

Abstract
Esophageal squamous cell carcinoma (ESCC) is one of the most aggressive human cancers, and novel treatment modalities are required. We investigated the therapeutic potential of the tumor necrosis factor-related apoptosis-inducing ligand (TRAIL/Apo2L) in combination with the proteasome inhibitor bortezomib (Velcade) on human ESCC cell lines. Bortezomib enhanced the susceptibility to TRAIL in 12 of the 15 ESCC cell lines tested, even though most showed low sensitivity to TRAIL as a single agent. The enhancement of TRAIL-induced apoptosis by bortezomib was caspase-dependent. Increased processing of caspase-8 often accompanied enhancement of TRAIL-induced apoptosis by bortezomib. However, the increased cell surface expression of death receptors observed upon bortezomib treatment did not seem to be crucial for this effect. For some ESCC, bortezomib treatment resulted in a more efficient recruitment of caspase-8 and the Fas-associated death domain (FADD) to the death-inducing signaling complex (DISC). Additional downregulation of the cellular FLICE-inhibitory protein long isoform (c-FLIP(L)) could cooperate in the activation of the extrinsic pathway in some cases. For other ESCC, the crucial effect of bortezomib treatment appeared to be increased signaling via the intrinsic apoptotic pathway on subsequent exposure to TRAIL. Thus bortezomib could sensitize ESCC to TRAIL apoptosis by multiple molecular mechanisms of action. Therefore the combination of bortezomib and TRAIL might be a novel therapeutic strategy for ESCC patients who fail to respond to standard chemoradiotherapy that predominantly targets the mitochondrial apoptotic pathway.
Keywords: apoptosis, esophageal cancer, proteasome inhibitor, TRAIL/Apo2L
Introduction
Esophageal squamous cell carcinoma (ESCC) is the third most common cancer of the digestive tract and the seventh leading cause of cancer-related deaths worldwide. ESCC is one of the most aggressive types of cancer (1, 2). In addition to the many difficulties of curative resection, postoperative recurrences are often resistant to the currently available radiochemotherapies. Hence, there is a need for a novel treatment modality involving anti-cancer drugs that selectively target cancer cells and circumvent treatment-resistant pathways for the management of ESCC.
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL/Apo2L) (3, 4) is a promising cytokine for anti-cancer therapy. TRAIL can bind to five receptors: TRAIL-R1 (DR4), TRAIL-R2 (DR5), TRAIL-R3 (decoy receptor [DcR] 1), and TRAIL-R4 (DcR2), which are located at the cell surface; and the secreted protein osteoprotegrin (OPG), which has a lower affinity to the ligand (5). However apoptosis in cancer cells is only induced by binding to death receptor DR4 and DR5 (5). DR4 and DR5 contain death domain (DD) motifs crucial for transmission of an apoptotic signal. DcR1 lacks both cytoplasmic and transmembrane domains, and DcR2 has a truncated death domain, so both are unable to trigger apoptotic death. DcR1 and DcR2 are thought to act as decoys and their expression is higher in normal tissues than in most cancer cells (6). More recently, DcR2 was proposed to act as a regulatory rather than a decoy receptor (7). The preligand assembly domains in DR5 and DcR2 could permit mixed complex formation as a means to negatively regulate apoptosis induction. These findings led to the proposal that TRAIL might be a novel cancer-targeting drug, and the DcRs could account for the resistance to TRAIL-mediated apoptosis in normal tissues.
Clinical trials to characterize the safety and efficacy of recombinant human TRAIL (rhTRAIL) and receptor-targeted agonist antibodies (Abs), both alone and in combination with other cancer therapies, are ongoing (8). The early trial data suggest that rhTRAIL is generally safe and provide preliminary evidence for potential antitumor activity. However, many tumor cell lines are either partially or completely resistant to TRAIL-mediated apoptosis in vitro despite the expression of TRAIL death receptors, thus many studies have explored how this resistance might be overcome in order to improve the clinical outcome (9). Resistance results from a number of factors including increased Akt activity (10, 11), the overexpression of caspase inhibitors (12, 13), Bcl-2 family members (14), or other molecules (12, 15) in the mitochondrial apoptotic pathway, and the overexpression of cellular FLICE-inhibitory protein (c-FLIP) (12, 16, 17). Combination therapies involving several conventional and novel chemotherapeutic drugs have been reported to increase TRAIL-mediated apoptosis in these resistant cells (9, 12, 18–22).
The inhibition of the proteasome is a novel approach for anti-cancer therapy. The proteasome inhibitor bortezomib (Velcade) was recently approved for the treatment of patients with multiple myeloma. The treatment of tumor cells with bortezomib results in multiple biological effects, including inhibition of the cell cycle, increased apoptosis, changes in cell adherence, and inhibition of nuclear factor-κB (NF-κB) activation. Therefore, bortezomib is currently being tested in clinical trials against a variety of solid tumors, and could provide an opportunity for exploring the interaction between proteasome inhibition and other apoptosis-inducing agents in vitro (23, 24). Many recent studies have shown that the treatment of tumor cells with proteasome inhibitors overcomes TRAIL resistance in various human solid cancers including prostate cancer (25), colon cancer (26), glioma (27), non-small-cell lung cancer (NSCLC) (28, 29), and hepatocellular carcinoma (30, 31). However, the molecular mechanism(s) underlying the effects of the combined treatment are largely unknown. In the current study, we examined the efficacy of combined treatment with bortezomib and TRAIL on apoptosis in a number of human ESCC cell lines, and assessed the molecular mechanisms underlying the effects of this combination.
Material and Methods
Tumor cell lines
The KE3, KE4, KE5, KE6, KE7, KE8, and KE10 ESCC cell lines were established in our facility from surgical sections taken at Kurume University Hospital, Japan, as previously described (32, 33). The TE8, TE9, and TE10 cell lines were a generous gift from T. Nishihira (Tohoku University, Sendai, Japan) (32–34). The YES1, YES2, YES3, YES5, and YES6 cell lines were generously provided by T. Murakami (Yamaguchi University, Ube, Japan) (33, 35). Cell lines were maintained in RPMI-1640 supplemented with 10% fetal bovine serum (FBS) and antibiotics (100 U/mL penicillin and 100 μg/mL streptomycin; Gibco, Invitrogen Co., Grand Island, NY) at 37°C in a humidified atmosphere of 5% CO2 and 95% air.
