

Abstract
Protease activity measurement has broad application in drug screening, diagnosis and disease staging, and molecular profiling. However, conventional immunopeptidemetric assays (IMPA) exhibit low fluorescence signal-to-noise ratios, preventing reliable measurements at lower concentrations in the clinically important pM∼nM range. Here, we demonstrated a highly sensitive measurement of protease activity using nanoplasmonic resonator (NPR). NPRs enhance Raman signals by 6.1×1010 times in a highly reproducible manner, enabling fast detection of proteolytically active Prostate Specific Antigen (paPSA) activities in real-time, at sensitivity level at 6 pM (0.2 ng/ml) with a dynamic range of 3 orders of magnitude. Experiments on extracellular fluid (ECF) from the paPSA-positive cells demonstrate specific detection in a complex bio-fluid background. This method offers a fast, sensitive, accurate, and one-step approach to detect the proteases activities in very small sample volumes.
Keywords: Plasmonic resonator, Surface Enhanced Raman Scattering, Sensing, Prostate Cancer, Protease
Originally developed in 1928, Raman spectroscopy has been used extensively to characterize molecular properties1. Surface-Enhanced Raman Spectroscopy (SERS) increases the Raman signal significantly2-5 through enhanced electromagnetic fields in close proximity to a surface. Additional enhancement can be obtained by utilizing molecular resonance Raman (RR) effect when the molecule was excited at its absorption band6. SERS measurements performed on dispersed metal nanoparticle aggregates, which is the mostly commonly used SERS substrate7, have demonstrated detection sensitivity up to single molecule level8-10. However, these measurement often suffer from poor reproducibility11. To improve the reproducibility, other methods including self-assembly of metallic colloidal nano-particles,12 nanosphere lithography (NSL) and metal film over nanosphere (MFON)13, electrochemical roughening of polished gold substrate,14 and periodic structured metallic substrate using electron-beam lithography,15 have been developed to fabricate SERS substrate consisting homogeneous features over large area with reproducible enhancement factors up to 108. Although these efforts lead to successful utilization of SERS analysis in many promising applications including gene and protein discrimination,16-18 bio-warfare agents detection19 and real-time glucose monitoring,20 lacking of the ability to fabricate SERS hot-spots at specific location limits application for very small sample volume. To overcome such limit, we recently developed tunable nanoplasmonic resonators (NPRs), consisting of thin SiO2 layer sandwiched between metallic nano-disks.21 The resonance frequency can be precisely tuned by varying the dielectric layer thickness and aspect-ratio of the NPRs. Individual NPRs can enhance the Raman intensity by a factor of 6.1×1010; among the largest values obtained for a single SERS substrate or nanoparticle. Fabricated using well established nanolithography processes, the NPR-based method enables producing SERS hot-spots at desired location in a much smaller dimension reproducibly, allowing multiplexed high throughput detection and lab-on-chip applications.
Prostate cancer biomarker Prostate Specific Antigen (PSA), a kallikrein (hK) family serine protease22, 23, is used as the model protease in this study. The commonly used prostate-specific antigen (PSA) blood test has being widely used for early diagnosis and management of prostate cancer, the leading male cancer.24, 25 However, serum PSA concentrations reflect the presence of benign prostatic hyperplasia (BPH) more often than cancer.26, 27 The lack of specificity causes a high false-positive rate and often leads to costly prostate needle biopsies for diagnosis and post-biopsy complications as well as considerable anxiety.22, 28, 29 Recent research has identified a family of highly specific peptides that can be cleaved by paPSA isoform in xenografts models30 and human samples31, 32 thus, measurement of paPSA protease activity from in vivo samples is possible and would be potentially valuable as a more specific screening agent for prostate cancer and in detection of recurrent disease. However, reported results based on immunopeptidemetric assays (IMPA) exhibit low fluorescence signal-to-noise ratios, preventing reliable measurements at lower concentrations in the clinically important range of 60-300 pM.31, 32 In addition, there is usually a limited number of prostate cancer cells (<1000) isolated from fine needle biopsy or circulating cell capture. No method exists that can perform a paPSA protease activity assay on a small number of cells for clinical staging. Therefore, a key goal of this work is to develop NPR-based method that allows specific and sensitive measurements of paPSA for prostate cancer detection in a very small sample volume.
