of stem cell genes HoxB4, Bmi-1 and c-Kit, however, OHT treated shDnmt1 cells continued to express high levels of these stem cell genes (figure 1D). OHT treated control cells activated expression of differentiation genes Mcsfr, Gmcsfr and F4/80, however, these genes remained relatively repressed in OHT treated shDnmt1 cells (figure 1D).
OHT treated control displayed extensive morphologic changes of macrophage differentiation (figure 1D). In contrast, OHT treated shDnmt1 cells continued to display morphologic immaturity, with most cells resembling untreated shDnmt1 cells (figure 1D).
OHT treatment substantially decreased the proliferation of control cells. OHT treatment also decreased the proliferation of shDnmt1 cells, but not to the same extent as the decrease in proliferation of control cells (OHT treated shDnmt1 cells continued to expand exponentially) (figure 1E).
Therefore, depletion of Dnmt1 prevented Pu.1 mediated repression of HoxB4 and other stem cell genes, prevented differentiation, and allowed hematopoietic precursor self-renewal in the presence of nuclear Pu.1.
Maintenance of hematopoietic precursor self-renewal by decitabine requires timing of treatment to prevent Pu.1 mediated stem cell gene repression
To further develop this observation, decitabine, the cytosine analogue that binds and depletes Dnmt1, was added to the PUER cells concurrent with the introduction of Pu.1 (concurrent with OHT) or 6 hours after introduction of Pu.1 (6 hours after OHT), that is, after the repression of HoxB4 had occurred.
OHT treatment of PUER caused rapid repression of the stem cell genes HoxB4, Bmi-1 and c-Kit coupled with activation of the differentiation genes F4/80, Gmcsfr, and Mcsfr. Decitabine addition concurrent with the OHT attenuated the repression of the stem cell genes and activation of differentiation genes. Decitabine addition 6 hours after the OHT did not significantly impede stem cell gene repression or the subsequent differentiation gene activation (figure 2A).
Figure 2. Maintenance of hematopoietic precursor (PUER) self-renewal by decitabine (DAC) 0.5 µM requires timing of decitabine treatment to prevent Pu.1 mediated stem cell gene repression.
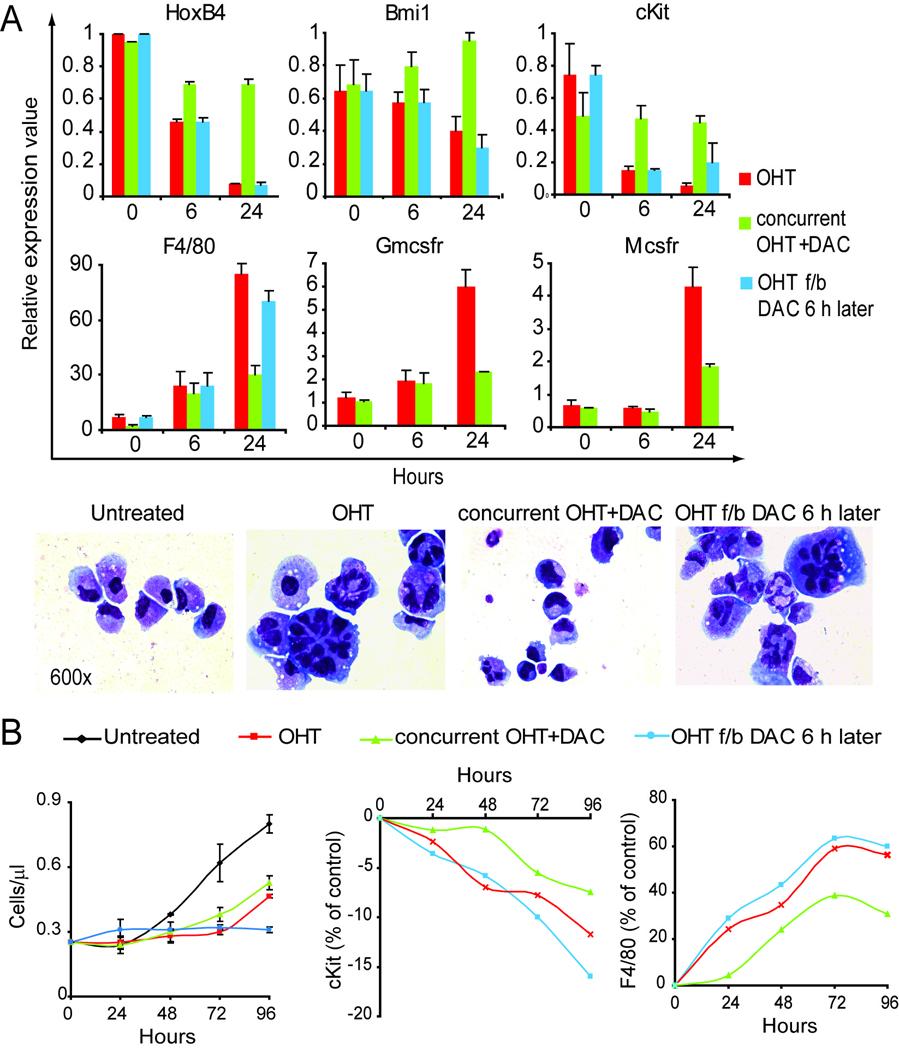
A) DAC addition concurrent with OHT (Pu.1), but not 6 hours after OHT, attenuated the repression of stem cell genes (HoxB4, Bmi-1 and c-Kit), and the activation of differentiation genes (F4/80, Gmcsfr, and Mcsfr ) compared to OHT alone. Gene expression by RQ-PCR, average of 4 independent experiments. Giemsa-stained cytospin preparations 48h after OHT. B) DAC addition concurrent with OHT (Pu.1) attenuated the decrease in proliferation, and protein expression changes, seen with OHT alone. Cells counts by automated cell counter, average of 3 independent experiments. Error bars = standard error. c-Kit and F4/80 protein expression measured by flow-cytometry.
