

Abstract
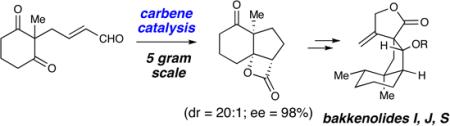 A general strategy for the catalytic asymmetric syntheses of the bakkenolides is reported. The key bond-forming step involves an N-heterocyclic carbene catalyzed desymmetrization of a 1,3-diketone to form three new bonds in one step with excellent enantio– and diastereoselectivity. This intramolecular reaction allows direct access to the hydrindane core of the bakkenolides family and enables a facile synthesis of these natural products.
A general strategy for the catalytic asymmetric syntheses of the bakkenolides is reported. The key bond-forming step involves an N-heterocyclic carbene catalyzed desymmetrization of a 1,3-diketone to form three new bonds in one step with excellent enantio– and diastereoselectivity. This intramolecular reaction allows direct access to the hydrindane core of the bakkenolides family and enables a facile synthesis of these natural products.
The bakkanes are a large class of sesquiterpene natural products containing a characteristic cisfused 6,5-bicyclic core.1 The first isolation of a bakkane was reported in 1968, and ensuing investigations have identified over fifty members in this family.2 The bakkenolides are biogenetically related to the eremophilanes, and the conversion of the initial decalin core to the 6,5 system is proposed to involve an intriguing oxidative ring contraction.3 These natural products possess a wide spectrum of biological activity including antifeedant effects, platelet aggregation inhibition, and potent inhibitory activity against a variety of tumor cell lines.4 The five contiguous stereocenters around the hydrindane core, including two quaternary carbons and the spiro γ-butyrolactone, present an inspiring challenge to the application of modern asymmetric methodology.
With their compact topology combined with useful biological activity, this natural product family has elicited considerable attention since the total synthesis of (±)-bakkenolide A by Evans.5 The majority of syntheses of the bakkenolides are racemic and have relied on efficient Diels–Alder or [2+2] cycloadditions to build the hydrindane core.6 The asymmetric synthetic approaches reported to date rely primarily on chiral pool technology. Recent asymmetric work by Silva begins with the Wieland–Miescher ketone and is highlighted by a novel ring contraction.7 Two examples reported previously start with carvone8 or employ (−)-N-methylephedrine reduction technology.6b The dearth of catalytic enantioselective approaches to these molecules to date is an opportunity to challenge the boundaries of asymmetric catalysis in the context of synthesis and provide new means to efficiently prepare optically active bakkanes.
The field of N-heterocyclic carbene catalysis has undergone significant development recently9 and its application in complex molecule synthesis is an emerging area.10 Carbene catalysis provides efficient access to a variety of nucleophilic species (e.g., acyl anion, homoenolate, enolate) with high levels of stereoselectivity and is particularly attractive for integration into synthetic strategies as a key bond-forming tactic. We report herein the efficient, enantioselective total syntheses of (−)-bakkenolides I, J and S utilizing a cascade homonenolate protonation/ intramolecular aldol/acylation sequence catalyzed by an N-heterocyclic carbene (NHC).
The key structural element in this family of natural products is the fused 6,5-bicyclic ring system that contains an angular methyl group flanked by tertiary and spiro quaternary carbon atoms. We envisaged the core of these molecules being synthesized using our recently reported NHC-catalyzed desymmetrization of 1,3-diketones.11 This strategy would allow us to control the formation of the key quaternary stereogenic center present in all the bakkenolides starting from readily available material and produce appropriate functional groups for further elaboration.
The synthesis of the required symmetric 1,3-diketone began with the palladium-catalyzed addition of 2-methy-1,3-cyclohexadione (1) to butadiene mono-epoxide. A subsequent TEMPO-catalyzed oxidation of the allylic alcohol furnished the α,β-unsaturated aldehyde (2, 59% for two steps). In the key bonding-forming event, the achiral aldehyde (2) underwent a tandem homoenolate protonation, intramolecular aldol addition, acylation in the presence of 5 mol % A and generated the desired β-lactone 3 in 69% yield.
