

Abstract
Current chemotherapy regimens against pancreatic cancer are met with little success as poor tumor vascularization significantly limits the delivery of oncological drugs. High-dose targeted drug delivery, through which a drug delivery vehicle releases a large payload upon tumor localization, is thus a promising alternative strategy against this lethal disease. Herein, we synthesize anti-CEA half-antibody conjugated lipid-polymer hybrid nanoparticles and characterize their ligand conjugation yields, physicochemical properties, and targeting ability against pancreatic cancer cells. Under the same drug loading, the half-antibody targeted nanoparticles show enhanced cancer killing effect compared to the corresponding non-targeted nanoparticles.
Keywords: Lipid-polymer nanoparticles, half-antibody, targeted drug delivery, pancreatic cancer, carcinoembryonic antigen
Introduction
Pancreatic cancer is the 4th leading cause of cancer-related death in the United States.1 Because of its elusive symptoms, pancreatic cancer patients are often diagnosed at an advanced stage, at which time the tumors are often unresectable and best addressed with chemotherapy. However, most chemotherapeutic regimens show little to no response against pancreatic cancer. Even the current first-line drug, gemcitabine, shows only modest improvement in patient survival.2 A recent study by Olive et al. suggests that low efficacy of chemotherapy against pancreatic cancer is due to the poor vascularization and inadequate drug perfusion at the tumoral sites.3 Such a clinical challenge calls for new and more effective drug delivery strategies that can enhance the drug availability at the tumor sites.
To address this fundamental issue in pancreatic cancer treatment, here we propose a targeted drug delivery platform consisting of a lipid-polymer hybrid nanoparticle (NP) to carry therapeutic payloads and a selectively reduced anti-CEA half-antibody (hAb) to target pancreatic cancer cells. It is hypothesized that the targeted NPs can preferentially bind to pancreatic cancer cells rather than normal cells. Given the high drug loading capacity of each lipid-polymer hybrid NP, the cancer cells are expected to be destroyed even though only few NPs are taken up by one cell.
The lipid-polymer hybrid NPs consist of a biodegradable and biocompartible hydrophobic polymeric core made of poly d,l-lactic-co-glycolic acid (PLGA), a monolayer of phospholipids, and an outer corona layer made of polyethylene glycol (PEG). Despite their complex structure, these hybrid NPs are synthesized in a simple, single-step fashion, which allows future scale-up production and cost-effective real-life applications.4 Several features make these NPs a promising drug delivery platform, including biocompatibility, biodegradability, sustained drug-release profiles, functionalizable surface, excellent stability in blood, and most importantly, high drug loading yield.5 These properties provide a basis for a stable, high-payload targeted drug delivery vehicle that can potentially maximize the chemotherapeutic efficacy against target cancer cells.
For the targeting ligand, we selected anti-CEA monoclonal antibody (mAb) as a basis because CEA is overexpressed in 90% of pancreatic tumors.6 In a study of using fluorophore-conjugated anti-CEA mAb for pancreatic cancer visualization, Kaushal et al. demonstrated that the mAb could effectively and safely deliver fluorescent dyes to subcutaneous, orthotopic, and metastatic human pancreatic cancer xenografts in nude mice.7 Several reports have also conjugated anti-CEA mAb with NPs for various applications.8-10 In the case of NP drug delivery, however, whole antibodies have several limitations. For instance, the large hydrodynamic size of antibodies significantly increases nonspecific uptake by the reticuloendothelial system (RES) and reduces the NP circulation half-life.11 In addition, the nonspecific nature of antibody conjugation with NPs, typically involving random carboxyl-amine reaction, could result in hindered binding sites of the antibody and particle dimerization. Smaller antibody variants, such as single-chain variable fragments (svFc).12 diabodies, and minibodies13, 14 have emerged in research settings to address these issues. Here we selectively reduced the disulfide bonds between the heavy chains of the anti-CEA mAb using low molar excess of reducing agent to produce anti-CEA half-antibodies (hAb). The resulting hAb possess intact binding sites and reactive thiol groups on their constant region. These hAb retain their targeting ability but are smaller in size and can be conjugated in a site-specific manner. Several reports have employed the hAb strategy to deliver imaging agents such as quantum dots and iron oxide.15, 16 To the best of our knowledge, this is the first report that optimizes such a strategy for high-dose targeted drug delivery.
In this study, we successfully synthesized and characterized anti-CEA hAb conjugated lipid-polymer hybrid NPs. We demonstrated the integrity and stability of the complex structure of the targeted NPs after being internalized by the target cells as well as their targeting specificity toward CEA-presenting pancreatic cancer cells in vitro. In addition, using paclitaxel as a model chemotherapy drug, we showed superior cytotoxicity of the targeted NPs against pancreatic cancer cells in comparison with their non-targeted counterparts.