OHT treatment of PUER caused rapid macrophage differentiation. Decitabine concurrent with OHT largely prevented morphologic differentiation, with most cells resembling cells without OHT. Decitabine 6 hours after the OHT resulted in more extensive changes of macrophage differentiation than cells treated with OHT alone (figure 2A).
OHT treatment of PUER caused a substantial decrease in proliferation compared to untreated cells. Decitabine concurrent with OHT also decreased cell proliferation compared to untreated cells, but not to the same extent as OHT alone. Decitabine 6 hours after the OHT terminated cell proliferation (figure 2B).
OHT treatment of PUER caused a decrease in c-Kit protein expression, and an increase in F4/80 protein expression as measured by flow-cytometry. Decitabine concurrent with OHT significantly attenuated the decrease in c-Kit expression and the increase in F4/80 expression produced by OHT alone. Decitabine 6 hours after the OHT resulted in c-Kit and F4/80 expression changes that resembled OHT alone (figure 2B).
Therefore, the ability of decitabine to maintain self-renewal of hematopoietic precursors in the presence of nuclear Pu.1 depends on timing of decitabine treatment in relationship to the Pu.1 differentiation stimulus.
Maintenance of human hematopoietic precursor self-renewal in the presence of G-CSF requires timing of decitabine treatment to prevent stem cell associated gene repression by G-CSF
Previously, we and others demonstrated that treating human CD34+ hematopoietic stem and progenitor cells (HSPC) with decitabine maintains or promotes their self-renewal in vitro such that decitabine can be used for ex vivo expansion of HSPC 8,9,24.
Human CD34+ cells express high levels of HOXB4 and other factors known to be associated with stem cell self-renewal such as the stem cell factor receptor (c-KIT) and the polycomb protein BMI-1 (figure 3A). Addition of the myeloid differentiation promoting cytokine G-CSF to these cells results in rapid repression of the stem cell genes HOXB4, BMI-1 and c-KIT, coupled with rapid activation of the differentiation genes PU.1, MCSFR and G-CSF receptor (G-CSFR) (figure 3A). Addition of decitabine concurrent with G-CSF significantly attenuated repression of the stem cell genes, and attenuated activation of the differentiation genes compared to G-CSF alone. Addition of decitabine 6 hours after the G-CSF did not impede stem cell gene repression or differentiation gene activation compared to G-CSF alone (figure 3A).