Results and Discussion
In this work, NPRs (Fig. 1a) were conjugated with a PSA protease-specific substrate peptide, which has the sequence R19-HSSKLQLAAAC30, 33, with the SERS molecule Rhodamine 19 (R19) at the N-terminus and cysteine at the C-terminus (Fig. 1b). The peptide has been identified as a highly specific peptides that can be cleaved by paPSA in vivo in xenografts models30 and human samples31, 32. The paPSA cleaves the peptide, leading to the release of the R19 moiety (Fig, 1c) and a subsequent decrease in the Raman scattering intensity in a dose- and time-dependent manner, and the PSA protease activity can be accurately quantified. It has been theoretically estimated that SERS enhancement is strongly localized to the vicinity of nanoparticle resonator surface (5-10 nm), which effectively eliminates the assay background noise from the Raman scattering substance in the surrounding fluids, or the R19 moieties that diffuse into the solution after protease cleavage. Because of this unique property, the assay can be performed in a simplified one-step format with no additional washing step required.
Figure 1.
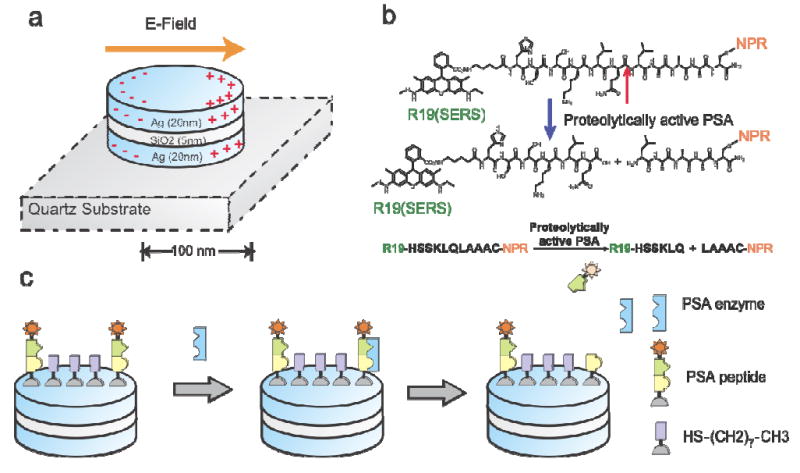
Schematic illustration of the working principle of detecting PSA protease activity using peptide-conjugated NPR SERS nanosensors. (a) NPRs exhibit a tunable plasmon resonance and highly enhanced local electromagnetic field through coupled plasmonic resonance. NPRs with a short axis of 150 nm and long axis of 200 nm were made of multi-stacks of silver and SiO2 layers with thicknesses of 25 nm and 5 nm, respectively. (b) The molecular structure of the biomarker that consists of Raman dye R19, PSA specific peptide sequence HSSKLQLAAAC, and cysteine. The peptide can be cleaved by PSA enzyme between HSSKLQ and LAAAC. (c) The detection scheme of NPR functionalized with peptide sequence HSSKLQLAAAC and the Raman dye R19 (star). The presence of the PSA enzyme will cleave the peptide sequence. After cleavage, the diffusion of the R19 (star) away from the surface will be monitored by the loss of the SERS signatures of the R19 moiety. Packing molecule octanethiol (HS-(CH2)2-CH3) was used to reduce the packing density of the reporting peptide on NPR surface, and thus, allows PSA enzyme to access the reporting peptide.
Under an optical microscope, the NPR arrays were distinctly visible due to the strong scattering of light at their resonant wavelength (Fig. 2a). The magnified views of NPR arrays that measured by Scanning Electron Microscope (SEM) and Atomic Force Microscope (AFM) are showing in Fig. 2b and 2c. The optical properties of the NPR were characterized by illuminating the NPRs with collimated light delivered by a multimode optical fiber from a 150W Xenon lamp (Thermo Oriel) and collecting the extinction spectra using a grating spectrometer (Triax 550, Jobin Yvon) with matched liquid nitrogen cooled CCD detector (CCD-3500, Jobin Yvon).21 The SiO2 layer, sandwiched between the Ag layers, enable precisely tuning of NPR resonance. As shown in Fig. 2d, the measured resonance peak of NPRs-peptide-R19 conjugates closely matches laser excitation wavelength at 532 nm and thus, maximizes the enhancement of Raman scattering. It is worthwhile to note at the laser excitation overlap with the R19 absorption band centered at 517 nm, the resulting resonant Raman scattering may also contribute to the enhanced Raman signal. For the SERS experiments, Raman spectra were measured using a modified inverted microscope (Axiovert 200, Zeiss) with a 50× objective in a backscattering configuration. As shown in Fig. 2e, NPR-based SERS substrate exhibits reproducible Raman spectrum with consistent enhancement factor at same order of magnitude. The variation of experimentally measured the SERS intensities obtained from 6 different NPR array are below 25% and it can be easily normalized in the experiment.