Experimental Section
Conjugation of fluorophore to anti-CEA antibody and PLGA polymer
Monoclonal antibody specific for CEA was acquired from Biodesign International (Saco ME). The antibody was labeled with Alexa Fluor 488 (Molecular Probes, Eugene, OR) fluorophores following a protocol provided by the manufacturer. Briefly, the monoclonal antibody was reconstituted at 2 mg/mL in 0.1M sodium bicarbonate. 500 uL of the antibody solution were added to the reactive dye and allowed to incubate for 1 hr at room temperature. The antibody-dye conjugates were separated from unconjugated free dyes via a purification column. Alexa Fluor 647 (Molecular Probes, Eugene, OR) was conjugated to carboxy-terminated PLGA polymer through activation by 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) and N-hydroxysuccinimide (NHS). Briefly, 40 mg of 0.16 dL/g PLGA (Durect Corp, Cuppertino, CA) dissolved in 1.6 mL of dimethylformamide (DMF) were mixed with 40 mg of EDC and 20 mg of NHS (Pierce, Rockford, IL) for 15 min. The 534 μL of 0.5 mg/mL Alexa Fluor 647 solutions were added to the mixture and incubated overnight. The unconjugated dyes were removed by cold methanol wash. The fluorophore-conjugated PLGA was then vacuumed dried and re-dissolved in acetonitrile.
Synthesis and characterization of anti-CEA hAb targeted lipid-polymer hybrid NPs
Lipid-polymer hybrid NPs were prepared via self-assembly of PLGA (Lactel, Pelham, AL), lecithin (soybean, refined, molecular weight: ∼330 Da; Alfa Aesar, Ward Hill, MA), carboxyl-terminated DSPE-PEG (1,2-distearoyl-sn-glycero-3-phosphoethanolamine- N-carboxy (polyethylene glycol)2000) and maleimide-terminated DSPE-PEG (Avanti Polar Lipids, Alabaster, AL) through a single-step nanoprecipitation method following a previously published protocol.4 Briefly, 0.12 mg of lecithin and 0.26 mg of DSPE-PEG (the molar ratio between DSPE-PEG-COOH and DSPE-PEG-maleimide is determined by the amount of anti-CEA hAb to be conjugated to the NP surface) were dissolved in 2 mL 4 wt% ethanol aqeous solution. In parallel, 1 mg of PLGA polymer was dissolved in 1 mL acetonitrile. The resulting PLGA solution was then added into the lipid solution dropwise under gentle stirring. The mixed solution was vortexed vigorously for 3 minutes followed by gentle stirring for 2 hours at room temperature. The remaining organic solvent and free molecules were removed by washing the NP solution three times using an Amicon Ultra-4 centrifugal filter (Millipore, Billerica, MA) with a molecular weight cut-off of 10 kDa. The NPs with maleimide moieties were then incubated with selectively reduced anti-CEA hAb for 4 hrs. Finally, the unconjugated antibodies were removed by centrifugation using 300 kDa Pall Nanosep® centrifugal device (Sigma-Aldrich, St. Louis, MO).
The cleaved antibodies were separated on a SDS-PAGE 3-8% Tri-Acetate 10-well mini gel in tri-acetate running buffer using NovexSureLockXcell Electrophoresis System (Invitrogen). The samples were run at 150V for 1 hr and the resulting polyacrylamide gel was stained in SimplyBlue™ (Invitrogen) overnight for visualization. To quantify the ligand conjugation, Alexa Fluor 488 fluorescence in the hAb-NP solution was measured and matched to a standardization curve. The resulting hAb content was converted to mole. The number of NPs per 1mg of PLGA was approximated under the assumptions of spherical structure and uniform polymer density. Mass of each NP was estimated based on a particle diameter of 83 nm and a PLGA density of 1.2 g/mL. Assuming no loss in PLGA, the number of NPs was derived by dividing 1mg by the mass of a single NP.
NP size (diameter, nm) and surface charge (zeta potential, mV) were measured by dynamic light scattering (DLS) using Nano-ZS, Model ZEN3600 (Malvern, UK). NPs (∼500 μg) were dispersed in water (∼1 mL) and measurements were performed in triplicates at room temperature. The morphology and particle size were further characterized using scanning electron microscopy (SEM). Samples for SEM were prepared by dropping 5 μL of NPs solutions onto a polished silicon wafer. After drying the droplet at room temperature overnight, the sample was coated with chromium and then imaged by SEM.
