

Abstract
We developed an informatic method to identify tandem mass spectra composed of chemically cross-linked peptides from those of linear peptides and to assign sequence to each of the two unique peptide sequences. For a given set of proteins the key software tool, xComb, combs through all theoretically feasible cross-linked peptides to create a database consisting of a subset of all combinations represented as peptide FASTA files. The xComb library of select theoretical cross-linked peptides may then be used as a database that is examined by a standard proteomic search engine to match tandem mass spectral datasets to identify cross-linked peptides. The database search may be conducted against as many as 50 proteins with a number of common proteomic search engines, e.g. Phenyx, Sequest, OMSSA, Mascot and X!Tandem. By searching against a peptide library of linearized, cross-linked peptides, rather than a linearized protein library, search times are decreased and the process is decoupled from any specific search engine. A further benefit of decoupling from the search engine is that protein cross-linking studies may be conducted with readily available informatics tools for which scoring routines already exist within the proteomic community.
Introduction
Cross-linking and mass spectrometry (CXMS) has become an essential tool for the analysis of protein-protein interactions and protein conformations 1-3. The general principle underlying this method is the covalent capture of juxtaposed amino acids using a variety of cross-linking reagents 4,5. Covalent chemical cross-links between two peptides formed during such experiments provide two highly valuable pieces of information: i) identification of interacting partners within a protein complex, and ii) spatial proximity of cross-linked amino acid residues. Together this information may be used to generate new or refine existing structural models of the composition of protein complexes and the relative juxtaposition of proteins within the complex. While conducting such experiments in vivo would be a desirable means to monitor protein activities6, most analyses are performed on two interacting proteins or multi-protein complexes with limited numbers of interacting proteins. The main reason for this failure to routinely map protein interactions in vivo is the low stoichiometry and low frequency of cross-linked peptides relative to unmodified ones which often necessitates enrichment steps 7-11 and specialized mass spectrometry 6,12. Finally, the most challenging part of such analysis remains the identification of cross-linked peptides present in a mixture of mostly linear peptides. While every proteomics lab is equipped with very sophisticated database search programs for the analysis of standard peptide dataset, none of these programs can be directly used as such for cross-link analysis. Additionally, our laboratory as well as others have come up with various strategies that are unfortunately not always freely available or hosted on public servers discouraging many users due to lack of privacy 6,13.
In order to make CMXS data analysis more generally applicable across laboratories, we developed a database processing tool which we refer to as xComb that may be used with any standard database search engine and is publicly available (http://phenyx.proteomics.washington.edu/CXDB/index.cgi). Up to 50 proteins may be input to xComb from which a concatenated, combinatorial database of a subset of all possible theoretical cross-linked peptides is created based on experiment-specific details. We believe that any database search engine may be used with xComb and we have confirmed use with the following including Phenyx, Sequest, OMSSA, Mascot and X!Tandem 14-18. This xComb strategy provides the user a tandem mass spectral scoring scheme with which they are already familiar and suits analysis of small to mid-size protein-protein complexes.
Rationale for xComb
The goal in developing xComb was to provide a generic means by which any standard proteomic database search engine could be used to identify and assign sequence to tandem mass spectra of cross-linked peptides. To our knowledge, only a single attempt has been made to this end by Rappsilber and colleagues 13 who used Mascot to search a database of linearized sequences of two “cross-linked” peptides where loss of a water molecule accounted for the chemically cross-linked bond between the two. To do this they developed (unreleased) software that constructs a chimeric protein sequence composed of all theoretically possible cross-linked amino acid combinations, between tryptic peptides from the proteins of interest, concatenated together as a linear sequence. Specifically, for a protein P with a peptide set [a, b, c] and a protein Q with peptide set [A, B, C], all permutations are built as a single protein P-protein Q sequence combination, i.e. caabacbbccCAABACBBCCcAaAbAcBaBbBcCaCbC. In order to account for the presence of cross-linkers (e.g. amine based reagents) that block enzymatic cleavage, their approach includes all peptides that contain missed cleavages. This chimeric protein sequence was then interrogated with Mascot by specifying the enzyme used and allowing a sufficient number of missed cleavages for the program to identify the built-in cross-link sequences. For example, one would include at least one missed cleavage to jump over the link made by the two linearized peptides for non-amine based cross-linkers and at least two to three missed cleavages for amine based cross-linkers.