Figure 2.
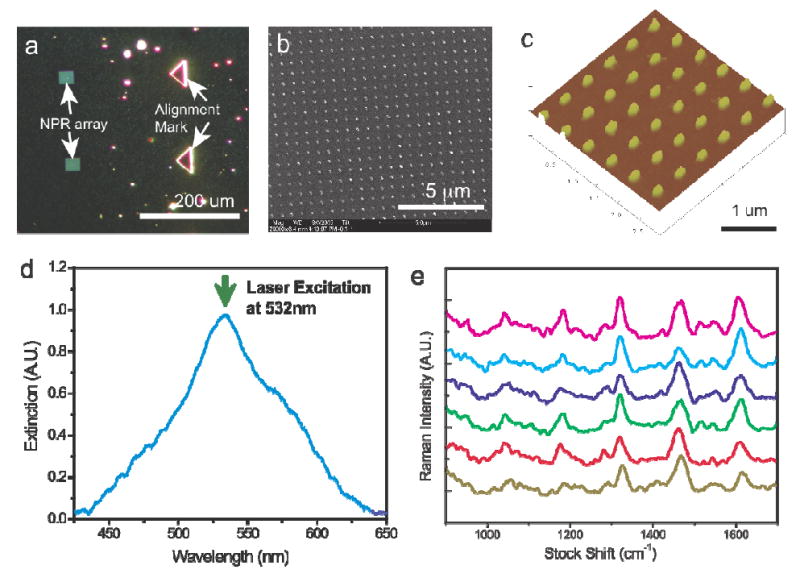
(a) Optical microscopic image of NPRs arrays fabricated using standard e-beam lithography and thin film deposition process. Fabricated NPRS arrays consists of 30 × 30 NPRs with 500 nm spacing. Multiple NPRs arrays and alignment mark can be conveniently fabricated on the same substrate. (b) Magnified image of NPR array measured by Scanning Electron Microscope. Using precision lithography methods, the NPR can be prepared in a controlled manner. (c) Image of NPRs measured at higher magnification using Atomic Force Microscope (AFM). (d) Measured extinction spectrum of an NPR-peptide-R19 conjugate array at a wavelength range of 425 to 650 nm. The resonance peak of the NPR has been tuned to closely match the laser excitation and Raman emission frequencies, and thus, maximize the overall enhancement of the Raman signal. (e) Reproducible Raman spectra of NPR-peptide-R19 conjugates measured from six different NPRs arrays. Integration time is 30 seconds.
The assay was performed by exposing the NPR-peptide-R19 nanosensor to the fluidic samples and the subsequent time-dependent R19 Raman spectra change was recorded at an interval of one minute and an integration time of 30 seconds. The Raman peak at 1316 cm-1 of SERS label molecule (R19) was monitored as the primary signature peak in this study, while the 1456 cm-1, 1526 cm-1, and 1597 cm-1 peaks were also monitored as additional references (Fig. 3). The time-resolved spectral measurement in the presence of PSA (Fig. 3a) is plotted in Fig. 3c. Considering that the SERS intensities are proportional to the remaining R19 on the NPR surface, the normalized SERS intensity change is a direct indicator of PSA activity. Despite the variation in the initial intensity of different Raman signature peaks, the normalized data all converged into the same curve, indicating the normalized SERS intensity change is a reliable measure to quantitatively determine the substrate peptide being cleaved. As shown in Fig. 3c, before PSA addition, the NPR nanosensor showed steady intensities for all the peaks over a 10 minute period. However, upon addition of PSA, a significant decrease in each Raman peak is observed in the first 10-12 minutes, indicating that the PSA protease was able to cleave the peptides on the NPR. At the endpoint of 30 min, the decrease of the Raman signal reaches a plateau at PSA concentration of 6 nM while at lower concentration level (∼pM), the signal continue to decrease at much lower rate. Another serine protease, granzyme B, was selected as a negative control (Fig. 3b,c). Within 50 minutes of recording, no substantial changes in SERS intensity was observed, even at concentrations up to 1 μM. This result demonstrates that the decrease of Raman signal in the PSA assay was based on a genuine enzymatic process, rather than a non-specific hydrolysis reaction or due to the displacement of the peptide from the surface by other components in the buffer.
Figure 3.