Agents and bortezomib/TRAIL treatment
Soluble rhTRAIL was purchased from Biomol (Plymouth Meeting, PA) and was cross-linked by adding an anti-6 × histidine monoclonal Ab (mAb; R&D Systems, Minneapolis, MN) at a concentration of 2 μg Ab/μg TRAIL. Bortezomib was obtained from Millennium Pharmaceuticals (Cambridge, MA). To determine the effects of TRAIL, bortezomib, and bortezomib/TRAIL, the cancer cells were plated (at 1–2 × 104 cells per well in a flat-bottomed 96-well plate, and 2–4 × 105 cells per well in a six-well plate) and incubated overnight. The cells were either untreated or treated with bortezomib, TRAIL, or both at the indicated concentrations and for the indicated time periods. For the inhibition of NF-κB or general caspase activity, SN50 (Biomol), Bay11-7085 (Calbiochem, San Diego, CA), or zVAD-fmk (R&D Systems) was added to the culture with pretreatment for 2 h at the indicated concentration.
Cell-viability assay and detection of apoptosis
After the incubation with bortezomib/TRAIL treatment, the viable cells were quantified by a 3-[4,5-dimethyiazol-2-yl-5]-[3-carboxymethyloxyphenyl]-2-[4-sulfophenyl]-2H tetrazolium (MTS) assay (Promega, Madison, WI), as described previously (36). The percentage decrease in cell number was calculated as follows: 1 – [absorbance of treated cells – absorbance of media]/[absorbance of untreated cells – absorbance of media] × 100%. For the detection of apoptosis, the cells were stained with hoechst33342 (Dojon, Kumamoto, Japan) and/or fluorescein isothiocynate (FITC)-coupled annexin-V/propidium iodide (PI) using Annexin-V-Fluos staining kit (Roche, Basel, Switzerland). Images were acquired with an IN Cell Analyzer 1000 (GE Healthcare, Piscataway, NJ), and cells with apoptotic morphology of nuclei (condensation/fragmentation) or annexin-V-positive cells were analyzed using the Developer Toolbox software (GE Healthcare). Apoptosis was also evaluated by measuring the enzyme activity of the executioner caspase-3. The caspase-3, caspase-8 and caspase-9 activities were estimated using the Caspase-Glo assay kit (Promega), as previously described (36). Each experiment was carried out in triplicate wells and repeated separately at least three times.
Flow-cytometric analysis of cell-surface expression of TRAIL receptors
After the blocking of nonspecific binding with mouse immunoglobulin G (IgG; Caltag, Burlingame, CA), the cells were stained with mouse anti-human TRAIL-R1, TRAIL-R2, TRAIL-R3, and TRAIL-R4 mAbs (R&D Systems) or isotype-matched mAbs, followed by phycoerythrin (PE)-labeled goat anti-mouse IgG polyclonal Ab (pAb; BD, Pharmingen, San Diego, CA) according to standard procedures. In the experiment shown in Fig. 5A, mouse anti-human TRAIL-R1 (HS101) and TRAIL-R2 (HS201) mAbs from Alexis (San Diego, CA) were used as the primary Ab. The HS201 mAb showed higher sensitivity against DR5 than the other mAb from R&D Systems in our staining conditions. Flow-cytometric analysis of stained cells was performed using the FACSCalibur system (BD Biosciences, Mountain View, CA), and the results were analyzed using CellQuest (BD Biosciences) and FlowJo (TreeStar Inc, San Carlos, CA) software. Three independent experiments were performed with single samples. The expression levels were evaluated based on the values corresponding to a ratio of the signal of the specific TRAIL receptor Ab and the negative isotype-matched control Ab.
FIGURE 5.
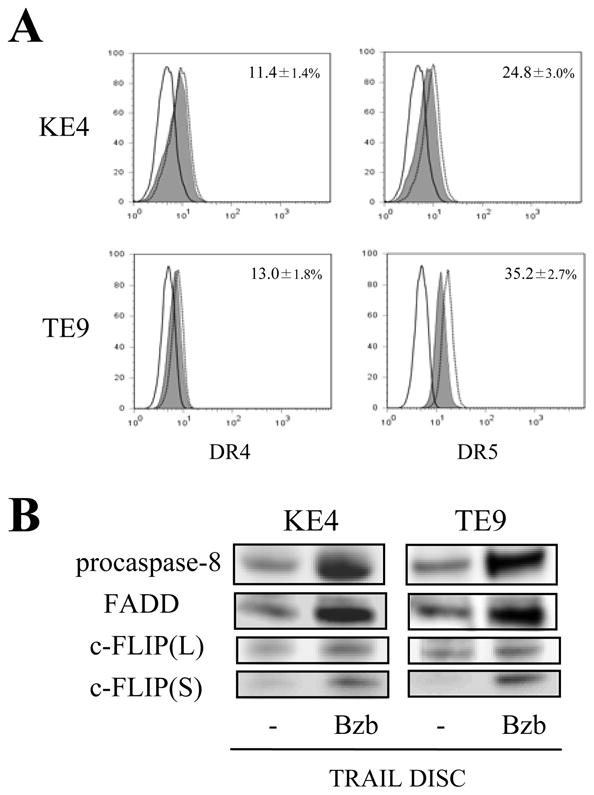
Effects of bortezomib on DISC formation upon TRAIL stimulation. KE4 and TE9 cells were incubated with or without bortezomib (Bzb; 100 nM) for 8 h. (A) Surface expression levels of DR4 and DR5 were analyzed by flow cytometry. Representative histograms from three independent experiments with similar results are shown (solid line, isotype control; shaded area, untreated; dashed line, bortezomib-treated). The percentage increase (average ± SD) of expression with bortezomib is presented as described in “Materials and methods”. (B, C) Cells were further incubated with His-tagged rhTRAIL cross-linked by anti-6 × histidine mAb (1 μg/ml) for 30 min. The cells were lysed and the precipitated DISC was analyzed by western blotting. Representative images from three independent experiments are shown in (B).
Evaluation of mitochondrial membrane depolarization
Cells were incubated with a serum-free culture medium containing 10 μM of 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolycarbocyanine iodide (JC-1) dye (Wako, Osaka, Japan) (36) for 15 min. After washing with phosphate-buffered saline (PBS), the stained cells were observed using an inverted fluorescence microscope (IX81, Olympus, Tokyo, Japan). The red and green fluorescence emissions were separately recorded and merged by DP manager software (Olympus).