Figure 3. Cell fate in response to decitabine (DAC) 0.5 µM treatment varies with timing in relationship to G-CSF differentiation stimulus.
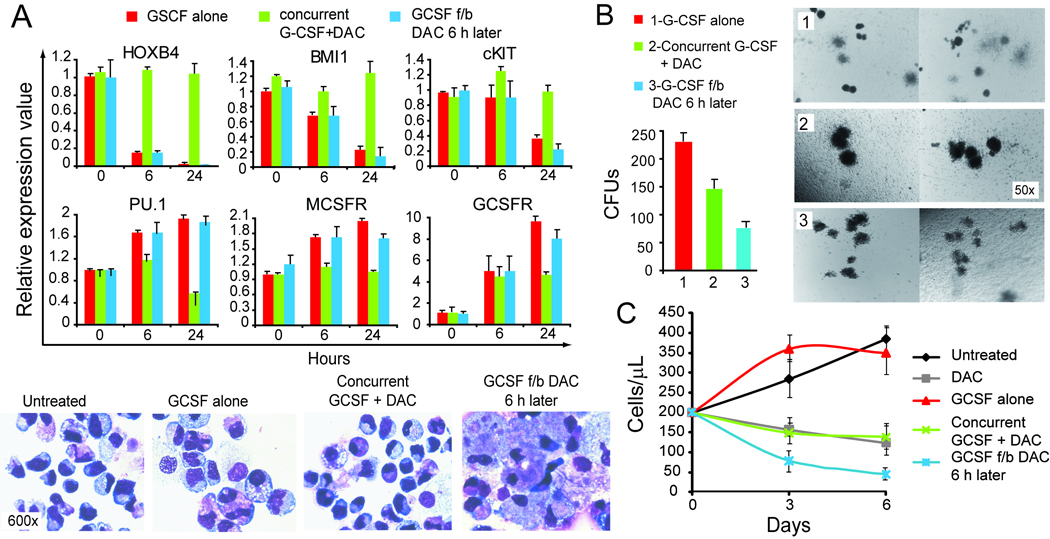
Human CD34+ hematopoietic precursors were cultured with SCF, TPO, FLT3, IL-3, IL-6 with or without G-CSF and decitabine. A) DAC concurrent with G-CSF, but not 6 hours after G-CSF, significantly attenuated repression of HOXB4, BMI1 and c-KIT, and attenuated activation of PU.1, MCSFR or GCSFR, compared to G-CSF alone. Gene expression by RQ-PCR, average of 4 independent experiments. Giemsa-stained cytospin preparations on day 7 after G-CSF addition. B) DAC addition concurrent with G-CSF decreased the number of progenitors but increased size of colonies formed. DAC 6h after G-CSF decreased both number and size of colonies. Cells harvested from liquid culture 9 days after addition of G-CSF. Cells plated into methyl-cellulose at 20,000 cells per ml of semi-solid media. Colonies counted/photographed Day 12 of semi-solid culture (n=4). C) DAC concurrent with G-CSF decreased cell proliferation compared to G-CSF alone, however, not to the same extent as DAC 6 hours after G-CSF. Cell counts by automated cell counter, average of 4 independent experiments. Error bars = standard error.
G-CSF induced morphologic differentiation of the cells (increased cell size, decreased nuclear-cytoplasmic ratio, chromatin clumping, increased granulation) (figure 3A). Decitabine addition concurrent with G-CSF largely prevented the morphologic differentiation seen with G-CSF alone (figure 3A). In contrast, decitabine addition 6 hours after the G-CSF produced more extensive morphologic changes of differentiation compared to cells treated with G-CSF alone (figure 3A).