Cellular targeting and internalization studies
The human pancreatic cell lines BxPC-3 and XPA-3 were maintained in RPMI (Gibco-BRL, Grand Island, NY) supplemented with 10% fetal calf albumin (FCS; Hyclone, Logan, UT), penicillin/streptomycin (Gibco-BRL), L-glutamine (Gibco-BRL), MEM nonessential amino acids (Gibco-BRL), sodium bicarbonate (Cellgro, Herndon, VA), and sodium pyruvate (Gibco-BRL). Both cell lines were cultured at 37°C with 5% CO2 and were plated in chamber slides (Cab-Tek II, eight wells; Nunc, Rochester, NY) with the aforementioned media. On the day of the experiment, cells were washed with pre-warmed PBS and incubated with pre-warmed RPMI media for 30 min before adding 100 μg of hAb-NPs or non-targeted NPs with the same amount of Alexa Fluor 647 dyes. The NPs were incubated with cells for 30 min at 37°C, washed with PBS 3 times, fixed with tissue fixative (Millipore, Bellerica, MA) for 30 min at room temperature, stained with 4′,6-diamidino-2-phenylindole (DAPI, nucleus staining), mounted in ProLong® Gold antifade reagent (Invitrogen), and imaged using deconvolution scanning fluorescence microscopy (DeltaVision System, Applied Precision, Issaquah, WA). Digital images of blue, green, and red fluorescence were acquired using a 40× and a 100× oil immersion objective and DAPI, FITC, and CY5 filters, respectively. Images were overlaid and deconvoluted using softWoRx software.
In vitro cytotoxicity study
To prepare drug-encapsulated NPs, 0.1 mg of paclitaxel (Sigma-Aldrich, St Louis, MO) was dissolved into the 1 mg of PLGA acetonitrile solution before the nanoprecipitation process. To quantify the loading yield of paclitaxel, the paclitaxel-loaded NPs were dissolved in a 70% acetonitrile aqueous solution and left on a vortex at room temperature for two days. Paclitaxel was quantified by high performance liquid chromatography (HPLC). The column used was μBONDAPAK 4.6×150 mm. The paclitaxel absorbance was measured by a UV/Vis detector at 230 nm in an acetonitrile/water gradient mobile phase (1 mL/min). The mobile phase acetonitrile/water ratio started at 50/50 at 0 min and increased to 100% acetonitrile over 10 min. Acetonitrile was then held for 4 min before returning to a 50/50 ratio in 14-25 min. Each set of data was measured with a calibration curve. For the cytotoxicity study, BxPC-3 cells were grown in 96-well plates at concentrations leading to 60% confluence in 24 h (40000 cell/cm2). NPs with different paclitaxel loading were incubated with the cells in RPMI for 1 hr. Then the cells were washed and supplemented with fresh media. Following 72 hrs of culture, cell viability was assessed with the ATPLite 1-step luminescence ATP detection assay (PerkinElmer, Waltham, MA) following a protocol provided by the manufacturer.
Results and Discussions
Figure 1 illustrates the synthesis scheme of anti-CEA hAb conjugated lipid-polymer hybrid NPs. Briefly, the maleimide-terminated hybrid NPs were self-assembled from their building blocks via a nanoprecipitation method following a previously described protocol.4 Tris (2-carboxyethyl) phosphine (TCEP) with a molar excess of 3× over the molar concentration of the anti-CEA mAb was used to selectively reduce the mAb into anti-CEA hAb. Subsequentially, the hAb were conjugated with the hybrid NPs through maleimide-thiol coupling. The resulting hAb-NPs were then filtered to remove excess antibodies (both uncleaved mAb and excess hAb) before further characterization and application.
Figure 1.
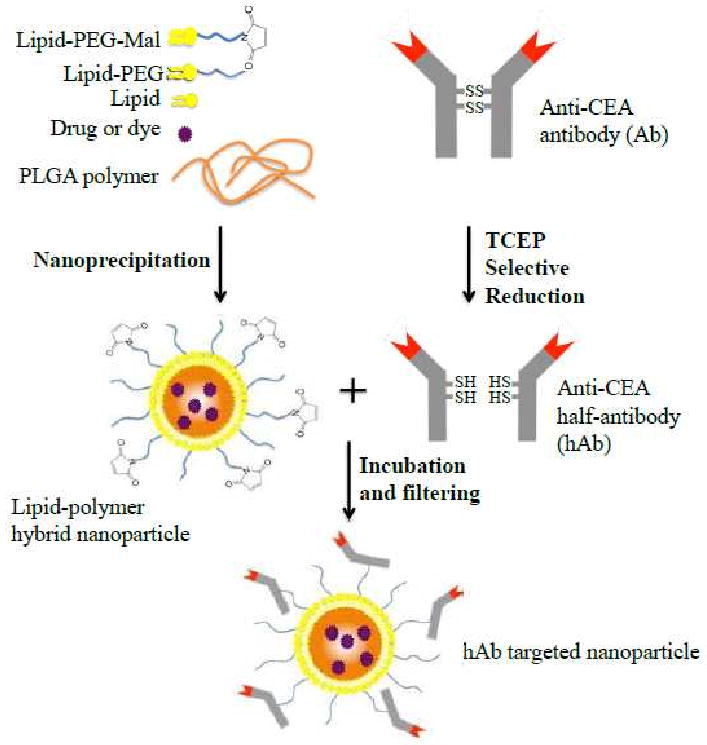
Schematic diagram of the synthesis of anti-CEA half-antibody (hAb) conjugated lipid-polymer hybrid NPs.