An interesting facet of this reaction is the isolation of the β-lactone product. In our 2007 work with this desymmetrizing aldol reaction,11 substrates with aromatic ketones readily decarboxylate under the reaction conditions to yield substituted cyclopentene rings.12 This highly selective desymmetrization reaction can be performed on a five gram scale and affords the tricyclic product with excellent levels of diastero– and enantioselectivity (>20:1 dr, 98% ee). The concentration was increased and the reaction temperature was lowered from 40 °C to 35 °C to accommodate larger reaction scales, which slightly improved both the yield and enantioselectivity.
With β-lactone 3 in hand, the installation of an oxygen atom directly to the C-9 position was required. As a prelude to this oxidation, the decarboxylation of 3 in the presence of silica gel13 followed by protection of the ketone using Noyori's protocol allowed for the successful installation of the ketal whereas typical Dean-Stark conditions resulted in poor yields.14 A hydroboration of the alkene occured from the convex face of 4, resulting in formation of the secondary alcohol in a high yield (75%) with >20:1 regio– and diastereoselection. The reformation of the ketone was facilitated with p-TsOH in acetone followed by protection of the secondary alcohol with TBSCl and imidazole (98% yield for two steps). A subsequent Wittig olefination of the sterically hindered ketone 6 successfully installed the exo-methylene subunit of 7 (81%).
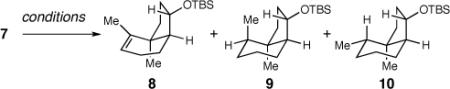
The reduction of the newly formed alkene within 7 to generate the necessary vicinal methyl substituents was not straightforward. A survey of numerous reduction conditions yielded at best a 1:1 ratio of the desired diastereomer 10 and 9. The use of Et3SiH in combination with Pd/C and H2 led to an increase in 10, but also promoted isomerization to the endocyclic olefin (8).15 This observation proved fortuitous since alkene 8 could be independently converted to 10 and 9 in a 6:1 ratio in the presence of Pd/C and hydrogen at elevated pressure. With these results in hand, we explored conditions to promote the isomerization cleanly since this reaction followed by the stereoselective reduction would be the superior route (7 to 8 to 10). Exposure to mild reduction conditions such as PdBaSO4 in the presence of H2 improved the ratio but the results were not satisfactory. Attempts at isomerization with elevated reaction temperatures also failed to improve the efficiency of the reaction. Finally, we found that 1 mol % of Crabtree's catalyst ([(cod)(pyr)(PCy3)Ir]PF6)16 successfully provided internal alkene 8 in 99% yield from 7 which allowed us to proceed with the Pd/C reduction to afford 10 (69%). Minimized structures of 8 and 10 (MM2 Spartan) suggest that there are subtle torsional effects that control the facial selectivity of this reduction. The undesired isomer 9 is higher in energy than 10 due to the axial methyl group, and this difference may direct a more late-type transition state for this hydrogenation.
The conversion of silyl ether 10 to ketone 11 was accomplished with TBAF followed by Dess-Martin periodinane (98%, two steps). An acylation of the kinetic enolate with Mander's reagent17 followed by trans-esterification with propargyl alcohol yielded β-keto propargyl ester 12 as a 9:1 mixture of diastereomers.18
At this juncture, the formation of the key butyrolactone using a Conia-ene reaction was investigated (Table 3).19 While this method was quite effective with β-ketoesters with the alkyne previously installed on the α-carbon, to our knowledge there are no reports of the cyclization occurring from the propargyl ester. Unfortunately, this route led to only minor amounts of product.
Table 3.