While conceptually powerful and thorough, we noticed some pitfalls when adopting this strategy. First, the search is not specific to the cross-link sequences only. As mentioned above, at least two to three missed cleavages have to be allowed during the search process. Consequently, peptide sequences of zero up to three missed cleavages are needlessly interrogated when in fact cross-linked sequences inherently contain at least one missed cleavage. This greatly decreases database search specificity towards only cross-linked peptide sequences (see Supplementary Figure 1A). Second, because of the combinatorial behavior of such a thorough strategy, a limited number of proteins should be used to prevent the number of possible peptide sequence matches from exceeding what the standard database search engine can accommodate with respect to false discovery rates. For example, Rappsilber and colleagues used a 125 protein database which in theory would contain 1-2 orders of magnitude more peptide sequences (approx. 3E8) than a database with 100,000 proteins after trypsin proteolysis and allowing for up to two missed cleavages (approx. 1E7). Third, because of the former two issues, special software is needed post-search to filter out all false-positives that arise from chimeric sequences. Thus, different filtering software would be required for each standard search engine used making the process less generic than desired. Fourth, because of the concatenated nature of the chimeric protein sequence, the position of each peptide discovered to be cross-linked is not immediately accessible requiring yet another filtering step (see Supplementary Figure 1A and 1B). For all these reasons, we developed a simplified strategy based on the same powerful linearization principle put forward by Rappsilber and colleagues. Our xComb approach alleviates some of the issues described above and furthermore the xComb database generation tool and all ancillary tools required to deploy this approach are publicly available.
Results and discussion
Description of the xComb strategy
Figure 1 describes the xComb strategy. First a theoretical cross-linked peptide database is created out of a list of desired proteins. Briefly, each protein is digested with trypsin (or the appropriate enzyme) by allowing up to three missed cleavages and these sequence cuts stored in a file. Missed cleavages are used to account for the peptides containing Lysine residues modified by the cross-linkers that block trypsin cleavage at these sites. Next, peptides are combined pair-wise to create all possible intra- and inter-protein cross-links. To increase the search specificity cross-linked peptides are filtered according to experiment-specific parameters specified by the user (e.g. cross-linker specificity, intra/inter specificity, protease used) a process which removes experimentally irrelevant theoretical sequences many of which contain redundant sequence information. This process is repeated until all intra- and inter-protein combinations have been performed. The cross-linked peptide sequence database is created by writing each peptide pair into a FASTA formatted file as a linearized sequence and permuted (see the next section for details) to allow for maximum coverage of fragment ions during the search. In addition, a detailed description of each peptide pair, such as the parent proteins involved and peptide sequence within the parent proteins, is added to the header for rapid data screening. The xComb generated database is then uploaded to a server for use by a standard database search engine.
Figure 1. Description of the xComb strategy.

A.1) All theoretically possible cross-linked amino acid combinations are built based on the input protein sequences. Irrelevant combinations are then filtered based on specific parameters defined by the user (e.g. cross-linker specificity). A.2) A cross-linked peptides fasta database is generated with each entry corresponding to one cross-linking combinations (both permutations are written). A header describes precisely both peptides (origin and position) for straightforward interpretation and validation. B) Product ion spectra are acquired on ≥+4 precursors only at high mass accuracy. Depending on the search engine used, raw tandem mass spectral data are first de-convoluted and re-written into a lower charge state form. C) Finally, tandem mass spectra can be searched against the cross-linked peptides database using any standard search engine (Sequest, Phenyx, Mascot, X!Tandem or OMSSA). A “do not cleave” enzyme is added and used to specifically search only the full length cross-linked sequences. The cross-link reagent is specified as a variable modification with the addition of a water molecule to account for loss of mass during the linearization process.
Here we present the test results for use of xComb with Phenyx, Sequest, OMSSA, Mascot and X!Tandem. Since the xComb database is created from digested peptides, not proteins, no enzyme needs to be specified during the search. In the case of Phenyx and OMSSA, an option for “do_not_cleave” or “whole protein” enzyme respectively is already present in the list of enzymes, while for Sequest, Mascot and X!Tandem a new enzyme was added to simulate the “do_not_cleave” mode. Thus, in the “do_not_cleave” mode all programs search the database without performing any enzymatic digestion and take each entry in its whole rendering the search extremely fast and specific to cross-linked peptide sequences only. A modification corresponding to the cross-linker and a water molecule is added to the search parameters to account for the peptide linearization process. Finally, for software not designed to cope with high charge state precursors and fragment ions (e.g. Mascot, X!Tandem) arising from cross-linked peptides, the original spectra are de-convoluted and re-written into a lower charge state form before submission to the search algorithm.
Why do we need two permutations?
