

Abstract
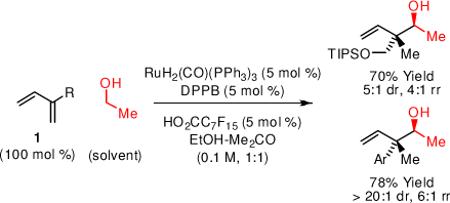 Under ruthenium catalyzed transfer hydrogenation conditions, direct C-C coupling of ethanol and 2-substituted dienes occurs to furnish products of hydro-hydroxyethylation: anti-configured neopentyl homoallylic alcohols. Identical adducts are generated from acetaldehyde under related conditions employing isopropanol as reductant.
Under ruthenium catalyzed transfer hydrogenation conditions, direct C-C coupling of ethanol and 2-substituted dienes occurs to furnish products of hydro-hydroxyethylation: anti-configured neopentyl homoallylic alcohols. Identical adducts are generated from acetaldehyde under related conditions employing isopropanol as reductant.
The majority of chemical commodities are made from rapidly depleting petrochemical feedstocks. The development of byproduct-free catalytic C-C bond forming processes that exploit abundant, renewable resources would assist in defining a sustainable paradigm for chemical production.1 With annual U.S. production now exceeding 10 billion gallons (2009),2 ethanol is vastly abundant, yet its direct use as a C2 building block in homogeneous catalytic C-C coupling is largely unexplored.3 Here, as part of a broad effort toward hydrogen-mediated C-C bond formations beyond hydroformylation,4 we report the direct C-C coupling of ethanol and 2-substituted dienes to furnish products of hydroxyethylation: a ruthenium catalyzed C-C bond forming transfer hydrogenation (Scheme 1). This method enables diastereoselective formation of all-carbon quaternary centers under catalytic conditions in the absence of pre-metallated nucleophiles or resulting stoichiometric metallic byproducts. To our knowledge, this process, which enables direct, byproduct-free access to anti-configured neopentyl homoallylic alcohols, has no stereoselective counterpart in conventional allylmetal chemistry.5
Scheme 1.

Carbonyl addition via ruthenium catalyzed C-C coupling of dienes.
aFor oxidative ruthenium catalyzed diene-alcohol C-C coupling to form β,γ-enones, see reference 7c.
In prior studies from our laboratory it was found that iridium and ruthenium catalysts promote hydrogen exchange between alcohols and π-unsaturated reactants to generate nucleophile-electrophile pairs that engage in byproduct-free C-C coupling.4 This new pattern of reactivity availed the opportunity to explore the direct activation of renewable alcohols, such as methanol and ethanol, as C1 and C2 building blocks in catalytic C-C couplings to π-unsaturated reactants. Our initial efforts toward direct activation of methanol proved unfruitful, likely due to the fact that methanol dehydrogenation is energetically demanding.6 Indeed, whereas attempted C-C couplings of methanol fail, corresponding reactions of paraformaldehyde employing isopropanol as terminal reductant proceed readily, as demonstrated in ruthenium catalyzed hydroxymethylations of 1,1-disubstituted allenes and 2-substituted dienes.7 b,d As ethanol dehydrogenation occurs more readily than the dehydrogenation of methanol (ΔH = +68 vs. +84 kJ/mol, respectively),6 the direct activation of ethanol in couplings to 2-substituted diene 1c was explored.7,8,9,10,11
Gratifyingly, using RuH(O2CC7F15)(CO)(dppb)(PPh3) as catalyst,12 a complex that was effective in related diene-formaldehyde couplings,7b a 78% isolated yield of C-C coupling product as a 6:1 mixture of constitutional isomers 2c and 3c. Notably, 2c appears as a single diastereomer. Thus, C-C coupling occurs at the 2-position of the diene resulting in diastereoselective formation of an all carbon quaternary center. In most cases, regioisomers 2c and 3c differ substantially in polarity and are easily separated by silica gel chromatography. Under these conditions, ethanol was coupled to dienes 1a-1i. With the exception of myrcene 1f and diene 1h, constitutional isomers 2 are the major products formed. Diastereoselectivities for adducts 2a-2i range between 4:1 to > 20:1 in favor of the indicated anti-isomer (Scheme 2). The stereochemical assignment of adducts 2a-2i is made in analogy to that determined for the product obtained from the coupling of benzyl alcohol to myrcene 1f.13
Scheme 2.