Investigation of Lactone Formation
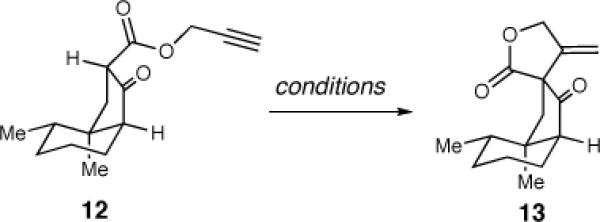 | |||
|---|---|---|---|
| entry | conditions | yielda | drb |
| 1 | 5 mol % (PPh3)AuCl, AgOTf, CH2Cl2 | <5 | – |
| 2 | 5 mol % [(PPh3)Au)3O]BF4, HOTf, DCE | <5 | – |
| 3 | Mn(OAc)3•H2O, EtOH | 70 | 20:1 |
Isolated yield;
As determined by 1H NMR spectroscopy.
After this approach proved unsuccessful, we resorted to the ring closure protocol and final synthetic sequence initially developed by Greene and Déspres.6b The exposure of 12 to Mn(OAc)3 promoted a highly-diastereoselective cyclization, thereby providing the spirocyclic lactone in good yield (70%) as a single diastereomer.20,21 A chemoselective reduction of the ketone using SmI2 provided β-hydroxy ester 14 in 89% yield and 20:1 dr. The relative stereochemistry of 14 was confirmed by X-ray analysis.22
The construction of the spiro lactone was high yielding and selective, but produced the undesired epimer at the C7 position. Fortunately, the β-hydroxy lactone skeleton of 14 allows for a possible retro-aldol/aldol process to access the desired diastereomer. Following the elegant prior reports on the bakkenolides using this strategy by Greene,23,6d the treatment of 14 with TBAF promotes a C7-C9 scission followed by an intramolecular aldol reaction between the intermediate lactone enolate and transient aldehyde to afford a 5:1 mixture of (−)-bakkenolide S (15) and 14 (72%).24 This thermodynamically-driven process favors 15 presumably due to the placement of the hydroxyl group on the convex face and then hydrogen bonding between this alcohol and the lactone carbonyl oxygen. Due to the challenges separating the bakkenolide S/14 mixture by standard chromatography, the transformation of alcohol 14 to (−)-bakkenolides I (16) and J (17) via bakkenolide S was accomplished by direct addition of either iso-butyryl chloride (69%) or iso-valeryl chloride (64%) to the unpurified TBAF reaction.
In summary, a catalytic enantioselective strategy for the syntheses of the bakkenolide core has been established. These studies demonstrate the utility of the intramolecular carbene-catalyzed desymmetrization reaction which provides the bakkane scaffold by forming three new bonds in a single step in 98% ee and >20:1 dr from a simple achiral precursor. The elaboration of the hydrindane architecture through Deprés and Greene's lactone closure and retro-adol sequence, the natural enantiomers of bakkenolide I, J and S. Since the successful carbene-catalysis strategy does not rely on starting from chiral pool materials, it provides the impetus for us to explore these compounds and related structural analogs in biological and medical settings. Our continued studies using carbene catalysis in total synthesis to construct target molecules with stereochemical control and expediency will be reported in due course.
Supplementary Material
Scheme 1.

Synthetic Approach
Scheme 2.

NHC-Catalyzed Desymmetrization
Scheme 3.

Construction of Olefin 7
Scheme 4.

Construction of Alcohol 14
Scheme 5.

Synthesis of Bakkenolides I, J, and S
Table 2.
Formation of Olefin 8
 | ||
|---|---|---|
| entry | conditions | 8:9:10a |
| 1 | Pd/C, H2, EtOH | 0:1:1 |
| 2 | PtO2, H2, MeOH | 0:2:1 |
| 3 | Pd/C, H2, PhH, Et3SiH | 1:1:2 |
| 4 | PdBaSO4, H2, EtOH | 2:1:1 |
| 5 | Pd/C, H2, toluene, 110 °Cb | NR |
| 6 | (cod)(pyr)(PCy3)IrPF6, H2 | 1:0:0c |
Ratio determined by 1H NMR spectroscopy;
Reaction exposed to H2 for 5 minutes and was then sealed;
99% isolated yield.