Western blot analysis
Western blotting was performed as described previously (17). Monoclonal antibodies for caspase-8, cleaved caspase-3 (Cell Signaling Technology, Beverly, MA), c-FLIP (Alexis), Fas-associated death domain (FADD), Bcl-2, Bax, XIAP, p27 (BD Transduction Laboratories, San Diego, CA), and β-actin (Sigma-Aldrich, St Louis, MO), or polyclonal Abs for caspase-9, PARP, Bcl-XL, Mcl-1 (Cell Signaling Technology), cIAP-1 (R&D), and p21 (Santa Cruz Biotechnology, Santa Cruz, CA) were used as primary Abs. These were followed by appropriate horseradish peroxidase (HRP)-conjugated secondary Abs (GE Healthcare). The Ab–protein complexes were detected using the SuperSignal West Femto Maximum Sensitivity Substrate (Pierce, Woburn, MA). The signals were analyzed using a LAS1000 image analyzer and mutigage software (Fuji Film, Tokyo, Japan).
c-FLIP knockdown by small-interfering RNA (siRNA) transfection
An siRNA pool consisting of four siRNAs specific for the c-FLIP gene (siGENOME SMARTpool reagent, M-003772-06) and control siRNA were purchased from Dharmacon (Lafayette, CO). Target sequences of the c-FLIP gene (RefSeq, NM_003879) are as follows; AAUAACUUCAGGCUCCAUA, GCUAUGAAGUCCAGAAAUU, GAUGUGUCCUCAUUAAUUU, and UAAAGAACAUCCACAGAAU. The transfection of these siRNAs was conducted at a final concentration of 100 nM using the Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA) following the manufacturer’s instructions. Each transfection experiment was performed at least in triplicate and repeated separately at least three times.
Immunoprecipitation of death- inducing signaling complex (DISC)
DISC analysis was performed as previously described with some modifications (19). Briefly, approximately 10 × 106 cells were treated with bortezomib or medium for 8 h, and then His-tagged rhTRAIL cross-linked by an anti-6 × histidine mAb was added to the cultures at a concentration of 1 μg/mL. After 30 min incubation with TRAIL, the cells were intensively washed with cold PBS, and cell lysates were prepared and subjected to immunoprecipitation with a protein G column (Pierce). The precipitated samples were tested for assembled DISC-components which contains protein complex with receptor-ligand (TRAIL) aggregation by western blot analysis as described above. HRP-conjugated anti-mouse IgG Ab (TrueBlot™; eBioscience, San Diego, CA) was used as the secondary Ab.
Results
TRAIL sensitization with bortezomib in ESCC cell lines
Most human ESCC cell lines were resistant or showed a low sensitivity to TRAIL-mediated apoptosis. The KE6 cell line was the only one to show more than 50% reduction of cell number following TRAIL treatment even at the high concentration, 500 ng/mL (Fig. 1A). All human ESCC expressed DR4 and/or DR5 on the cell surface. Although the sensitivities could have been related to the surface expression levels of these TRAIL receptors or the balance between DR4/DR5 and DcR2, there seemed to be no clear direct correlation between level of surface receptor expression and sensitivity to TRAIL (Table I).
FIGURE 1.

Enhancement of sensitivity to TRAIL in ESCC cell lines. (A) 15 ESCC cell lines were seeded on 96-well plates, and treated with TRAIL at 20, 100, and 500 ng/mL for 18 h. (B) The cells were treated with bortezomib (100 nM) or medium alone for 2 h, and subjected to a further 16–18 h of incubation with or without the addition of TRAIL (100 ng/mL). (C) KE4, TE8, and TE9 cells were incubated in the various concentrations of bortezomib (◆ 0, □ 5, ▲ 20, and ○ 100 nM) and TRAIL as described in (B) in the presence or absence of zVAD-fmk (100 μM). Cell viability was assessed by an MTS assay, and the percentage decrease in cell number (±SD) compared to the medium control is shown in (A–C). (D) TE9 cells were also stained by annexin-V-FITC (green), PI (red), and hoechst33342 (blue) at 6 h and 18 h after the addition of TRAIL. Representative images with bortezomib (100 nM) and TRAIL (500 ng/mL) are shown with the values (±SD) of % apoptotic cells as described in the “Material and methods”. B, botrezomib; T, TRAIL.
Table I.
Expression levels of TRAIL receptors
| KE3 | KE4 | KE5 | KE6 | KE8 | KE10 | TE8 | TE9 | TE10 | YES1 | YES2 | YES5 | YES6 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DR4 | - | 3.0 | 1.3 | 1.5 | 3.2 | 2.1 | 1.5 | 3.6 | 1.3 | 1.2 | 1.3 | 1.3 | 1.4 | 2.8 |
| Bzb | 2.9 | 1.6 | N.T. | N.T. | 2.4 | N.T. | 2.5 | 1.4 | 1.1 | N.T. | 1.4 | N.T. | N.T. | |
| DR5 | - | 1.6 | 1.1 | 1.5 | 1.5 | 1.4 | 1.6 | 1.7 | 1.3 | 1.0 | 1.3 | 1.2 | 1.3 | 1.7 |
| Bzb | 2.0 | 1.3 | N.T. | N.T. | 3.3 | N.T. | 2.6 | 1.7 | 1.1 | N.T. | 1.5 | N.T. | N.T. | |
| DcR1 | - | 1.0 | 1.0 | 1.0 | 1.1 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 |
| Bzb | 1.0 | 1.0 | N.T. | N.T. | 1.1 | N.T. | 1.0 | 1.0 | 1.0 | N.T. | 1.0 | N.T. | N.T. | |
| DcR2 | - | 1.2 | 1.0 | 1.1 | 1.0 | 1.6 | 1.1 | 3.2 | 1.0 | 1.1 | 1.5 | 1.1 | 1.3 | 1.4 |
| Bzb | 1.5 | 1.0 | N.T. | N.T. | 2.5 | N.T. | 6.8 | 1.1 | 1.4 | N.T. | 2.1 | N.T. | N.T. |
Constitutive surface expression of TRAIL receptors was examined in 13 ESCC lines. In 7 cell lines, effect of bortezomib (100 nM) on the receptor expression was also tested after 18 h treatment. Cells were stained with anti-DR4, anti-DR5, anti-DcR1, and anti-DcR2 mAbs, and subjected to flow-cytometric analysis. The values indicate the relative surface-expression levels of receptors, as assessed by mean channel fluorescence (MFI), compared with the negative control (set at 1.0). Representative results from three independent experiments with similar results are shown. Bzb, bortezomib; N.T., not tested.
We then tested whether the proteasome inhibitor bortezomib could influence TRAIL sensitivity in 15 human ESCC cell lines. Bortezomib at a concentration of 100 nM was added to the cultures 2 h before treatment with TRAIL (100 ng/mL) for 16–18 h. All of the cell lines showed decrease in cell numbers when treated with a combination of TRAIL and bortezomib compared with TRAIL alone. Furthermore, in 12 of the cell lines (that is, all except TE10, YES2, and YES3) this combination resulted in a greater reduction in cell number than additive effects of the individual agents (Fig. 1B). These results indicate that bortezomib could cooperate with TRAIL and increase the susceptibility of ESCC cell lines to TRAIL-mediated apoptotis.