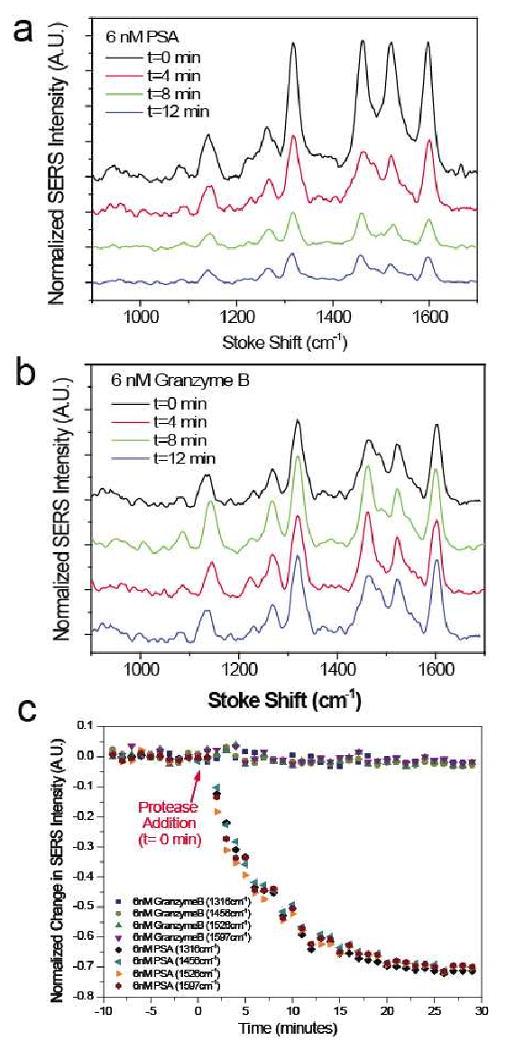
Real-time kinetic measurement of PSA protease activity. (a) SERS spectra for 6 nM PSA incubation taken over 30 minutes with an integration time of 30 seconds. (b) SERS spectra for the negative control, 6 nM granzyme B, taken over 30 minutes. (c) Time-resolved measurements of relative change in Raman peak intensity at 1316 cm-1, 1456 cm-1, 1526 cm-1, and 1597 cm-1 before and after addition of PSA protease. Negative time represents time before protease addition.
The sensitivity of the NPR nanosensor was evaluated by measuring SERS intensity change of set of sample with PSA enzyme concentration ranging from 6 nM to 6 pM. The absolute value of normalized SERS intensity change is shown in Fig. 4a. As expected, the rate of decrease in the Raman signal was proportional to the concentration of PSA, which is explained by a reduced rate of peptide cleavage when the PSA concentrations decreased. The proteolytic activity eventually reaches an equilibrium stage at 30 minutes as indicated by each Raman signal reaching a plateau. The absolute value of normalized decrease in SERS intensity versus various concentrations, after 30 minutes of enzyme addition, is plotted in Fig. 4b. The dynamic range for detection, in the current assay setup, was from 6 pM to 6 nM. PSA concentration higher than 6 nM does not exhibit a distinct difference with the given detection time. As shown in Fig. 4c, the monitored Raman peak intensity exhibits distinguishable decay characteristics at different PSA concentrations during the initial 4 minutes of sampling. Thus, by fitting the decaying rate, reliable assays can possibly be accomplished in 4 minutes.
Figure 4.
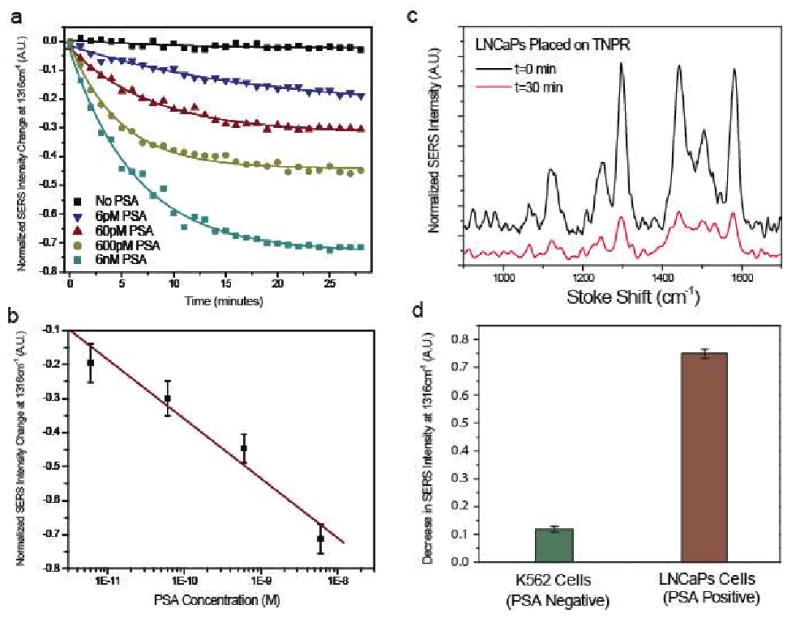
Time-resolved measurements of PSA activity by varying the active PSA concentration. (a) Normalized SERS intensity change for 1316 cm-1 peak at active PSA concentration from 6 pM to 6 nM. The decreasing of SERS intensity can be clearly measured while no significant change can be observed in the control experiment with no active PSA protease. (b) Concentration dependence of normalized SERS intensity change obtained at 30 min. (c) Protease activity measurement obtained from unprocessed extracellular fluid (ECF): Raman spectra obtained at the beginning of exposing LNCaP cells (positive control) ECF to NPR nanosensors (t = 0 min) and after 30 minutes. (d) Normalized change of SERS intensity at 1316 cm-1 peak indicating PSA proteolytic activity in the fluid extracted from LNCaP and K562 cell lines.