Acknowledgments
Support for this work was provided by NIGMS (RO1GM73072), Amgen, the Sloan Foundation, GlaxoSmithKline and AstraZeneca. E.M.P is a recipient of a 2008-2009 ACS Division of Organic Chemistry fellowship. We thank Drs. Steve Wittenberger and Thad Franczyk for insightful discussions.
Footnotes
Supporting Information Available: Experimental procedures and spectral data for all new compounds, (PDF). This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.(a) Silva LF. Synthesis. 2001:671. [Google Scholar]; (b) Brocksom TJ, Brocksom U, Constantino MG. Quim. Nova. 2008;31:937. [Google Scholar]
- 2.(a) Abe N, Onoda R, Shirahat K, Kato T, Woods MC, Kitahara Y. Tetrahedron Lett. 1968:369–373. [Google Scholar]; (b) Wu TS, Kao MS, Wu PL, Lin FW, Shi LS, Liou MJ, Li CY. Chem. Pharm. Bull. 1999;47:375–382. doi: 10.1248/cpb.47.375. [DOI] [PubMed] [Google Scholar]
- 3.(a) ref 2.; (b) Marshall JA, Cohen GM. J. Am. Chem. Soc. 1971;36:877–882. [Google Scholar]
- 4.(a) Jamieson GR, Reid EH, Turner BP, Jamieson AT. Phytochemistry. 1976;15:1713–1715. [Google Scholar]; (b) Kano K, Hayashi K, Mitsuhashi H. Chem. Pharm. Bull. 1982;30:1198–1203. [Google Scholar]
- 5.Evans DA, Sims CL, Andrews GC. J. Am. Chem. Soc. 1977;99:5453–5461. [Google Scholar]
- 6.(a) Greene AE, Deprés JP, Coelho F, Brocksom TJ. J. Org. Chem. 1985;50:3943–3945. [Google Scholar]; (b) Greene AE, Coelho F, Deprés JP, Brocksom TJ. Tetrahedron Lett. 1988;29:5661–5662. [Google Scholar]; (c) Back TG, Nava-Salgado VO, Payne JE. J. Org. Chem. 2001;66:4361–4368. doi: 10.1021/jo015624h. [DOI] [PubMed] [Google Scholar]; (d) Hamelin O, Deprés J-P, Greene AE, Tinant B, Declercq J-P. J. Am. Chem. Soc. 1996;118:9992–9993. [Google Scholar]; (e) Hamelin O, Deprés J-P, Heidenhain S, Greene AE. Nat. Prod. Lett. 1997;10:99–103. [Google Scholar]; (f) Hamelin O, Wang Y, Deprés J-P, Greene AE. Angew. Chem. Int. Ed. 2000;39:4314–4316. doi: 10.1002/1521-3773(20001201)39:23<4314::AID-ANIE4314>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]; (g) Brocksom TJ, Coelho F, Depres JP, Greene AE, de Lima MEF, Hamelin O, Hartmann B, Kanazawa AM, Wang YY. J. Am. Chem. Soc. 2002;124:15313–15325. doi: 10.1021/ja0208456. [DOI] [PubMed] [Google Scholar]
- 7.Carneiro VMT, Ferraz HMC, Vieira TO, Ishikawa EE, Silva LF. J. Org. Chem. 2010;75:2877–2882. doi: 10.1021/jo100108b. [DOI] [PubMed] [Google Scholar]
- 8.(a) Jiang CH, Bhattacharyya A, Sha CK. Org. Lett. 2007;9:3241–3243. doi: 10.1021/ol071124k. [DOI] [PubMed] [Google Scholar]; (b) Hartmann B, Kanazawa AM, Deprés J-P, Greene AE. Tetrahedron Lett. 1993;34:3875–3876. [Google Scholar]