Cooperative effect of bortezomib and TRAIL on caspase activities
To assess the mechanisms underlying the effects of bortezomib on TRAIL sensitization, we further analyzed three cell lines (KE4, TE8, and TE9) that were refractory to TRAIL as a single agent and showed great enhancement of TRAIL sensitivity in the presence of bortezomib. This enhancement was blocked by the pan-caspase inhibitor zVAD-fmk in all three cell lines (Fig. 1C). Figure 1D also shows in TE9 cells that the combined treatment resulted in a significant induction of apoptotic cell death as determined by annexin-V and nuclear staining, which was completely inhibited by zVAD-fmk, indicating that this process was caspase dependent. Similar profiles were detected in KE4 and TE8 cells (data not shown).
The initial critical step in TRAIL signaling after the aggregation of the receptor with its cognate ligands is thought to be the dimerization and activation of pro-caspase-8 enzyme activity in the DISC (37), resulting in the subsequent accumulation of the cleaved form of caspase-8 (38, 39). The apoptotic signal can then diverge into two pathways. The first involves the direct activation of caspase-3 without the involvement of mitochondria (termed as extrinsic apoptosis pathway). The second involves perturbations at the mitochondria due to truncated Bid generation, resulting in the formation of apoptosomes and activation of caspase-9 (intrinsic or mitochondrial pathway) with subsequent activation of caspase-3. This caspase-based signaling was subjected to western blot analysis (Fig. 2A). With TRAIL alone, caspase-8 showed little activation and processing into cleaved forms (43/41 and 18 kDa), while the combined treatment greatly enhanced the processing of caspase-8 in the KE4 and TE9 cell lines. An increased activation of caspase-9 and caspase-3 by the accumulation of cleaved 37/35 and 19/17 kDa forms respectively, as well as increased PARP cleavage was also observed with the combined treatment. In contrast to the KE4 and TE9 cell lines, the TE8 cell line showed similar cleavage of caspase-8 following treatment with TRAIL alone or the combined treatment. However caspase-9 and caspase-3 were only significantly processed in TE8 following the combined treatment. Mitochondrial function as assessed by JC-1 dye staining showed that treatment with both drugs together, but not with either drug alone, produced significant depolarization of the mitochondrial membrane in the TE8 cell line. This indicated that only the combined treatment could efficiently elicit activation of the mitochondria-dependent apoptosis pathway in TE8 cells (Fig. 2B). A quantitative analysis of caspase enzyme activities upon 2 h TRAIL stimulation after bortezomib pretreatment was consistent with the western blot data in all three cell lines (Fig. 2C). Taken together, these findings suggest that amplification of caspase-8 activation might be important to trigger the complementary enhancement of cell death between bortezomib and TRAIL in the KE4 and TE9 cell lines. By contrast, the major molecular mechanism of bortezomib sensitization to TRAIL-mediated apoptosis in TE8 appeared to be further downstream at the mitochondrial level, indicating that multiple molecular mechanism(s) of TRAIL sensitization by bortezomib could occur even among ESCC isolated from different individual patients.
FIGURE 2.

Caspase activities with bortezomib/TRAIL treatment in KE4, TE8, and TE9 cells. Cells were incubated in the presence or absence of bortezomib (100 nM for KE4 and TE9 cells, and 20 nM for TE8 cells) for 2 h prior to the addition of vehicle or TRAIL (100 ng/mL). (A) After a further 6 h, cell extracts were prepared, and western blotting was performed with anti-caspase-8, anti-caspase-9, anti-cleaved caspase-3, anti-PARP, and anti-β-actin Abs. (B) Cells were incubated with JC-1 dye during the last 15 min of culture. Mitochondrial depolarization was evaluated as indicated by a decrease in the red-to-green fluorescence intensity ratio. Representative results for TE8 cells are shown. (C) Cells were incubated in the presence or absence of bortezomib for 6–8 h. TRAIL was added for a further 2 h (500 ng/mL), and the enzyme activities of caspase-8, caspase-9, and caspase-3 in the cells were measured as described in the “Materials and methods”. B, bortezomib; T, TRAIL; CL, cleaved form.
Upregulation of death receptors by treatment with bortezomib and its partial contribution to TRAIL-induced apoptosis
The surface expression levels of TRAIL receptors were tested after overnight treatment with bortezomib in seven of the ESCC cell lines: five cell lines (KE3, KE4, KE8, TE8, and TE9) that showed increased TRAIL sensitivity with bortezomib, and two cell lines (TE10 and YES2) that showed no circumvention of TRAIL resistance with bortezomib (Table I). Increased cell surface levels of DR5 were seen on all of the cell lines following bortzomib treatment, although the upregulation of DR5 in the TE10 cell line was marginal. Four of the cell lines (KE4, KE8, TE9, and YES2) showed increased DR4 expression according to flow-cytometric analysis. By contrast, the surface expression of DcR2 was also increased significantly in five cell lines (KE3, KE8, TE8, TE10, and YES2) and marginally in one cell line (TE9). The TE8 cell line showed marked enhancement of TRAIL sensitivity with bortezomib treatment, despite an increase of DcR2 expression that was significantly greater than that of DR4 and/or DR5. This finding was consistent with several previous studies that failed to show a good correlation between expression of decoy receptors and TRAIL sensitivity (40–42). To further investigate any contribution of death receptor upregulation to TRAIL sensitization by bortezomib, a “wash kill” experiment was performed, as previously reported by Koschny et al. (27, 31, 43). KE4 and TE9 cells were pretreated with TRAIL in order to occupy the TRAIL receptors already present on the cell surface, followed by thorough washing to remove all unbound TRAIL after 1 h. The cells were then treated with bortezomib either alone or in combination with TRAIL for 12 h, and were subjected to an apoptosis assay (Fig. 3A). Pretreatment with TRAIL followed by the addition of bortezomib without further TRAIL, which activated the preexisting but not the upregulated receptors after bortezomib treatment, significantly enhanced the activity of caspase-3 although to a lesser extent than when further TRAIL was added. These findings suggest that the upregulation of DR4 and DR5 partially contributed to, but was not crucial for, the TRAIL sensitization by bortezomib in both KE4 and TE9 cells.
FIGURE 3.