In addition to protease activity measurements of purified PSA, measurements for PSA protease activity in extracellular fluid (ECF) from live cell culture was performed (Fig. 4d). It is well known that LNCaP cells secrete PSA and have recently been used in xenografts to evaluate in vivo PSA concentration30. For comparison, a K562 cell line, which does not secrete PSA into the ECF, was used as a negative control. LNCaP ECF showed a significant change in SERS signal, while K562 exhibits very low PSA enzymatic activity (Fig. 4d). By correlating the normalized decrease in Raman signal with Fig. 3b, it was determined that the LNCaP media had an elevated amount of paPSA concentration (Fig. 4d).
Summary
Compare with fluorescence-based assay, the NPR-based method offers several advantages. Strongly localized Raman enhancement can substantially amply the signal and also effectively reduce background noise. Therefore, it allows one-step and label free detection of protease activity with sensitivity at 6 pM, and dynamic range of 3 orders of magnitude. It should note PSA is considered a weak protease and other proteases would allow even better sensitivity. Second, it allows accurate measurement with very small sample volume of 15 pL (Supplemental Information). Third, fabricated using well-established nano-lithography process, NPR-based method is highly reproducible and thus, allowing quantitative assessment of protease activity. Finally, NPRs can be spatially arranged in a microarray format to achieve multiplexed measurements with broad applications, by measuring all know proteases in unprocessed biological samples without complex sample processing and purification steps. The multiplexity of the substrate peptide spatially can be achieved with microarrayer with industry standard protocols, when combined with NPR nanoarray clusters arranged in microarray format. Even if each of the 500+ proteases are cross-interrogated by 10 different substrate peptides, the array still has an easily manageable feature number of less than ten thousand34.
Materials and Methods
NPR Fabrication
The NPR was patterned on quartz substrates (HOYA Corp.) by electron beam lithography (EBL) (Nanowriter Series EBL 100, Leica Microsystems). A 30 nm thick indium-tin-oxide (ITO) under-layer was sputtered on the substrate to prevent charging effects during the EBL process. 100 nm-thick polymathylmethacrylate (PMMA, MicroChem Corp.) films spin-coated on the ITO-quartz glass was used as a positive photoresist. After exposure, the patterns were developed using a 1:3 ratio of MIBK and IPA mixture followed by multilayer deposition of metal and dielectric materials using electron beam evaporation (Mark 40, CHA) and standard lift-off procedures. We fabricated three layered Ag/SiO2/Ag NPR arrays with each silver and SiO2 layer thickness equal to 25 nm and 5 nm, respectively. The geometry of the fabricated NPR was examined by atomic force microscopy, and the total thickness of the NPR is confirmed to be 55 nm.
Device design and characterization
The optical properties of the NPR were characterized by illuminating the NPRs with collimated light delivered by a multimode optical fiber from a 150W Xenon lamp (Thermo Oriel) and collecting the extinction spectra using a grating spectrometer (Triax 550, Jobin Yvon) with matched liquid nitrogen cooled CCD detector (CCD-3500, Jobin Yvon). For the SERS experiments, Raman spectra were measured using a modified inverted microscope (Axiovert 200, Zeiss) with a 50× objective in a backscattering configuration. Baseline subtraction was applied to remove the fluorescence background of the measured spectra. The spectra were then smoothed in Matlab using the Savitsky-Golay method with a second-order polynomial and window size of 9. To correct the possible influence due to the fluctuation of illumination intensity, frequency dependence of Raman scattering, and the variation of initial packing density of the report molecules, the change in SERS intensity was normalized to the average intensity before protease addition. The normalized SERS intensity change is defined as
where I0 is the average SERS intensity before the addition of the protease and It is the SERS intensity measured at the given time t.