Ruthenium catalyzed coupling of ethanol to 2-substituted dienes 1a-1i.a
aCited yields are of material isolated by silica gel chromatography. Conditions: (a) 80 °C, 20 h; (b) 90 °C, 20 h; (c) 90 °C, 40 h; (d) 100 °C, 40 h. See Supporting Information for detailed experimental procedures.
For most C-C bond forming transfer hydrogenations developed in our laboratory,4,7 carbonyl addition is possible from the alcohol or aldehyde oxidation level. In the latter case, a stoichiometric reductant such as isopropanol or formic acid is required. Accordingly, it was found that the reductive coupling of dienes 1a-1i to acetaldehyde can be conducted using the same ruthenium catalyst under essentially identical conditions employing isopropanolacetone (1:1) as solvent to furnish an equivalent set of adducts 2a-2i with similar trends in regio- and diastereoselectivity 2a-2i (Scheme 3).
Scheme 3.

Ruthenium catalyzed coupling of acetaldehyde to 2-substituted dienes 1a-1i.a
aConditions: (a) 90 °C, 20 h; (b) 100 °C, 40 h. Otherwise, as described in Table 1.
Our collective data reveal that regioselectivity varies in response to steric features of the aldehyde, with small aldehydes such as formaldehyde7b delivering the greatest proportion of isomers 2. These data are consistent with a Curtin-Hammett scenario involving rapid interconversion of isomeric π-allyls A and B.14 Here, the relative energies of the competing transition structures for carbonyl addition determine regioselectivity. If one presumes a chair-like transition structure, additions from π-allyl B by way of (E)- and (Z)-σ-allyls B are likely disfavored due to strain arising from non-bonded interactions between the pseudo-axially oriented “R-substituent” with groups attached to the ruthenium center. Similarly, the transition state for carbonyl addition from π-allyl A by way of the (Z)-σ-allyl A is likely disfavored due to pseudo-axial orientation of the “R-substituent”. In contrast to all other pathways, carbonyl addition from the (E)-σ-allyl A involves pseudo-equatorial placement of the “R-substituent”, which directs preferential formation of anti-isomers 2. Only through rapid interconversion of π-allyls and σ-allyls A and B can the minimum energy pathway en route to anti-isomers 2 be traversed (Scheme 4).
Scheme 4.

Regio- and diastereoselective hydro-hydroxyethylation via selective carbonyl addition from isomeric ruthenium π-allyl and σ-allyl intermediates.a
In summary, we report a direct catalytic C-C coupling of ethanol, an abundant, renewable alcohol, which results in the diastereoselective formation of anti-configured homoallylic alcohols possessing all carbon quaternary centers. These studies represent an important step toward the long-term objective of defining catalytic systems for the byproduct-free C-C coupling of abundant alcohols (methanol and ethanol) to α-olefins.15 Future studies will focus on the development related hydro-hydroxyalkylations, including enantioselective variants of the process described herein.
Supplementary Material
Acknowledgments
Acknowledgment is made to the Robert A. Welch Foundation and the NIH-NIGMS (RO1-GM069445) for partial support of this research.
Footnotes
Supporting Information Available. Spectral data for all new compounds (1H NMR, 13C NMR, IR, HRMS). This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.For selected reviews on sustainable chemical synthesis, see: Anastas P, Eghbali N. Chem. Soc. Rev. 2010;39:301. doi: 10.1039/b918763b. Sheldon RA. Green Chem. 2007;9:1273. Clark JH. J. Chem. Tech. Biotech. 2007;82:603. Lange J-P. ChemSusChem. 2009;2:587. doi: 10.1002/cssc.200900003.