(A) Contribution of bortezomib-induced upregulation of death receptors to TRAIL-induced apoptosis. KE4 and TE9 cells were incubated with TRAIL at 500 ng/mL for 1 h and then washed five times with medium to remove unbound TRAIL. Cells were left untreated or sensitized with bortezomib (100 nM) for 12 h, either with or without the further addition of TRAIL (500 ng/mL). Apoptotic cells were evaluated by an active caspase-3 assay as described in the “Materials and methods”. (B) Expression of c-FLIP and FADD proteins following bortezomib treatment. Cells were incubated in the presence or absence of various concentrations of bortezomib for 18 h. Cell extracts were then prepared, and western blotting was performed with anti-c-FLIP, anti-FADD, and anti-β-actin Abs. B, bortezomib (Bzb); T, TRAIL.
Effects of c-FLIP modification by bortezomib on TRAIL sensitivity
We previously reported that the reduction of c-FLIP expression correlated with TRAIL sensitization in mouse and human renal cancer cell lines (16, 17). Western blot analysis of whole cell lysates revealed that c-FLIP long isoform (c-FLIP(L)) expression was reduced in the KE4 cell line, and increased in the TE8 cell line, with no significant change seen in the TE9 cell line following bortezomib treatment (Fig. 3B). By contrast, the c-FLIP short isoform (c-FLIP(S)) was elevated in all the cell lines tested. The total cellular level of FADD expression did not change at the various concentrations of bortezomib tested in all three cell lines. To evaluate the involvement of c-FLIP expression in TRAIL-mediated apoptosis, we used c-FLIP siRNAs to block c-FLIP function. c-FLIP-targeting siRNAs and control siRNAs were transfected into the KE4, TE8, and TE9 cell lines. TE8 had undetectable amounts of both c-FLIP(L) and c-FLIP(S) proteins (data not shown). However, protein levels of c-FLIP(L) were reduced on average (±standard deviation [SD]) by 73.2±11.2% in KE4 and 84.1±9.5% in TE9. By contrast, c-FLIP(S) levels in KE4 and TE9 cells were not obviously changed by the siRNAtreatment (Fig. 4A). Interestingly these reductions of c-FLIP(L) using siRNA demonstrated that c-FLIP(L) could protect the two ESCC lines from TRAIL apoptosis (Fig. 4B). Furthermore, these findings suggest that reduced levels of c-FLIP(L) observed in KE4 cells in response to bortezomib were likely to be of importance for increasing caspase-8 activation on exposure to TRAIL.
FIGURE 4.

Enhanced susceptibility to TRAIL-induced apoptosis in bortezomib-treated cells following specific downregulation of c-FLIP with siRNAs. KE4 and TE9 cells were transfected with siRNAs targeting c-FLIP or control (Ctrl) siRNAs, and cultured for 48 h. (A) Cells were lysed and analyzed for c-FLIP and β-actin expression by western blotting. (B) Transfected cells were incubated in the presence or absence of bortezomib (100 nM) for 2 h, and for a further 10–12 h with the addition of TRAIL at various concentrations. % apoptotic cells (±SD) were assessed after hoechst33342 nuclear staining as described in the “Materials and methods” (Ctrl siRNA: ◇ vehicle, ◆ bortezomib, c-FLIP siRNA: □ vehicle, ■ bortezomib). B, bortezomib.
Enhancement of DISC formation upon TRAIL stimulation by bortezomib pretreatment
Since our results suggested that increased caspase-8 activation might be crucial for the combined treatment of the KE4 and TE9 cell lines, we analyzed the effects of bortezomib on DISC assembly upon TRAIL stimulation in KE4 and TE9 cells. After 8 h pretreatment with bortezomib, the cell surface levels of expression of DR4, as assessed by changes in mean channel fluorescene (MFI), were only marginally increased (KE4, 11.4±1.4%; TE9, 13.0±1.8%). Also, only slight increases in cell surface levels of DR5 (KE4, 24.8±3.0%; TE9, 35.2±2.7%) were observed (Fig. 5A). Following immunoprecipitation, DISC components were subjected to western blot analysis. As shown in Fig. 5B, significantly enhanced recruitment of FADD at the DISC was observed in both the KE4 and TE9 cell lines following bortezomib pretreatment (fold increase in densitometric analysis: KE4, 3.13±0.77; TE9, 3.40±0.23). In addition, caspase-8 was significantly recruited at the DISC in these cells. Although bortezomib treatment did seem to also result in increases of c-FLIP at the DISC, which might be due in part to increased expression level of c-FLIP(S) with bortezomib treatment (Fig. 3B), these changes did not counteract the increased activation of caspase-8. These findings suggest that bortezomib preferentially favored recruitment of FADD and procaspase-8 to this DISC, and this increase may be sufficient to subsequently increased triggering of the extrinsic apoptotic pathway.
Bortezomib-induced changes in other proteins involved in apoptosis
Since in the TE8 cell line bortezomib did not enhance caspase-8 activation in response to TRAIL, effects on the intrinsic apoptotic pathway could be involved in enhanced TRAIL-induced apoptosis in this case. Numerous proteins have been reported to block or promote the apoptotic cascade of TRAIL (9). Interestingly inhibitors of NF-κB other than bortezomib did not sensitize ESCC to TRAIL apoptosis (data not shown), in agreement with previous reports showing that blocking NF-κB activation is not always critical for the sensitization of tumor cells to TRAIL (17, 30, 44, 45). We therefore tested the expression of apoptosis-related proteins by western blotting after bortezomib treatment of the KE4, TE8, and TE9 cell lines (Fig. 6). There were no major changes in the levels of Bcl-2, Bcl-XL, Bax, or XIAP in any of the three cell lines. Interestingly levels of cIAP-1 were reduced to some extent in the TE8 cell line, which would be consistent with bortezomib affecting downstream apoptotic signaling in this particular ESCC. However, Mcl-1, p21 and p27 were increased following bortezomib treatment in all ESCC tested. This would be expected since the levels of all of these proteins are tightly controlled by the proteasome. Since Mcl-1 is reported to be a inhibitor of mitochondrial-mediated TRAIL apoptosis (15), the observed increases could argue against a crucial contribution of intrinsic apoptotic signaling following bortezomib exposure for ESCC. Bortezomib-induced increases in p21 and p27 could be accompanied with cell cycle arrest at G2/M phase in ESCC (data not shown) that may also contribute to TRAIL sensitization as previously described (25). However, increases in p21 and p27 also occurred in ESCC that remain resistant to bortezomib and TRAIL apoptosis (data not shown). Therefore, the importance of cell-cycle inhibition by bortezomib on TRAIL sensitization should be investigated in future studies.
FIGURE 6.

Effects of bortezomib on the expression of various proteins involved in apoptosis or cell cycle arrest. KE4, TE8, and TE9 cells were treated with various concentrations of bortezomib (Bzb) for 18 h, and the cell extracts were then prepared. The given proteins were detected using western blot analysis.