Peptide conjugation to NPR and Applying Sample
The peptide attaches to the NPR via the thiol group on cysteine. The PSA substrate peptides were mixed with octanethiol at a 1:3 ratio and at a concentration of 50 μM. Octanethiol, with a SAM chain length of 1-2 nm, was used as packing material to manage the distance among the PSA substrate peptides and help to erect the PSA substrate peptide for optimal spatial presentation. Thus, the octanethiol SAM improves access for the protease to bind to the peptides. The peptide solution was incubated with the NPR substrate for 24 hours to ensure a well-ordered self-assembled monolayer (SAM) on the NPR metallic surface. During incubation the sample was kept at 20°C. Before incubation with PSA enzyme, the sample was washed repeatedly with deionized water to ensure that all unattached peptides were removed. Upon washing, the sample was incubated with sample in a Tris-HCl, pH 8.0, 100mM NaCl, and 0.1mM EDTA buffer. Incubation was performed by placing a 100μL drop of the sample on the NPR array. SERS measurements were performed directly after incubation at 1 minute intervals.
Cell Culture And Clinical Semen Samples
LNCaP cells actively secrete PSA into ECF, and the human CML cells K562 are negative for PSA. Both cell lines are maintained in RPMI-1640 with 10% FBS and 1× Pen/Strep, at 37°C with 5% CO2. 10×106 cells were cultured overnight in 10ml fresh media. Media from both cultures were collected and PSA activity was measured by fluorescent methods as described before, and calibrated against commercial PSA (Calbiochem, San Diego, CA). Briefly, the PSA-binding peptides and derivatives with a spacer were chemically synthesized and used to prepare an affinity column, which was used to fractionate PSA in seminal plasma.
Purified PSA preparation and proteolytic reaction
Proteolytically active PSA was purified to homogeneity from human seminal plasma by column chromatography, eliminating all known PSA complexes and retaining its protease fraction. Cleavage of the substrate peptide immobilized on the NPR nanosensor is performed in a buffer of 50 mM Tris-HCl, pH 8.0, 100 mM NaCl, and 0.1 mM EDTA, and the reaction was monitored in real-time in 37°C. Protease inhibitors (to prevent PSA and Granzyme B degradation) are obtained from CalBiochem and added to the reaction following the manufacturer's instructions, so that the final reaction solution contains 5 μM AEBSF, 4.2 nM Aprotinin, 200 nM Elastatinal and 10 nM GGACK. The concentration of proteolytically active PSA in the PSA reagent has been prepared with a wide range of concentration from 6 pM to 6 nM.
Supplementary Material
Acknowledgments
We thank Dr. R. Oulton for useful discussion and help with the manuscript. This work is supported by NSF Nano-scale Science and Engineering Center (CMMI-0751621), NSFSST/Collaborative Research Program (DMI-0427679), and National Institutes of Health (NIH) through NIH Roadmap for Medical Research (PN2EY018228). F. Chen is supported by NHLBI/NIH HL078534 and NCI/NIH R1CA95393-01, DARPA, and UCSF Prostate Cancer SPORE award (NIH Grant P50 CA89520). JAE and Y were supported by P01 CA072006. This work was performed under the auspices of the U.S. Dept. of Energy, at the University of California/Lawrence Berkeley National Laboratory under contract no. DE-AC03-76SF00098.