- 2.Renewable Fuels Association (RFA) http://www.ethanolrfa.org/ (accessed March 1, 2010)
- 3.Ethanol carbonylation to form propionic acid under the conditions of homogenous catalysis has been described: Patil RP, Kelkar AA, Chaudhari RV. J. Mol. Catal. 1988;47:87. Ubale RS, Kelkar AA, Chaudhari RV. J. Mol. Catal. A: Chem. 1997;118:9.
- 4.For selected reviews on C-C bond forming hydrogenation and transfer hydrogenation, see: Shibahara F, Krische MJ. Chem. Lett. 2008;37:1102. doi: 10.1246/cl.2008.1102. Patman RL, Bower JF, Kim IS, Krische MJ. Aldrichim. Acta. 2008;41:95. Bower JF, Kim IS, Patman RL, Krische MJ. Angew. Chem. Int. Ed. 2009;48:34. doi: 10.1002/anie.200802938.
- 5.For isolated examples of stereoselective carbonyl allylboration to furnish syn-configured neopentyl homoallylic alcohols, see: Ely RJ, Morken JP. J. Am. Chem. Soc. 2010;132:2534. doi: 10.1021/ja910750b. reference 13.
- 6.For methanol dehydrogenation, ΔH = +84 kJ/mol. For ethanol dehydrogenation, ΔH = +68 kJ/mol: Qian M, Liauw MA, Emig G. Appl. Catal. A: Gen. 2003;238:211. Lin W-H, Chang H-F. Catal. Today. 2004;97:181.
- 7.For ruthenium catalyzed C-C bond forming transfer hydrogenation developed in our laboratory, see: Shibahara F, Bower JF, Krische MJ. J. Am. Chem. Soc. 2008;130:6338. doi: 10.1021/ja801213x. Smejkal T, Han H, Breit B, Krische MJ. J. Am. Chem. Soc. 2009;131:10366. doi: 10.1021/ja904124b. Shibahara F, Bower JF, Krische MJ. J. Am. Chem. Soc. 2008;130:14120. doi: 10.1021/ja805356j. Ngai M-Y, Skucas E, Krische MJ. Org. Lett. 2008;10:2705. doi: 10.1021/ol800836v. Skucas E, Zbieg JR, Krische MJ. J. Am. Chem. Soc. 2009;131:5054. doi: 10.1021/ja900827p. Grant CD, Krische MJ. Org. Lett. 2009;11:4485. doi: 10.1021/ol9018562. Zbieg JR, McInturff EL. Org. Lett. 2009;11 doi: 10.1021/ol1007235. ASAP-10.1021/ol1007235. Patman RL, Chaulagain MR, Williams VM, Krische MJ. J. Am. Chem. Soc. 2009;131:2066. doi: 10.1021/ja809456u. Williams VM, Leung JC, Patman RL, Krische MJ. Tetrahedron. 2009;65:5024. doi: 10.1016/j.tet.2009.03.068. Patman RL, Williams VM, Bower JF, Krische MJ. Angew. Chem. Int. Ed. 2008;47:5220. doi: 10.1002/anie.200801359.
- 8.For related catalytic C-C couplings that occur by way of nucleophilic ruthenium π-allyls, see: Tsuji Y, Mukai T, Kondo T, Watanabe Y. J. Organomet. Chem. 1989;369:C51. Kondo T, Ono H, Satake N, Mitsudo T.-a, Watanabe Y. Organometallics. 1995;14:1945. Kondo T, Hiraishi N, Morisaki Y, Wada K, Watanabe Y, Mitsudo T.-a. Organometallics. 1998;17:2131. Yu C-M, Lee S, Hong Y-T, Yoon S-K. Tetrahedron Lett. 2004;45:6557. Omura S, Fukuyama T, Horiguchi J, Murakami Y, Ryu I. J. Am. Chem. Soc. 2008;130:14094. doi: 10.1021/ja806929y.