Decitabine addition concurrent with G-CSF decreased the number of progenitors present on day 9 (9 days after addition of G-CSF) of liquid culture (progenitors were assayed by plating cells harvested from liquid culture into semi-solid media) compared to G-CSF alone. However, the colonies formed were larger, consistent with more immature character of the progenitors (figure 3B). In contrast, decitabine addition 6 hours after the G-CSF (after HOXB4 repression and PU.1 activation) produced a greater decrease in colony number (decreased progenitors) and colony size (increased maturity of progenitors) compared to cells treated with concurrent decitabine and G-CSF or G-CSF alone (figure 3B).
Decitabine addition concurrent with G-CSF decreased cell proliferation compared to G-CSF alone, however, not to the same extent as decitabine addition 6 hours after G-CSF (figure 3C).
Therefore, the effects of decitabine treatment on human hematopoietic cell fate depend on timing of treatment in relationship to the cytokine differentiation stimulus.
Discussion
The repression of HOXB4 and other stem cell genes that occurs early after a differentiation stimulus is necessary for differentiation to proceed. This suggests a mechanistic basis for the observation that drugs that antagonize transcription repression can increase hematopoietic stem cell self-renewal 8–10,19,20,24–26. DNMT1 depletion by decitabine, prior to or concurrent with a differentiation inducing stimulus, by preventing the repression of stem cell genes by the differentiation stimulus, maintains stem cell self-renewal. Consistent with the proposed mechanism, treatment with decitabine shortly after a differentiation stimulus (after transition through the early repression phase) does not maintain self-renewal and instead augments differentiation.
An alternative model to explain the ability of decitabine to increase hematopoietic stem cell self-renewal is that decitabine treatment to deplete DNMT1 directly reactivates a stem cell program in the treated cells. The different cell fate produced by treating cells with decitabine before versus after a differentiation stimulus is more consistent with the proposed model than this alternative. Emphasizing that the cell fate consequences of decitabine treatment are cell context dependent, decitabine treatment induces terminal differentiation of leukemia cells 1–5, including the demonstrably self-renewing sub-set of leukemia initiating cells (supporting manuscript), in contrast to its effect on normal hematopoietic stem cells.
Another question is whether the differentiation altering effects of decitabine can be attributed to DNMT1 depletion rather than other effects of the drug. Decitabine is a cytosine analogue, therefore, as per the class effect of nucleoside analogues, it can induce DNA damage and cytotoxicity. However, decitabine is rapidly cleaved and degraded by hydrolysis 27. Hence, decitabine is substantially less efficient at impeding DNA replication machinery and terminating DNA strand elongation than an equi-molar concentration of cytosine arabinoside (ara-C), the cytosine analogue that is the mainstay of cytotoxic AML therapy 28,29. We have demonstrated that the concentrations of decitabine used here deplete DNMT1 without causing measurable DNA damage or apoptosis in normal human hematopoietic precursors (supporting manuscript). Others have demonstrated that decitabine depletes the maintenance DNA methyl-transferase DNMT1 11, but not the de novo DNA methyl-transferases DNMT3A and DNMT3B 11, consistent with the S-phase specificity of decitabine. The similar shifts in differentiation produced by HDAC inhibitors and decitabine is also consistent with a mechanism that relates to antagonism of transcription repression. Finally, conditional deletion of DNMT1 by genetic methods demonstrates that it has a role in regulating hematopoietic cell fate. In a murine model of inducible knock-out of Dnmt1 in adult mice, defects in the hematopoietic stem cell compartment and in progenitors suggested that Dnmt1 is required for hematopoietic stem cell self-renewal and differentiation 30. In another genetic study, Dnmt1 was partially depleted using a hypomorphic Dnmt1 allele 31. An increase in hematopoietic stem cells and deficiencies in lineage-committed progenitors were noted 31.
Although the murine conditional Dnmt1 knock-out models demonstrate a role for Dnmt1 in hematopoietic differentiation, these genetic models differ from DNMT1 depletion by decitabine treatment in important respects: DNMT1 depletion by decitabine is transient, with recovery of DNMT1 protein in normal hematopoietic precursors to close to pre-treatment levels within 96 hours of decitabine exposure (supporting manuscript); the transient DNMT1 depletion by decitabine although substantial is not complete 3,11; DNMT1 depletion by decitabine is S-phase dependent, and is therefore more extensive in actively cycling cells such as hematopoietic precursors than in supporting tissue such as stroma.