Synthesis of anti-CEA hAb
Since the extent of antibody reduction governs the conjugation efficiency as well as the targeting ability, the selective reduction process was optimized using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). To confirm the production of hAb and to optimize the selective reduction process, anti-CEA mAb were mixed with different molar excess of TCEP for 4 hrs and then separated and visualized. Figure 2A showed the SDS-PAGE results of reduced anti-CEA mAb under various molar excess of TCEP. Using the reference molecular weight ladder, the whole mAb (∼130kDa) and three distinct cleavage products - hAb (∼67kDa), heavy-chain fragments (∼46kDa), and light-chain fragments (∼21kDa) - were clearly identified. The presence of four distinct bands suggests that the disulfide bonds between the two heavy-chains were preferentially cleaved resulting in the formation of hAb. If all disulfide bonds in the mAb had been cleaved equally, two additional bands would have been observed, corresponding to double-heavy-chain fragments (∼92kDa) and single-light-chain-double-heavy-chain fragments (∼113kDa). Figure 2A also revealed the extent of mAb cleavage under different TCEP concentrations. It was observed that under 30× molar excess of TCEP the mAb were completely cleaved to heavy-chain and light-chain fragments. These fragments showed no binding affinity toward CEA-presenting pancreatic cancer cells (data not shown). We found that 3× molar excess of TCEP would maximize the yield of hAb and minimize the production of non-targeting fragments that compete with hAb for maleimide conjugation sites on the hybrid NP surface.
Figure 2.
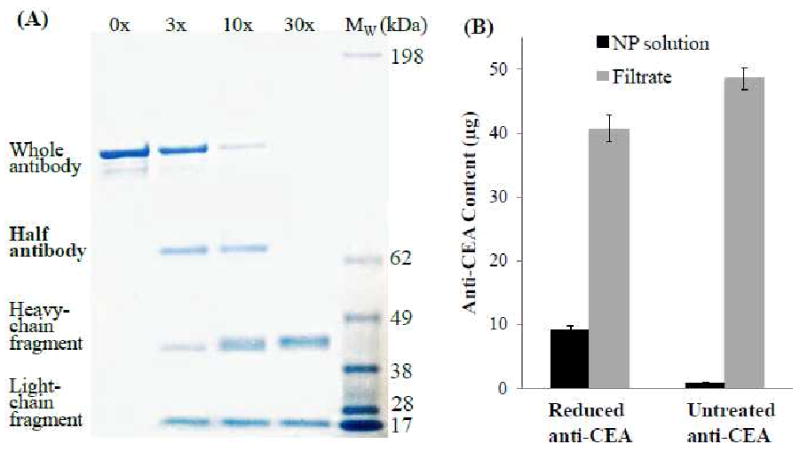
(A) Gel electrophoresis results of anti-CEA antibody reduction at TCEP (reducing agent) concentration of 0×, 3×, 10× and 30× over the concentration of the antibody, respectively. The rightmost lane represents a standard molecular weight ladder. (B) Quantifying the conjugation yield of 1 mg of maleimide-terminated hybrid NPs with 50 μg of anti-CEA antibody reduced by 3× TCEP versus 50 μg of untreated anti-CEA antibody.
Synthesis and Characterization of hAb-NP conjugates
The resulting hAb was conjugated to the hybrid NPs through maleimide-thiol coupling and the reaction was confirmed and quantified using fluorescence measurements (Figure 2B). Before TCEP reduction, anti-CEA mAb were covalently labeled with Alex-488 fluorophores. In the study, 50 μg of reduced anti-CEA mAb (3× molar excess of TCEP) were incubated with 1 mg of maleimide-terminated hybrid NPs (molar ratio: mAb/NP ≈ 150/1) for 4 hrs before the solution was filtered through a membrane with a molecular weight cutoff of 300 kDa. Fluorescence readings of the NP solution and the corresponding filtrate indicate that approximately 9.7 μg (or 19.4%) of selectively reduced anti-CEA mAb were conjugated to 1 mg of NPs, which translates to a 32:1 ligand-to-particle molar ratio (see experimental section for this estimation). To confirm that the retained fluorescence was due to maleimide-thiol conjugation rather than non-specific absorption of uncleaved anti-CEA mAb, noting that intact mAb do not react with the maleimide-terminated NPs, the NP solution was then mixed with 50 μg of untreated anti-CEA mAb. It was found that less than 1% (0.47 μg) of the mAb was retained in the NP solution. This control experiment suggests that the filter membrane used in this study was sufficient enough to remove all free mAb or their fragments from the NP solution and negligible amount of mAb non-specifically absorbs onto the NP surface.