- 9.(a) Enders D, Balensiefer T. Acc. Chem. Res. 2004;37:534–541. doi: 10.1021/ar030050j. [DOI] [PubMed] [Google Scholar]; (b) Nair V, Bindu S, Sreekumar V. Angew. Chem. Int. Ed. 2004;43:5130–5135. doi: 10.1002/anie.200301714. [DOI] [PubMed] [Google Scholar]; (c) Marion N, Diez-Gonzalez S, Nolan IP. Angew. Chem. Int. Ed. 2007;46:2988–3000. doi: 10.1002/anie.200603380. [DOI] [PubMed] [Google Scholar]; (d) Phillips EM, Chan A, Scheidt KA. Aldrichimica Acta. 2009;42:55–66. [PMC free article] [PubMed] [Google Scholar]
- 10.For total syntheses employing carbene catalysis, see: Trost BM, Shuey CD, Dininno F. J. Am. Chem. Soc. 1979;101:1284–1285. Harrington PE, Tius MA. J. Am. Chem. Soc. 2001;123:8509–8514. doi: 10.1021/ja011242h. Takikawa H, Suzuki K. Org. Lett. 2007;9:2713–2716. doi: 10.1021/ol070929p. Koyama Y, Yamaguchi R, Suzuki K. Angew. Chem. Int. Ed. 2008;47:1084–1087. doi: 10.1002/anie.200704625. Nicolaou KC, Nold AL, Li HM. Angew. Chem. Int. Ed. 2009;48:5860–5863. doi: 10.1002/anie.200902509.
- 11.Wadamoto M, Phillips EM, Reynolds TE, Scheidt KA. J. Am. Chem. Soc. 2007;129:10098–10099. doi: 10.1021/ja073987e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.For the first report of this decarboxylation in carbene-catalyzed reactions to yield cyclopentenes (although prurportedly through a homoenolate and not enolate pathway), see, Nair V, Vellalath S, Poonoth S, Suresh E. J. Am. Chem. Soc. 2006;128:8736–8737. doi: 10.1021/ja0625677.
- 13.Danheiser RL, Nowick JS. J. Org. Chem. 1991;56:1176–1185. [Google Scholar]
- 14.Tsunoda T, Suzuki M, Noyori R. Tetrahedron Lett. 1980;21:1357–1358. [Google Scholar]
- 15.Yeung YY, Chein RJ, Corey EJ. J. Am. Chem. Soc. 2007;129:10346–10347. doi: 10.1021/ja0742434. [DOI] [PubMed] [Google Scholar]
- 16.Krel M, Lallemand JY, Guillou C. Synlett. 2005:2043–2046. [Google Scholar]
- 17.Mander LN, Sethi SP. Tetrahedron Lett. 1983;24:5425–5428. [Google Scholar]
- 18.DeCicco CP, Buckle RN. J. Org. Chem. 1992;57:1005–1008. [Google Scholar]
- 19.Kennedy-Smith JJ, Staben ST, Toste FD. J. Am. Chem. Soc. 2004;126:4526–4527. doi: 10.1021/ja049487s. [DOI] [PubMed] [Google Scholar]
- 20.See reference 6 for the use of Mn(OAc)3 ring closures in the synthesis of the bakkenolides. For a review on Mn(III)-based reactions, see: Snider BB. Chem. Rev. 1996;96:339–363. doi: 10.1021/cr950026m.
- 21.For select references on α-carbonyl radical cyclizations relevant to the bakkenolides, see: Sha CK, Chiu RT, Yang CF, Yao NT, Tseng WH, Liao FL, Wang SL. J. Am. Chem. Soc. 1997;119:4130–4135. Sha CK, Santhosh KC, Lih SH. J. Org. Chem. 1998;63:2699–2704. doi: 10.1021/jo9723570.
- 22.See supporting information.
- 23.Hamelin O, Wang Y, Deprés JP, Greene AE. Angew. Chem. Int. Ed. 2000;39:4314–4316. doi: 10.1002/1521-3773(20001201)39:23<4314::AID-ANIE4314>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 24.The synthesis of bakkenolide S using this sequence was reported by Deprés and Greene (see reference 6g) but without any characterization data of the final product.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.