Footnotes
Supporting Information Available: Peptide synthesis, instrumentation setup, and estimation of the detection volume. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Ferraro John R, N K, Brown Chris W. Introductory Raman Spectroscopy. 2nd. Elsevier Science; 2003. p. 434. [Google Scholar]
- 2.Yonzon CR, Haynes CL, Zhang X, Walsh JT, Jr, Van Duyne RP. A Glucose Biosensor Based on Surface-Enhanced Raman Scattering: Improved Partition Layer, Temporal Stability, Reversibility, and Resistance to Serum Protein Interference. Anal Chem. 2004;76:78–85. doi: 10.1021/ac035134k. [DOI] [PubMed] [Google Scholar]
- 3.Grow AE, Wood LL, Claycomb JL, Thompson PA. New Biochip Technology for Label-Free Detection of Pathogens and Their Toxins. J Microbiol Methods. 2003;53:221–33. doi: 10.1016/s0167-7012(03)00026-5. [DOI] [PubMed] [Google Scholar]
- 4.Nithipatikom K, McCoy MJ, Hawi SR, Nakamoto K, Adar F, Campbell WB. Characterization and Application of Raman Labels for Confocal Raman Microspectroscopic Detection of Cellular Proteins in Single Cells. Anal Biochem. 2003;322:198–207. doi: 10.1016/j.ab.2003.07.020. [DOI] [PubMed] [Google Scholar]
- 5.Jackson JB, Halas NJ. Surface-Enhanced Raman Scattering on Tunable Plasmonic Nanoparticle Substrates. P Natl Acad Sci USA. 2004;101:17930–17935. doi: 10.1073/pnas.0408319102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hildebrandt P, Stockburger M. Surface-Enhanced Resonance Raman-Spectroscopy of Rhodamine-6g Adsorbed on Colloidal Silver. J Phys Chem-Us. 1984;88:5935–5944. [Google Scholar]
- 7.Stiles PL, Dieringer JA, Shah NC, Van Duyne RR. Surface-Enhanced Raman Spectroscopy. Annu Rev Anal Chem. 2008;1:601–626. doi: 10.1146/annurev.anchem.1.031207.112814. [DOI] [PubMed] [Google Scholar]
- 8.Kneipp K, Kneipp H, Itzkan I, Dasari RR, Feld MS. Surface-Enhanced Raman Scattering: A New Tool for Biomedical Spectroscopy. Curr Sci. 1999;77:915–924. [Google Scholar]
- 9.Nie S, Emory SR. Probing Single Molecules and Single Nanoparticles by Surface-Enhanced Raman Scattering. Science. 1997;275:1102–6. doi: 10.1126/science.275.5303.1102. [DOI] [PubMed] [Google Scholar]
- 10.Le Ru EC, Blackie E, Meyer M, Etchegoin PG. Surface Enhanced Raman Scattering Enhancement Factors: A Comprehensive Study. J Phys Chem C. 2007;111:13794–13803. [Google Scholar]
- 11.Moskovits M. Surface-Enhanced Spectroscopy. Rev Mod Phys. 1985;57:783–826. [Google Scholar]
- 12.Freeman RG, Grabar KC, Allison KJ, Bright RM, Davis JA, Guthrie AP, Hommer MB, Jackson MA, Smith PC, Walter DG, Natan MJ. Self-Assembled Metal Colloid Monolayers - an Approach to Sers Substrates. Science. 1995;267:1629–1632. doi: 10.1126/science.267.5204.1629. [DOI] [PubMed] [Google Scholar]
- 13.Haynes CL, Van Duyne RP. Plasmon-Sampled Surface-Enhanced Raman Excitation Spectroscopy. J Phys Chem B. 2003;107:7426–7433. doi: 10.1021/jp050508u. [DOI] [PubMed] [Google Scholar]
- 14.Sylvia JM, Janni JA, Klein JD, Spencer KM. Surface-Enhanced Raman Detection of 1,4-Dinitrotoluene Impurity Vapor as a Marker to Locate Landmines. Anal Chem. 2000;72:5834–5840. doi: 10.1021/ac0006573. [DOI] [PubMed] [Google Scholar]
- 15.Kahl M, Voges E, Kostrewa S, Viets C, Hill W. Periodically Structured Metallic Substrates for Sers. Sens Actuators, B-Chem. 1998;51:285–291. [Google Scholar]
- 16.Cao YC, Jin RC, Nam JM, Thaxton CS, Mirkin CA. Raman Dye-Labeled Nanoparticle Probes for Proteins. J Am Chem Soc. 2003;125:14676–14677. doi: 10.1021/ja0366235. [DOI] [PubMed] [Google Scholar]
- 17.Cao YWC, Jin RC, Mirkin CA. Nanoparticles with Raman Spectroscopic Fingerprints for DNA and Rna Detection. Science. 2002;297:1536–1540. doi: 10.1126/science.297.5586.1536. [DOI] [PubMed] [Google Scholar]