The NP diameter and surface zeta potential in water were compared before and after the hAb conjugation using dynamic light scattering (DLS). The results showed a slight increase in particle size (83 nm to 95 nm) and a less negative zeta potential (-61 mV to -55 mV) following the hAb conjugation (Figure 3A). The size increase corresponds to the coating of the hAb around the NP surface. The increase in zeta potential, on the other hand, is likely due to the shielding of the negatively charged NP surface by the nearly neutral anti-CEA hAb moieties. Scanning electron microscopy (SEM) images (Figure 3B) of the hAb-conjugated hybrid NPs revealed their spherical structures with an averaged diameter of sub-100 nm, which was consistent with the DLS results.
Figure 3.

(A) Size and surface zeta potential of the hybrid NPs before and after hAb conjugation determined by dynamic light scattering. (B) SEM image of hAb targeted lipid-polymer hybrid NPs.
Targeting ability of hAb-NPs
After having synthesized the hAb-NPs and characterized their physicochemical properties, we next examined their targeting ability, particularly against CEA-presenting pancreatic cancer cells. We began by confirming the concurrent delivery of the lipid shell and the PLGA core of the targeted NPs using a dual-fluorophore system. One concern of using the lipid-polymer hybrid NPs stems from the integrity of their core-shell structure, especially when the shell is conjugated with targeting moieties. That is, the hAb conjugated lipid shell may detach from the PLGA core during the delivery process, therefore the hAb cannot target the drug loaded PLGA core to the sites of action. To test this possibility, the anti-CEA hAb and the PLGA core were labeled with Alexa-488 (green) and Alexa-647 (red), respectively. Targeted NPs simultaneously containing these two dyes were incubated with CEA-presenting human pancreatic adenocarcinoma cells (BxPC-3) and subsequently imaged using deconvolution scanning fluorescence microscopy. The matching overlay of the green and red fluorescence (Figures 4A-C) from the same NPs at corresponding excitation wavelengths suggests that neither the lipid shell nor the targeting hAb ligands was striped from the polymeric core following cell binding and particle internalization.
Figure 4.
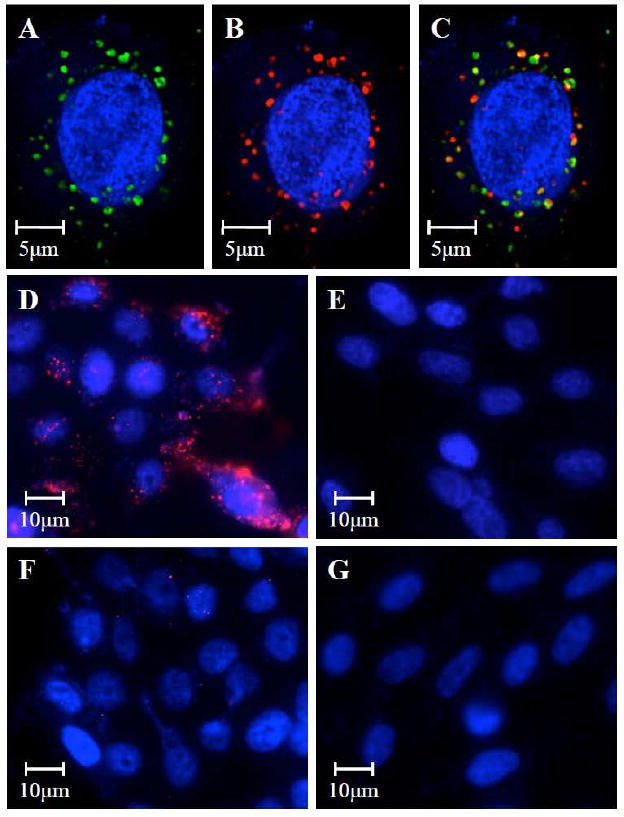
Scanning fluorescence microscopy images demonstrated the co-delivery of (A) the lipid shell to which the hAb conjugated (visualized with green Alexa Fluor 488 dyes) and (B) the PLGA polymeric core (visualized with red Alexa Fluor 647 dyes) to CEA-positive BxPC-3 cells (nucleus stained in blue with DAPI). (C) The overlay of the green and red fluorescence confirmed the integrity of the lipid-polymer hybrid NP structure after being internalized by the target cells. Targeting specificity was confirmed by incubating (D) CEA-positive BxPC-3 cells with anti-CEA hAb targeted NPs, (E) CEA-negative XPA-3 cells with anti-CEA hAb targeted NPs, (F) CEA-positive BxPC-3 cells with non-targeted NPs, and (G) CEA-negative XPA-3 cells with non-targeted NPs.