- 9.For selected reviews of ruthenium catalyzed C-C coupling beyond olefin metathesis, see: Trost BM, Toste FD, Pinkerton AB. Chem. Rev. 2001;101:2067. doi: 10.1021/cr000666b. Kondo T, Mitsudo T.-a. Curr. Org. Chem. 2002;6:1163. Dérien S, Monnier F, Dixneuf PH. Top. Organomet. Chem. 2004;11:1.
- 10.For catalytic intermolecular diene-aldehyde reductive coupling, see: Kimura M, Ezoe A, Shibata K, Tamaru Y. J. Am. Chem. Soc. 1998;120:4033. Takimoto M, Hiraga Y, Sato Y, Mori M. Tetrahedron Lett. 1998;39:4543. Kimura M, Fujimatsu H, Ezoe A, Shibata K, Shimizu M, Matsumoto S, Tamaru Y. Angew. Chem. Int. Ed. 1999;38:397. doi: 10.1002/(SICI)1521-3773(19990201)38:3<397::AID-ANIE397>3.0.CO;2-Y. Kimura M, Shibata K, Koudahashi Y, Tamaru Y. Tetrahedron Lett. 2000;41:6789. Kimura M, Ezoe A, Tanaka S, Tamaru Y. Angew. Chem. Int. Ed. 2001;40:3600. doi: 10.1002/1521-3773(20011001)40:19<3600::aid-anie3600>3.0.co;2-n. Loh T-P, Song H-Y, Zhou Y. Org. Lett. 2002;4:2715. doi: 10.1021/ol026216i. Sato Y, Sawaki R, Saito N, Mori M. J. Org. Chem. 2002;67:656. doi: 10.1021/jo0106086. Jang H-Y, Huddleston RR, Krische MJ. Angew. Chem. Int. Ed. 2003;42:4074. doi: 10.1002/anie.200351986. Bareille L, Le Gendre P, Moïse C. Chem. Commun. 2005:775. doi: 10.1039/b414322a. Kimura M, Ezoe A, Mori M, Iwata K, Tamaru Y. J. Am. Chem. Soc. 2006;128:8559. doi: 10.1021/ja0608904. Yang Y, Zhu S-F, Duan H-F, Zhou C-Y, Wang L-X, Zhou Q-L. J. Am. Chem. Soc. 2007;129:2248. doi: 10.1021/ja0693183. Sato Y, Hinata Y, Seki R, Oonishi Y, Saito N. Org. Lett. 2007;9:5597. doi: 10.1021/ol702543m.
- 11.For a recent review encompassing nickel-catalyzed diene-aldehyde reductive coupling, see: Kimuara M, Tamaru Y. Top. Curr. Chem. 2007;279:173.
- 12.RuH(O2CC7F15)(CO)(dppb)(PPh3) is prepared in situ from RuH2(CO)(PPh3)3, HO2CC7F15 and dppb: Dobson A, Robinson SR, Uttley MF. J. Chem. Soc., Dalton Trans. 1974:370.
- 13.Denmark SE, Fu J. J. Am. Chem. Soc. 2001;123:9488. doi: 10.1021/ja016552e. [DOI] [PubMed] [Google Scholar]
- 14.For isomerization of ruthenium π-allyls, see: Xue P, Bi S, Sung HHY, Williams ID, Lin Z, Jia G. Organometallics. 2004;23:4735. reference 7e.
- 15.For related amine-mediated “hydro-aminoalkylation” of α-olefins, see: Herzon SB, Hartwig JF. J. Am. Chem. Soc. 2008;130:14940. doi: 10.1021/ja806367e. Herzon SB, Hartwig JF. J. Am. Chem. Soc. 2007;129:6690. doi: 10.1021/ja0718366. references cited therein.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.