The findings here suggest that DNMT1, a key chromatin modifying enzyme involved in transcription repression, is required for the early repression of stem cell genes that occurs in response to a differentiation stimulus. This repression step is necessary for differentiation to proceed to the next level. The varied cell fate consequences of decitabine treatment reflect this differentiation phase and cell context dependent role of the DNMT1 molecular target of therapy.
Acknowledgements
We gratefully acknowledge the following gifts: Mary Laughlin and Nick Greco at the Abraham J and Phyllis Katz Cord Blood Foundation and Cleveland Cord Blood Center for cord blood samples; Dr Brian Bolwell for Departmental support and resources; Grant support: YS - Scott Hamilton CARES Foundation, NIH (U54HL090513, 1R01CA138858), Dept. of Defense (PR081404); JPM - NIH (R01HL082983; U54RR019391; K24HL077522), AA&MDS Foundation, Robert Duggan Cancer Research Foundation.
Footnotes
Disclosure of Conflicts of Interest:
YS receives consulting fees from HemaQuest, a company developing treatments for blood diseases such as thalassemia and sickle cell disease.
Reference List
- 1.Jones PA, Taylor SM. Cellular differentiation, cytidine analogs and DNA methylation. Cell. 1980;20:85–93. doi: 10.1016/0092-8674(80)90237-8. [DOI] [PubMed] [Google Scholar]
- 2.Pinto A, Attadia V, Fusco A, Ferrara F, Spada OA, Di Fiore PP. 5-Aza-2'-deoxycytidine induces terminal differentiation of leukemic blasts from patients with acute myeloid leukemias. Blood. 1984;64:922–929. [PubMed] [Google Scholar]
- 3.Creusot F, Acs G, Christman JK. Inhibition of DNA methyltransferase and induction of Friend erythroleukemia cell differentiation by 5-azacytidine and 5-aza-2'-deoxycytidine. J.Biol.Chem. 1982;257:2041–2048. [PubMed] [Google Scholar]
- 4.Nowak D, Stewart D, Koeffler HP. Differentiation therapy of leukemia: 3 decades of development. Blood. 2009;113:3655–3665. doi: 10.1182/blood-2009-01-198911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Niitsu N, Hayashi Y, Sugita K, Honma Y. Sensitization by 5-aza-2'-deoxycytidine of leukaemia cells with MLL abnormalities to induction of differentiation by all-trans retinoic acid and 1alpha,25-dihydroxyvitamin D3. Br.J.Haematol. 2001;112:315–326. doi: 10.1046/j.1365-2141.2001.02523.x. [DOI] [PubMed] [Google Scholar]
- 6.Saunthararajah Y, Hillery CA, Lavelle D, Molokie R, Dorn L, Bressler L, Gavazova S, Chen YH, Hoffman R, Desimone J. Effects of 5-aza-2'-deoxycytidine on fetal hemoglobin levels, red cell adhesion, and hematopoietic differentiation in patients with sickle cell disease. Blood. 2003;102:3865–3870. doi: 10.1182/blood-2003-05-1738. [DOI] [PubMed] [Google Scholar]
- 7.Saunthararajah Y, Molokie R, Saraf S, Sidhwani S, Gowhari M, Vara S, Lavelle D, DeSimone J. Clinical effectiveness of decitabine in severe sickle cell disease. Br.J.Haematol. 2008;141:126–129. doi: 10.1111/j.1365-2141.2008.07027.x. [DOI] [PubMed] [Google Scholar]
- 8.Milhem M, Mahmud N, Lavelle D, Araki H, Desimone J, Saunthararajah Y, Hoffman R. Modification of hematopoietic stem cell fate by 5aza 2'deoxycytidine and trichostatin A. Blood. 2004;103:4102–4110. doi: 10.1182/blood-2003-07-2431. [DOI] [PubMed] [Google Scholar]