The targeting specificity of the hAb-NPs was evaluated by incubating BxPC-3 and a non-CEA-presenting human pancreatic cancer cell line (XPA-3) with hAb-NPs and non-targeted hybrid NPs without hAb conjugation. As shown in Figure 4D-G, the hAb-NPs effectively bound to the CEA positive BxPC-3 cells (Figure 4D) but remained stealthy to the CEA negative XPA-3 cells (Figures 4E). In contrast, the non-targeted hybrid NPs showed negligible cellular uptake by either BxPC-3 cells (Figures 4F) or XPA-3 cells (Figures 4G). This selective targeting could only be attributed to the complementary binding to the carcinoembryonic antigens by the anti-CEA hAb. Such a mechanism could enhance the cancer cell killing effect through receptor-mediated endocytosis which facilitates particle internalization17, 18. After a single hAb-NP was taken up by a target cell, it may release enough drug payloads to show therapeutic activity.
Cytotoxicity of paclitaxel-loaded hAb-NPs
To examine the in vitro cellular cytotoxicity of the anti-CEA hAb targeted NPs against pancreatic cancer cells, we chose paclitaxel as a model hydrophobic chemotherapy drug. Paclitaxel is a widely used anticancer drug and has shown great therapeutic efficacy against pancreatic cancer.19 An additional reason of choosing paclitaxel is that the drug can be readily loaded into the hydrophobic PLGA core of the hybrid NPs with high and controllable loading yields. In the study, hAb-NPs with various paclitaxel loading yields were incubated with BxPC-3 cells. The resulting cellular cytotoxicity was measured by an ATP cellular viability assay and was normalized to that of BxPC-3 cells treated by PBS buffer. Non-targeted hybrid NPs with the corresponding paclitaxel loading yields were used in parallel as a control. The actual paclitaxel loading of different NP formulations were quantified using high performance liquid chromatography (HPLC) following a published protocol.20 The results showed that paclitaxel loading yield can be precisely tuned by varying the initial drug input during NP preparation process (Figure 5 inset). Here up to ∼38 μg of paclitaxel were encapsulated into 1 mg of the hybrid NP for the cytotoxicity study. This 3.8 wt% drug loading yield can be converted to roughly 10,000 paclitaxel drug molecules per NP, calculating from the diameter of the NP (83 nm), PLGA density (1.2 g/mL) and the molecular weight of paclitaxel (854 Da). Speculatively, a few drug-loaded NPs taken up by one cancer cell may be able to provide a sufficiently high dose of paclitaxel that can kill the cancer cell. It is worth noticing that the loading yield of paclitaxel can be further improved by tuning the formulation parameters of the hybrid NPs but this is beyond the focus of this study.
Figure 5.
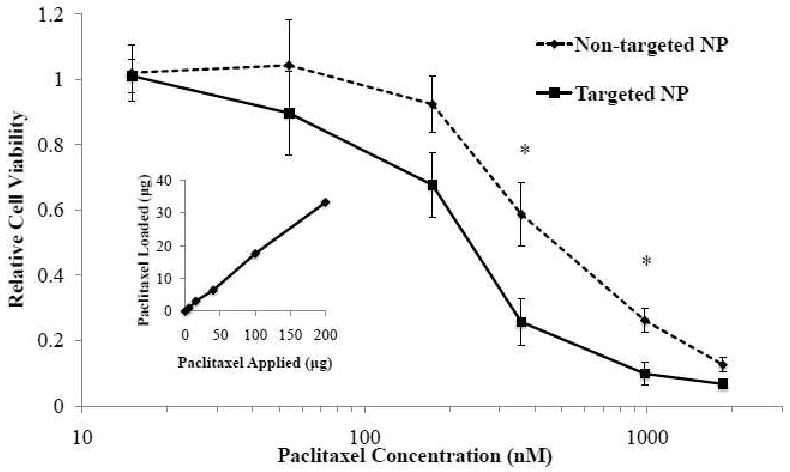
Relative viability of BxPC-3 cells incubated paclitaxel-loaded hAb targeted NPs (solid line) and non-targeted NPs (dashed line) with various paclitaxel loading yields using ATP assay. The results were normalized to controls without NP treatment. ANOVA test: *p<0.01 (n=8). Inset: paclitaxel loading vs. paclitaxel applied during NP preparation per 1mg of NPs.