Discussion
The effects of TRAIL have not been studied as intensively in ESCC as in other cancers (20–22, 46, 47). However, the relative resistance of ESCC to TRAIL-mediated apoptosis seen in the current study was consistent with a previous report by Kondo et al. (20). Several previous reports showed that combination treatment with agents such as cisplatin, gefitinib, and taurolidine could overcome TRAIL resistance in ESCC (20–22, 46). However, to our knowledge the effects of proteasome inhibitors on TRAIL sensitivity of ESCC have not been previously reported. Our data clearly indicated that bortezomib could enhance the susceptibility to TRAILin most ESCC lines tested.
Interestingly, the molecular mechanism(s) underlying bortezomib sensitization to TRAIL seemed to differ among individual ESCC. Thus, in the KE4 and TE9 cell lines bortezomib treatment resulted in increased caspase-8 activation following TRAIL exposure as described in many previous reports (25, 28, 29, 36), and this amplified caspase-8 activation seemed crucial for the bortezomib sensitization. Liu et al. reported that bortezomib alone induced apoptosis with caspase-8 activation in human NSCLC cells (28). However no activation of caspase-8 following bortezomib treatment alone was observed in our current study with ESCC. The activation of caspase-8 might have been partially due to an increase in the surface expression of DR4 and/or DR5. However, our “wash kill” experiments indicated that proteasome inhibitor-mediated upregulation of DR4 and DR5 was not crucial for TRAIL sensitization, consistent with previous reports for leukemias, gliomas and hepatocellular carcinomas (27, 30, 31, 43).
Recently, Ganten et al. reported that caspase-8 and FADD were more efficiently recruited to the DISC of hepatocellular carcinoma cells pretreated with another proteasome inhibitor, MG132, despite the presence of fewer death receptors and more c-FLIP in the DISC (30). We also observed significantly enhanced recruitment of caspase-8 and FADD to the DISC in both the KE4 and TE9 cell lines. In previous studies bortezomib has also been reported to reduce cellular levels of c-FLIP (17, 27, 31) in some cancer cells, whereas others have not observed changes in c-FLIP (29, 48). In our current study, the effect of bortezomib on c-FLIP(L) expression varied between individual ESCC lines. Since bortezomib could facilitate a greatly enhanced recruitment of caspase-8 and FADD to the DISC, this may be its dominant effect in promoting apoptotic signaling, thus overriding any concomitant inhibitory effects of c-FLIP. Recent findings suggest that the glycosylation state of the TRAIL receptors can influence their ability to active caspase-8 (49). Furthermore, the ubiquitination of caspase-8 can also influence the strength of the apoptotic signal (50). Therefore the effects of bortezomib on these processes would certainly be of interest for future studies.
Proteasome inhibition has also been reported to induce multiple molecular changes influencing components of the mitochondrial apoptosis signaling pathway thus promoting TRAIL-mediated apoptosis (23, 24, 26, 44). However others have questioned the importance of these effects for TRAIL-mediated apoptosis (36). Our findings indicate that the relative importance of bortezomib effects on components of the mitochondrial pathway for promoting TRAIL-mediated apoptosis of ESCC can vary, even between individual patients tumor samples. Taken together, our findings indicate that the mechanism of TRAIL sensitization with bortezomib typically involved increased activation of both caspase-8 and caspase-9. This amplified apoptotic signaling could override any bortezomib-induced increase in antiapoptotic proteins such as Mcl-1 or c-FLIP. In addition, the p53 gene is mutated in KE4 and TE8 (33), and a truncated form of p53 is expressed in TE9 as a result of a frameshift accompanied by functional inactivation of the p53 protein (34). Therefore, bortezomib sensitization to TRAIL-mediated apoptosis is independent of p53 in these ESCC.
In conclusion, TRAIL-mediated apoptosis was efficiently promoted in ESCC by combined therapy with rhTRAIL and bortezomib, which could activate both the extrinsic and intrinsic pathways of apoptosis. Combined TRAIL therapy could therefore be a novel therapeutic strategy for use in ESCC patients who fail to respond to standard chemoradiotherapy that mainly triggers the intrinsic apoptotic pathway.
Acknowledgments
This study was supported by Grant-in-Aid for Scientific Research from JSPS, and by “High-Tech Research Center” Project for Private Universities: matching fund subsidy from MEXT. This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract N01-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government. This Research was supported [in part] by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health.
References
- 1.Enzinger PC, Ilson DH, Kelsen DP. Chemotherapy in esophageal ca cer. Semin Oncol. 1999;26(5 Suppl 15):12–20. [PubMed] [Google Scholar]
- 2.Enzinger PC, Mayer RJ. Esophageal cancer. N Engl J Med. 2003;349(23):2241–52. doi: 10.1056/NEJMra035010. [DOI] [PubMed] [Google Scholar]
- 3.Wiley SR, Schooley K, Smolak PJ, et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity. 1995;3(6):673–82. doi: 10.1016/1074-7613(95)90057-8. [DOI] [PubMed] [Google Scholar]
- 4.Pitti RM, Marsters SA, Ruppert S, Donahue CJ, Moore A, Ashkenazi A. Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J Biol Chem. 1996;271(22):12687–90. doi: 10.1074/jbc.271.22.12687. [DOI] [PubMed] [Google Scholar]
- 5.Wang S, El-Deiry WS. TRAIL and apoptosis induction by TNF-family death receptors. Oncogene. 2003;22(53):8628–33. doi: 10.1038/sj.onc.1207232. [DOI] [PubMed] [Google Scholar]