- 9.Suzuki M, Harashima A, Okochi A, Yamamoto M, Nakamura S, Motoda R, Yamasaki F, Orita K. 5-Azacytidine supports the long-term repopulating activity of cord blood CD34(+) cells. Am.J.Hematol. 2004;77:313–315. doi: 10.1002/ajh.20178. [DOI] [PubMed] [Google Scholar]
- 10.Chung YS, Kim HJ, Kim TM, Hong SH, Kwon KR, An S, Park JH, Lee S, Oh IH. Undifferentiated hematopoietic cells are characterized by a genome-wide undermethylation dip around the transcription start site and a hierarchical epigenetic plasticity. Blood. 2009;114:4968–4978. doi: 10.1182/blood-2009-01-197780. [DOI] [PubMed] [Google Scholar]
- 11.Ghoshal K, Datta J, Majumder S, Bai S, Kutay H, Motiwala T, Jacob ST. 5-Azadeoxycytidine induces selective degradation of DNA methyltransferase 1 by a proteasomal pathway that requires the KEN box, bromo-adjacent homology domain, and nuclear localization signal. Mol.Cell Biol. 2005;25:4727–4741. doi: 10.1128/MCB.25.11.4727-4741.2005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12.Zucker KE, Riggs AD, Smith SS. Purification of human DNA (cytosine-5-)-methyltransferase. J.Cell Biochem. 1985;29:337–349. doi: 10.1002/jcb.240290407. [DOI] [PubMed] [Google Scholar]
- 13.Kohn KW, Aladjem MI, Weinstein JN, Pommier Y. Chromatin challenges during DNA replication: a systems representation. Mol.Biol.Cell. 2008;19:1–7. doi: 10.1091/mbc.E07-06-0528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang J, Hevi S, Kurash JK, Lei H, Gay F, Bajko J, Su H, Sun W, Chang H, Xu G, Gaudet F, Li E, Chen T. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat.Genet. 2009;41:125–129. doi: 10.1038/ng.268. [DOI] [PubMed] [Google Scholar]
- 15.Haaf T. The effects of 5-azacytidine and 5-azadeoxycytidine on chromosome structure and function: implications for methylation-associated cellular processes. Pharmacol.Ther. 1995;65:19–46. doi: 10.1016/0163-7258(94)00053-6. [DOI] [PubMed] [Google Scholar]
- 16.Kosugi H, Towatari M, Hatano S, Kitamura K, Kiyoi H, Kinoshita T, Tanimoto M, Murate T, Kawashima K, Saito H, Naoe T. Histone deacetylase inhibitors are the potent inducer/enhancer of differentiation in acute myeloid leukemia: a new approach to anti-leukemia therapy. Leukemia. 1999;13:1316–1324. doi: 10.1038/sj.leu.2401508. [DOI] [PubMed] [Google Scholar]
- 17.Wang J, Saunthararajah Y, Redner RL, Liu JM. Inhibitors of histone deacetylase relieve ETO-mediated repression and induce differentiation of AML1-ETO leukemia cells. Cancer Res. 1999;59:2766–2769. [PubMed] [Google Scholar]
- 18.Desimone J. Approaches to the reactivation of hemoglobin F as a treatment for sickle cell disease. Clin.Adv.Hematol.Oncol. 2004;2:23–24. [PubMed] [Google Scholar]
- 19.Bug G, Gul H, Schwarz K, Pfeifer H, Kampfmann M, Zheng X, Beissert T, Boehrer S, Hoelzer D, Ottmann OG, Ruthardt M. Valproic acid stimulates proliferation and self-renewal of hematopoietic stem cells. Cancer Res. 2005;65:2537–2541. doi: 10.1158/0008-5472.CAN-04-3011. [DOI] [PubMed] [Google Scholar]
- 20.De Felice L, Tatarelli C, Mascolo MG, Gregorj C, Agostini F, Fiorini R, Gelmetti V, Pascale S, Padula F, Petrucci MT, Arcese W, Nervi C. Histone deacetylase inhibitor valproic acid enhances the cytokine-induced expansion of human hematopoietic stem cells. Cancer Res. 2005;65:1505–1513. doi: 10.1158/0008-5472.CAN-04-3063. [DOI] [PubMed] [Google Scholar]