After the paclitaxel-loaded hybrid NPs (both targeted and non-targeted) were prepared, the BxPC3 cells were incubated with the NPs loaded with different doses of paclitaxel for 1 hr. Then the excess NPs were washed in PBS buffer and the cells were cultured in fresh media for up to 72 hrs before they were subject to ATP assay. The luminescence signals of all samples were normalized to that of the cells in the absence of NPs to calculate the relative cellular viability of each circumstance. The results showed that the hAb targeted NPs had enhanced cytotoxicitiy at all levels of paclitaxel dosage, especially at 359 nM (Figure 5), where the targeted NPs and non-targeted NPs resulted in 25.7±7.2% and 58.7±9.9% relative cellular viability, respectively. ANOVA test showed that the difference in relative viability of the targeted and non-targeted NPs was significant at both 359 nM and 984 nM (p<0.01) of paclitaxel loading. An IC50 of 251 nM and 526 nM were obtained for the hAb-NPs and non-targeted NPs, respectively, which translated to a more than 2-fold increase in therapeutic efficacy due to the anti-CEA hAb targeting. It needs to be noted that without drug loading, neither the targeted NPs nor the non-targeted NPs showed any cytotoxicity against the cells (data not shown). The biocompatibility of these NPs is consistent with previous findings on lipid-polymer hybrid NPs5. Therefore, the hAb-conjugated NPs can be safely used as a drug delivery vehicle for in vivo applications.
Conclusions
In summary, we have successfully synthesized a targeted NP drug delivery platform specific to pancreatic cancer cells using FDA-approved biomaterials and selectively reduced anti-CEA half-antibodies. Ligand-to-particle molar ratio, particle size, surface charge, and drug loading yield have been characterized and could be controlled for specific therapeutic applications. In addition, we have demonstrated the targeting specificity of the synthesized targeted NPs as well as their enhanced cellular cytotoxicity against target cells compared to their non-targeted counterparts. This platform has great therapeutic potentials as it can effectively deliver a variety of chemotherapeutic drugs to pancreatic tumors in a targeted manner. Additional in vivo studies are required to examine the pharmacokinetics, biodistribution and therapeutic efficacy of the hAb targeted NPs before further applications can be considered.
Acknowledgments
This work is supported by the National Institute of Health grant U54CA119335 and the University of California San Diego.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Bria E, Milella M, Gelibter A, Cuppone F, Pino MS, Ruggeri EM, Carlini P, Nistico C, Terzoli E, Cognetti F, Giannarelli D. Gemcitabine-based combinations for inoperable pancreatic cancer: have we made real progress? A meta-analysis of 20 phase 3 trials. Cancer. 2007;110:525–533. doi: 10.1002/cncr.22809. [DOI] [PubMed] [Google Scholar]
- 3.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, Frese KK, Denicola G, Feig C, Combs C, Winter SP, Ireland-Zecchini H, Reichelt S, Howat WJ, Chang A, Dhara M, Wang L, Ruckert F, Grutzmann R, Pilarsky C, Izeradjene K, Hingorani SR, Huang P, Davies SE, Plunkett W, Egorin M, Hruban RH, Whitebread N, McGovern K, Adams J, Iacobuzio-Donahue C, Griffiths J, Tuveson DA. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang L, Chan JM, Gu FX, Rhee JW, Wang AZ, Radovic-Moreno AF, Alexis F, Langer R, Farokhzad OC. Self-assembled lipid-polymer hybrid nanoparticles: A robust drug delivery platform. ACS Nano. 2008;2:1696–1702. doi: 10.1021/nn800275r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan JM, Zhang L, Yuet KP, Liao G, Rhee JW, Langer R, Farokhzad OC. PLGA-lecithin-PEG core-shell nanoparticles for controlled drug delivery. Biomaterials. 2009;30:1627–1634. doi: 10.1016/j.biomaterials.2008.12.013. [DOI] [PubMed] [Google Scholar]