- 6.Kim K, Fisher MJ, Xu SQ, el-Deiry WS. Molecular determinants of response to TRAIL in killing of normal and cancer cells. Clin Cancer Res. 2000;6(2):335–46. [PubMed] [Google Scholar]
- 7.Clancy L, Mruk K, Archer K, et al. Preligand assembly domain-mediated ligand-independent association between TRAIL receptor 4 (TR4) and TR2 regulates TRAIL-induced apoptosis. Proc Natl Acad Sci U S A. 2005;102(50):18099–104. doi: 10.1073/pnas.0507329102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ashkenazi A. Targeting the extrinsic apoptosis pathway in cancer. Cytokine Growth Factor Rev. 2008;19(3–4):325–31. doi: 10.1016/j.cytogfr.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 9.Thorburn A, Behbakht K, Ford H. TRAIL receptor-targeted therapeutics: resistance mechanisms and strategies to avoid them. Drug Resist Updat. 2008;11(1–2):17–24. doi: 10.1016/j.drup.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bortul R, Tazzari PL, Cappellini A, et al. Constitutively active Akt1 protects HL60 leukemia cells from TRAIL-induced apoptosis through a mechanism involving NF-kappaB activation and cFLIP(L) up-regulation. Leukemia. 2003;17(2):379–89. doi: 10.1038/sj.leu.2402793. [DOI] [PubMed] [Google Scholar]
- 11.Elrod HA, Lin YD, Yue P, et al. The alkylphospholipid perifosine induces apoptosis of human lung cancer cells requiring inhibition of Akt and activation of the extrinsic apoptotic pathway. Mol Cancer Ther. 2007;6(7):2029–38. doi: 10.1158/1535-7163.MCT-07-0004. [DOI] [PubMed] [Google Scholar]
- 12.Ricci MS, Kim SH, Ogi K, et al. Reduction of TRAIL-induced Mcl-1 and cIAP2 by c-Myc or sorafenib sensitizes resistant human cancer cells to TRAIL-induced death. Cancer Cell. 2007;12(1):66–80. doi: 10.1016/j.ccr.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 13.Cummins JM, Kohli M, Rago C, Kinzler KW, Vogelstein B, Bunz F. X-linked inhibitor of apoptosis protein (XIAP) is a nonredundant modulator of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis in human cancer cells. Cancer Res. 2004;64(9):3006–8. doi: 10.1158/0008-5472.can-04-0046. [DOI] [PubMed] [Google Scholar]
- 14.Fulda S, Meyer E, Debatin KM. Inhibition of TRAIL-induced apoptosis by Bcl-2 overexpression. Oncogene. 2002;21(15):2283–94. doi: 10.1038/sj.onc.1205258. [DOI] [PubMed] [Google Scholar]
- 15.Kim SH, Ricci MS, El-Deiry WS. Mcl-1: a gateway to TRAIL sensitization. Cancer Res. 2008;68(7):2062–4. doi: 10.1158/0008-5472.CAN-07-6278. [DOI] [PubMed] [Google Scholar]
- 16.Brooks AD, Sayers TJ. Reduction of the antiapoptotic protein cFLIP enhances the susceptibility of human renal cancer cells to TRAIL apoptosis. Cancer Immunol Immunother. 2005;54(5):499–505. doi: 10.1007/s00262-004-0595-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sayers TJ, Brooks AD, Koh CY, et al. The proteasome inhibitor PS-341 sensitizes neoplastic cells to TRAIL-mediated apoptosis by reducing levels of c-FLIP. Blood. 2003;102(1):303–10. doi: 10.1182/blood-2002-09-2975. [DOI] [PubMed] [Google Scholar]
- 18.Ganten TM, Haas TL, Sykora J, et al. Enhanced caspase-8 recruitment to and activation at the DISC is critical for sensitisation of human hepatocellular carcinoma cells to TRAIL-induced apoptosis by chemotherapeutic drugs. Cell Death Differ. 2004;11 (Suppl 1):S86–96. doi: 10.1038/sj.cdd.4401437. [DOI] [PubMed] [Google Scholar]
- 19.Lacour S, Micheau O, Hammann A, et al. Chemotherapy enhances TNF-related apoptosis-inducing ligand DISC assembly in HT29 human colon cancer cells. Oncogene. 2003;22(12):1807–16. doi: 10.1038/sj.onc.1206127. [DOI] [PubMed] [Google Scholar]
- 20.Kondo K, Yamasaki S, Sugie T, et al. Cisplatin-dependent upregulation of death receptors 4 and 5 augments induction of apoptosis by TNF-related apoptosis-inducing ligand against esophageal squamous cell carcinoma. Int J Cancer. 2006;118(1):230–42. doi: 10.1002/ijc.21283. [DOI] [PubMed] [Google Scholar]
- 21.Tsai WS, Yeow WS, Chua A, et al. Enhancement of Apo2L/TRAIL-mediated cytotoxicity in esophageal cancer cells by cisplatin. Mol Cancer Ther. 2006;5(12):2977–90. doi: 10.1158/1535-7163.MCT-05-0514. [DOI] [PubMed] [Google Scholar]
- 22.Teraishi F, Kagawa S, Watanabe T, et al. ZD1839 (Gefitinib, ‘Iressa’), an epidermal growth factor receptor-tyrosine kinase inhibitor, enhances the anti-cancer effects of TRAIL in human esophageal squamous cell carcinoma. FEBS Lett. 2005;579(19):4069–75. doi: 10.1016/j.febslet.2005.06.031. [DOI] [PubMed] [Google Scholar]
- 23.Adams J. The development of proteasome inhibitors as anticancer drugs. Cancer Cell. 2004;5(5):417–21. doi: 10.1016/s1535-6108(04)00120-5. [DOI] [PubMed] [Google Scholar]
- 24.Orlowski RZ, Kuhn DJ. Proteasome inhibitors in cancer therapy: lessons from the first decade. Clin Cancer Res. 2008;14(6):1649–57. doi: 10.1158/1078-0432.CCR-07-2218. [DOI] [PubMed] [Google Scholar]
- 25.Lashinger LM, Zhu K, Williams SA, Shrader M, Dinney CPN, McConkey DJ. Bortezomib abolishes tumor necrosis factor-related apoptosis-inducing ligand resistance via a p21-dependent mechanism in human bladder and prostate cancer cells. Cancer Research. 2005;65(11):4902–8. doi: 10.1158/0008-5472.CAN-04-3701. [DOI] [PubMed] [Google Scholar]
- 26.Nikrad M, Johnson T, Puthalalath H, Coultas L, Adams J, Kraft AS. The proteasome inhibitor bortezomib sensitizes cells to killing by death receptor ligand TRAIL via BH3-only proteins Bik and Bim. Molecular Cancer Therapeutics. 2005;4(3):443–9. doi: 10.1158/1535-7163.MCT-04-0260. [DOI] [PubMed] [Google Scholar]
- 27.Koschny R, Holland H, Sykora J, et al. Bortezomib sensitizes primary human astrocytoma cells of WHO grades I to IV for tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis. Clinical Cancer Research. 2007;13(11):3403–12. doi: 10.1158/1078-0432.CCR-07-0251. [DOI] [PubMed] [Google Scholar]