- 18.Vo-Dinh T, Yan F, Wabuyele MB. Surface-Enhanced Raman Scattering for Medical Diagnostics and Biological Imaging. J Raman Spectrosc. 2005;36:640–647. [Google Scholar]
- 19.Stuart DA, Biggs KB, Van Duyne RP. Surface-Enhanced Raman Spectroscopy of Half-Mustard Agent. Analyst. 2006;131:568–572. doi: 10.1039/b513326b. [DOI] [PubMed] [Google Scholar]
- 20.Lyandres O, Shah NC, Yonzon CR, Walsh JT, Glucksberg MR, Van Duyne RP. Real-Time Glucose Sensing by Surface-Enhanced Raman Spectroscopy in Bovine Plasma Facilitated by a Mixed Decanethiol/Mercaptohexanol Partition Layer. Anal Chem. 2005;77:6134–6139. doi: 10.1021/ac051357u. [DOI] [PubMed] [Google Scholar]
- 21.Su K, D S, Steel MJ, Xiong Y, Sun C, Zhang X. Raman Enhancement Factor of Single Tunable Nano-Plasmonic Resonator. J Phys Chem B. 2006;110:3964–3968. doi: 10.1021/jp055566u. [DOI] [PubMed] [Google Scholar]
- 22.Denmeade SR, Isaacs JT. The Role of Prostate-Specific Antigen in the Clinical Evaluation of Prostatic Disease. BJU Int. 2004;93 1:10–5. doi: 10.1111/j.1464-410x.2003.04634.x. [DOI] [PubMed] [Google Scholar]
- 23.Clements JA, Willemsen NM, Myers SA, Dong Y. The Tissue Kallikrein Family of Serine Proteases: Functional Roles in Human Disease and Potential as Clinical Biomarkers. Crit Rev Clin Lab Sci. 2004;41:265–312. doi: 10.1080/10408360490471931. [DOI] [PubMed] [Google Scholar]
- 24.Gronberg H. Prostate Cancer Epidemiology. Lancet. 2003;361:859–64. doi: 10.1016/S0140-6736(03)12713-4. [DOI] [PubMed] [Google Scholar]
- 25.Denmeade SR, Isaacs JT. A History of Prostate Cancer Treatment. Nat Rev Cancer. 2002;2:389–96. doi: 10.1038/nrc801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Caplan A, Kratz A. Prostate-Specific Antigen and the Early Diagnosis of Prostate Cancer. Am J Clin Pathol. 2002;117(Suppl):S104–8. doi: 10.1309/C4UN-12LK-43HP-JXY3. [DOI] [PubMed] [Google Scholar]
- 27.Canto EI, Shariat SF, Slawin KM. Molecular Diagnosis of Prostate Cancer. Curr Urol Rep. 2004;5:203–11. doi: 10.1007/s11934-004-0038-2. [DOI] [PubMed] [Google Scholar]
- 28.Gretzer MB, Partin AW. Psa Markers in Prostate Cancer Detection. Urol Clin North Am. 2003;30:677–86. doi: 10.1016/s0094-0143(03)00057-0. [DOI] [PubMed] [Google Scholar]
- 29.Haese A, Graefen M, Huland H, Lilja H. Prostate-Specific Antigen and Related Isoforms in the Diagnosis and Management of Prostate Cancer. Curr Urol Rep. 2004;5:231–40. doi: 10.1007/s11934-004-0042-6. [DOI] [PubMed] [Google Scholar]
- 30.Denmeade SR, Jakobsen CM, Janssen S, Khan SR, Garrett ES, Lilja H, Christensen SB, Isaacs JT. Prostate-Specific Antigen-Activated Thapsigargin Prodrug as Targeted Therapy for Prostate Cancer. J Natl Cancer Inst. 2003;95:990–1000. doi: 10.1093/jnci/95.13.990. [DOI] [PubMed] [Google Scholar]
- 31.Wu P, Stenman UH, Pakkala M, Narvanen A, Leinonen J. Separation of Enzymatically Active and Inactive Prostate-Specific Antigen (PSA) by Peptide Affinity Chromatography. Prostate. 2004;58:345–53. doi: 10.1002/pros.10337. [DOI] [PubMed] [Google Scholar]
- 32.Wu P, Zhu L, Stenman UH, Leinonen J. Immunopeptidometric Assay for Enzymatically Active Prostate-Specific Antigen. Clin Chem. 2004;50:125–9. doi: 10.1373/clinchem.2003.026146. [DOI] [PubMed] [Google Scholar]
- 33.Denmeade SR, Lou W, Lovgren J, Malm J, Lilja H, Isaacs JT. Specific and Efficient Peptide Substrates for Assaying the Proteolytic Activity of Prostate-Specific Antigen. Cancer Res. 1997;57:4924–4930. [PMC free article] [PubMed] [Google Scholar]
- 34.Vo-Dinh T, Stokes DL, Griffin GD, Volkan M, Kim UJ, Simon MI. Surface-Enhanced Raman Scattering (Sers) Method and Instrumentation for Genomics and Biomedical Analysis. J Raman Spectrosc. 1999;30:785–793. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.