- 21.Walsh JC, DeKoter RP, Lee HJ, Smith ED, Lancki DW, Gurish MF, Friend DS, Stevens RL, Anastasi J, Singh H. Cooperative and antagonistic interplay between PU.1 and GATA-2 in the specification of myeloid cell fates. Immunity. 2002;17:665–676. doi: 10.1016/s1074-7613(02)00452-1. [DOI] [PubMed] [Google Scholar]
- 22.Sauvageau G, Thorsteinsdottir U, Eaves CJ, Lawrence HJ, Largman C, Lansdorp PM, Humphries RK. Overexpression of HOXB4 in hematopoietic cells causes the selective expansion of more primitive populations in vitro and in vivo. Genes Dev. 1995;9:1753–1765. doi: 10.1101/gad.9.14.1753. [DOI] [PubMed] [Google Scholar]
- 23.Iwasaki H, Akashi K. Myeloid lineage commitment from the hematopoietic stem cell. Immunity. 2007;26:726–740. doi: 10.1016/j.immuni.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 24.Araki H, Mahmud N, Milhem M, Nunez R, Xu M, Beam CA, Hoffman R. Expansion of human umbilical cord blood SCID-repopulating cells using chromatin-modifying agents. Exp.Hematol. 2006;34:140–149. doi: 10.1016/j.exphem.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 25.Young JC, Wu S, Hansteen G, Du C, Sambucetti L, Remiszewski S, O'Farrell AM, Hill B, Lavau C, Murray LJ. Inhibitors of histone deacetylases promote hematopoietic stem cell self-renewal. Cytotherapy. 2004;6:328–336. doi: 10.1080/14653240410004899. [DOI] [PubMed] [Google Scholar]
- 26.Lee JH, Hart SR, Skalnik DG. Histone deacetylase activity is required for embryonic stem cell differentiation. Genesis. 2004;38:32–38. doi: 10.1002/gene.10250. [DOI] [PubMed] [Google Scholar]
- 27.Rogstad DK, Herring JL, Theruvathu JA, Burdzy A, Perry CC, Neidigh JW, Sowers LC. Chemical decomposition of 5-aza-2'-deoxycytidine (Decitabine): kinetic analyses and identification of products by NMR, HPLC, and mass spectrometry. Chem.Res.Toxicol. 2009;22:1194–1204. doi: 10.1021/tx900131u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Covey JM, D'Incalci M, Tilchen EJ, Zaharko DS, Kohn KW. Differences in DNA damage produced by incorporation of 5-aza-2'-deoxycytidine or 5,6-dihydro-5-azacytidine into DNA of mammalian cells. Cancer Res. 1986;46:5511–5517. [PubMed] [Google Scholar]
- 29.Schermelleh L, Haemmer A, Spada F, Rosing N, Meilinger D, Rothbauer U, Cardoso MC, Leonhardt H. Dynamics of Dnmt1 interaction with the replication machinery and its role in postreplicative maintenance of DNA methylation. Nucleic Acids Res. 2007;35:4301–4312. doi: 10.1093/nar/gkm432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trowbridge JJ, Snow JW, Kim J, Orkin SH. DNA methyltransferase 1 is essential for and uniquely regulates hematopoietic stem and progenitor cells. Cell Stem Cell. 2009;5:442–449. doi: 10.1016/j.stem.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Broske AM, Vockentanz L, Kharazi S, Huska MR, Mancini E, Scheller M, Kuhl C, Enns A, Prinz M, Jaenisch R, Nerlov C, Leutz A, Andrade-Navarro MA, Jacobsen SE, Rosenbauer F. DNA methylation protects hematopoietic stem cell multipotency from myeloerythroid restriction. Nat.Genet. 2009;41:1207–1215. doi: 10.1038/ng.463. [DOI] [PubMed] [Google Scholar]