- 28.Liu X, Yue P, Chen S, et al. The proteasome inhibitor PS-341 (bortezomib) up-regulates DR5 expression leading to induction of apoptosis and enhancement of TRAIL-induced apoptosis despite up-regulation of c-FLIP and survivin expression in human NSCLC cells. Cancer Research. 2007;67(10):4981–8. doi: 10.1158/0008-5472.CAN-06-4274. [DOI] [PubMed] [Google Scholar]
- 29.Voortman J, Resende TP, Abou El Hassan MAI, Giaccone G, Kruyt FAE. TRAIL therapy in non-small cell lung cancer cells: Sensitization to death receptor-mediated apoptosis by proteasome inhibitor bortezomib. Molecular Cancer Therapeutics. 2007;6(7):2103–12. doi: 10.1158/1535-7163.MCT-07-0167. [DOI] [PubMed] [Google Scholar]
- 30.Ganten TM, Koschny R, Haas TL, et al. Proteasome inhibition sensitizes hepatocellular carcinoma cells, but not human hepatocytes, to TRAIL. Hepatology. 2005;42(3):588–97. doi: 10.1002/hep.20807. [DOI] [PubMed] [Google Scholar]
- 31.Koschny R, Ganten TM, Sykora J, et al. TRAIL/bortezomib cotreatment is potentially hepatotoxic but induces cancer-specific apoptosis within a therapeutic window. Hepatology. 2007;45(3):649–58. doi: 10.1002/hep.21555. [DOI] [PubMed] [Google Scholar]
- 32.Nakao M, Yamana H, Imai Y, et al. HLA A2601-restricted CTLs recognize a peptide antigen expressed on squamous cell carcinoma. Cancer Res. 1995;55(19):4248–52. [PubMed] [Google Scholar]
- 33.Fujii T, Kato S, Yamana H, et al. Expression of G1 cell cycle markers and the effect of adenovirus-mediated overexpression of p21Waf-1 in squamous cell carcinoma of the esophagus. Int J Oncol. 2001;18(1):157–63. [PubMed] [Google Scholar]
- 34.Barnas C, Martel-Planche G, Furukawa Y, Hollstein M, Montesano R, Hainaut P. Inactivation of the p53 protein in cell lines derived from human esophageal cancers. Int J Cancer. 1997;71(1):79–87. doi: 10.1002/(sici)1097-0215(19970328)71:1<79::aid-ijc14>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 35.lizuka N, Miyamoto K, Tangoku A, et al. Downregulation of intracellular nm23-H1 prevents cisplatin-induced DNA damage in oesophageal cancer cells: possible association with Na(+), K(+)-ATPase. Br J Cancer. 2000;83(9):1209–15. doi: 10.1054/bjoc.2000.1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shanker A, Brooks AD, Tristan CA, et al. Treating metastatic solid tumors with bortezomib and a tumor necrosis factor-related apoptosis-inducing ligand receptor agonist antibody. Journal of the National Cancer Institute. 2008;100(9):649–62. doi: 10.1093/jnci/djn113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kischkel FC, Hellbardt S, Behrmann I, et al. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. Embo J. 1995;14(22):5579–88. doi: 10.1002/j.1460-2075.1995.tb00245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Donepudi M, Mac Sweeney A, Briand C, Grutter MG. Insights into the regulatory mechanism for caspase-8 activation. Mol Cell. 2003;11(2):543–9. doi: 10.1016/s1097-2765(03)00059-5. [DOI] [PubMed] [Google Scholar]
- 39.Boatright KM, Renatus M, Scott FL, et al. A unified model for apical caspase activation. Mol Cell. 2003;11(2):529–41. doi: 10.1016/s1097-2765(03)00051-0. [DOI] [PubMed] [Google Scholar]
- 40.Griffith TS, Lynch DH. TRAIL: a molecule with multiple receptors and control mechanisms. Curr Opin Immunol. 1998;10(5):559–63. doi: 10.1016/s0952-7915(98)80224-0. [DOI] [PubMed] [Google Scholar]
- 41.Zhang XD, Franco A, Myers K, Gray C, Nguyen T, Hersey P. Relation of TNF-related apoptosis-inducing ligand (TRAIL) receptor and FLICE-inhibitory protein expression to TRAIL-induced apoptosis of melanoma. Cancer Res. 1999;59(11):2747–53. [PubMed] [Google Scholar]
- 42.Leverkus M, Neumann M, Mengling T, et al. Regulation of tumor necrosis factor-related apoptosis-inducing ligand sensitivity in primary and transformed human keratinocytes. Cancer Res. 2000;60(3):553–9. [PubMed] [Google Scholar]
- 43.Koschny R, Sykora J, Walczak H, et al. Bortezomib-mediated up-regulation of TRAIL-R1 and TRAIL-R2 is not necessary for but contributes to sensitization of primary human glioma cells to TRAIL [3] Clinical Cancer Research. 2007;13(21):6541–2. [Google Scholar]
- 44.Leverkus M, Sprick MR, Wachter T, et al. Proteasome inhibition results in TRAIL sensitization of primary keratinocytes by removing the resistance-mediating block of effector caspase maturation. Mol Cell Biol. 2003;23(3):777–90. doi: 10.1128/MCB.23.3.777-790.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nencioni A, Wille L, Dal Bello G, et al. Cooperative cytotoxicity of proteasome inhibitors and tumor necrosis factor-related apoptosis-inducing ligand in chemoresistant Bcl-2-overexpressing cells. Clinical Cancer Research. 2005;11(11):4259–65. doi: 10.1158/1078-0432.CCR-04-2496. [DOI] [PubMed] [Google Scholar]
- 46.Daigeler A, Chromik AM, Geisler A, et al. Synergistic apoptotic effects of taurolidine and TRAIL on squamous carcinoma cells of the esophagus. Int J Oncol. 2008;32(6):1205–20. doi: 10.3892/ijo_32_6_1205. [DOI] [PubMed] [Google Scholar]
- 47.Kim K, Nakagawa H, Fei P, Rustgi AK, El-Deiry WS. Targeting Bcl-xL in esophageal squamous cancer to sensitize to chemotherapy plus TRAIL-induced apoptosis while normal epithelial cells are protected by blockade of caspase 9. Cell Death Differ. 2004;11(5):583–7. doi: 10.1038/sj.cdd.4401388. [DOI] [PubMed] [Google Scholar]
- 48.Johnson TR, Stone K, Nikrad M, et al. The proteasome inhibitor PS-341 overcomes TRAIL resistance in Bax and caspase 9-negative or Bcl-xL overexpressing cells. Oncogene. 2003;22(32):4953–63. doi: 10.1038/sj.onc.1206656. [DOI] [PubMed] [Google Scholar]
- 49.Wagner KW, Punnoose EA, Januario T, et al. Death-receptor O-glycosylation controls tumor-cell sensitivity to the proapoptotic ligand Apo2L/TRAIL. Nat Med. 2007;13(9):1070–7. doi: 10.1038/nm1627. [DOI] [PubMed] [Google Scholar]
- 50.Jin Z, Li Y, Pitti R, et al. Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell. 2009;137(4):721–35. doi: 10.1016/j.cell.2009.03.015. [DOI] [PubMed] [Google Scholar]