- 6.Ona FV, Zamcheck N, Dhar P, Moore T, Kupchik HZ. Carcinoembryonic antigen (CEA) in the diagnosis of pancreatic cancer. Cancer. 1973;31:324–327. doi: 10.1002/1097-0142(197302)31:2<324::aid-cncr2820310208>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 7.Kaushal S, McElroy MK, Luiken GA, Talamini MA, Moossa AR, Hoffman RM, Bouvet M. Fluorophore-conjugated anti-CEA antibody for the intraoperative imaging of pancreatic and colorectal cancer. J Gastrointest Surg. 2008;12:1938–1950. doi: 10.1007/s11605-008-0581-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tiefenauer LX, Kuhne G, Andres RY. Antibody-magnetite nanoparticles: in vitro characterization of a potential tumor-specific contrast agent for magnetic resonance imaging. Bioconjug Chem. 1993;4:347–352. doi: 10.1021/bc00023a007. [DOI] [PubMed] [Google Scholar]
- 9.Maruyama K, Takahashi N, Tagawa T, Nagaike K, Iwatsuru M. Immunoliposomes bearing polyethyleneglycol-coupled Fab' fragment show prolonged circulation time and high extravasation into targeted solid tumors in vivo. FEBS Lett. 1997;413:177–180. doi: 10.1016/s0014-5793(97)00905-8. [DOI] [PubMed] [Google Scholar]
- 10.Pan J, Yang Q. Antibody-functionalized magnetic nanoparticles for the detection of carcinoembryonic antigen using a flow-injection electrochemical device. Anal Bioanal Chem. 2007;388:279–286. doi: 10.1007/s00216-007-1224-0. [DOI] [PubMed] [Google Scholar]
- 11.Mastrobattista E, Koning GA, Storm G. Immunoliposomes for the targeted delivery of antitumor drugs. Adv Drug Deliv Rev. 1999;40:103–127. doi: 10.1016/s0169-409x(99)00043-5. [DOI] [PubMed] [Google Scholar]
- 12.Jackson H, Bacon L, Pedley RB, Derbyshire E, Field A, Osbourn J, Allen D. Antigen specificity and tumour targeting efficiency of a human carcinoembryonic antigen-specific scFv and affinity-matured derivatives. Br J Cancer. 1998;78:181–188. doi: 10.1038/bjc.1998.462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yazaki PJ, Shively L, Clark C, Cheung CW, Le W, Szpikowska B, Shively JE, Raubitschek AA, Wu AM. Mammalian expression and hollow fiber bioreactor production of recombinant anti-CEA diabody and minibody for clinical applications. J Immunol Methods. 2001;253:195–208. doi: 10.1016/s0022-1759(01)00388-x. [DOI] [PubMed] [Google Scholar]
- 14.Yazaki PJ, Wu AM, Tsai SW, Williams LE, Ikler DN, Wong JY, Shively JE, Raubitschek AA. Tumor targeting of radiometal labeled anti-CEA recombinant T84.66 diabody and t84.66 minibody: comparison to radioiodinated fragments. Bioconjug Chem. 2001;12:220–228. doi: 10.1021/bc000092h. [DOI] [PubMed] [Google Scholar]
- 15.Xing Y, Chaudry Q, Shen C, Kong KY, Zhau HE, Chung LW, Petros JA, O'Regan RM, Yezhelyev MV, Simons JW, Wang MD, Nie S. Bioconjugated quantum dots for multiplexed and quantitative immunohistochemistry. Nat Protoc. 2007;2:1152–1165. doi: 10.1038/nprot.2007.107. [DOI] [PubMed] [Google Scholar]
- 16.Yoon TJ, Yu KN, Kim E, Kim JS, Kim BG, Yun SH, Sohn BH, Cho MH, Lee JK, Park SB. Specific targeting, cell sorting, and bioimaging with smart magnetic silica core-shell nanomaterials. Small. 2006;2:209–215. doi: 10.1002/smll.200500360. [DOI] [PubMed] [Google Scholar]
- 17.Goldstein JL, Anderson RG, Brown MS. Coated pits, coated vesicles, and receptor-mediated endocytosis. Nature. 1979;279:679–685. doi: 10.1038/279679a0. [DOI] [PubMed] [Google Scholar]
- 18.Jiang W, Kim BY, Rutka JT, Chan WC. Nanoparticle-mediated cellular response is size-dependent. Nat Nanotechnol. 2008;3:145–150. doi: 10.1038/nnano.2008.30. [DOI] [PubMed] [Google Scholar]
- 19.Kim YJ, Bang S, Park JY, Park SW, Chung JB, Song SY. Phase II study of 5-fluorouracil and paclitaxel in patients with gemcitabine-refractory pancreatic cancer. Cancer Chemo Pharma. 2009;63:529–533. doi: 10.1007/s00280-008-0822-7. [DOI] [PubMed] [Google Scholar]
- 20.Zhang L, Radovic-Moreno AF, Alexis F, Gu FX, Basto PA, Bagalkot V, Jon S, Langer RS, Farokhzad OC. Co-delivery of hydrophobic and hydrophilic drugs from nanoparticle-aptamer bioconjugates. ChemMedChem. 2007;2:1268–1271. doi: 10.1002/cmdc.200700121. [DOI] [PubMed] [Google Scholar]