As shown by Rappsilber and colleagues a cross-linked peptide composed of peptides α and β can be linearized (i.e. both peptides are concatenated back to back into a single long sequence) in two different ways: αβ or βα. Fragment ion spectra from these two cross-linked peptides are a combination of fragment ions from the two corresponding linearized versions. Just as is the case with matching peptide sequences to tandem mass spectra derived from simultaneous fragmentation of two isobaric precursor ions 19, both sets of fragment ions can be matched to sequence independently. For example, if we consider the cross-linked peptide shown in Figure 2, fragmentation occurring on the N-terminal side of the peptide α and before the cross-linked amino acid generates a b-ion containing only a portion of the peptide α plus a complimentary y-ion containing all peptide β plus the remaining portion of peptide α. If this fragmentation occurs on the N-terminal side of peptide β and before the cross-linked amino acid, another b-ion is formed belonging this time to peptide β with a complimentary y-ion composed of peptide α and the remaining part of peptide β. Therefore, two b-ion series - bαβ-ions and bβα-ions – are generated. Identically, two y-ion series – yαβ-ions and yβα-ions – may also be generated. As shown in Figure 2 when linearizing a peptide like αβ, the bαβ-ions series runs from the N-terminus of peptide α to its cross-linked residue and from the cross-linked residue in peptide β to its C-terminus. Similarly, the yαβ-ion series runs from the C-terminus of peptide β through its cross-linked residues and from the cross-linked residue in peptide α to its N-terminus. Thus, if only one geometry is considered, all fragment ions occurring between the cross-linked residues in the two peptides are lost (e.g. residues not underlined in Figure 2). However, these fragment ions (complimentary bβα-ion and yβα-ion series) are identified in the second permutation represented by peptide βα. It is therefore crucial that both permutations are present in the cross-link database for 100% theoretical coverage of fragment ions. If the cross-linked amino acid residues occur in the middle of both cross-linked peptides, the fragment ions are equally distributed between both permutations and most likely both will be identified by a standard search algorithm. However in certain cases, as exemplified in Supplementary Figure 2, depending on the position of the cross-link, one of the two permutations will have most of the identified fragment ions assigned to it and the second will be less likely to be identified with a good score. Finally, as shown in Supplementary Figure 2 the extent of fragment ion coverage allows the user to assign the position of the cross-linked amino acids. In an ideal scenario, all fragment ions would be identified until they reach the position of the cross-linked amino acid for both b- and y-ion series and in both permutations providing unambiguous identification of cross-linked amino acids. In practice, unambiguous identification of cross-linked sites is not always achieved but rather refined to a small region with more than one possibility.
Figure 2. Description of the linearization process.

Two peptides cross-linked together can be considered as two unique linearized forms of both sequences that cover 100% of the fragment ions. These two concatenated sequences may be directly interpreted by a standard search engine to cover all fragment ions present in the spectrum.
Important metrics when using xComb
As mentioned previously, the xComb strategy is well suited for protein complexes from 1-20 unique protein sequences. Because of the combinatorial essence of such a concatenated theoretical database, a limited number of unique protein sequences should be interrogated in one search. This artificial limit may be exceeded, but maintaining it avoids over-loading the search engine process with an unrealistic number of peptide sequences that must be interrogated simultaneously. As exemplified in Supplementary Figure 3A, the total number of possible database entries (combinations) out of 30 proteins would roughly yield the same number of peptides (10 million) as a tryptic digest (with up to two missed cleavages) of 100,000 proteins, a number present in a complex organism proteome. However, if experimental specificity is taken into account (e.g. cross-linker specificity, minimum peptide length) then a larger number of proteins may be considered. Supplementary Figure 3B shows the number of possible combinations for different numbers of total proteins using cross-linkers with five different chemical specificities. At 50 proteins, roughly 10 million sequences are present in the xComb generated cross-link database, but 20 more proteins may be included when available experimental criteria are included. Because of the previously mentioned reason and also because of the size of the FASTA file generated (see Supplementary Figure 4), we currently consider 50 proteins the rational upper limit for this strategy. Finally, high mass accuracy plays an important role in the search specificity. While for a search with a small protein complex (n<10) high mass accuracy is less critical, with large protein complexes (n>10) it becomes an important factor especially if available for both precursor and product ions (see Supplementary Table 1).
General search strategy
Because of the low efficiency of most cross-link reactions, only a small proportion of available proteins are cross-linked. Thus proteolysis of any set of cross-linked proteins produces a complex mixture of different types of peptides such as: i) linear peptides occurring from regular non-cross linked peptides, ii) dead-end peptides (or Type 0 cross-links) occurring from a failure of the cross-linker to react at both ends, iii) intra-peptide cross-links (or Type 1 cross-links) where a bridge is formed within the same peptide, iv) inter-peptide cross-links within same protein molecule (or Type 2 cross-links), and finally v) inter-peptide cross-links between two separate protein molecules (or Type 2cross-links [inter-protein]. The Type 0, 1, 2 crosslink nomenclature used here is as put forward by Schilling et al. 20. In order to increase the search specificity and simplify the results, we propose a general multi-layer search strategy described below as the basis for the basic xComb strategy:
Round 0 (optional): Remove all peak list files for precursor ions with charge state lower than +4 because the vast majority of cross-links ionize with a charge state of +4 and higher (Approx. 80% reduction).
Round 1: Search the tandem MS data set for linear peptides and Type 0 cross-links using a standard protein sequence database using the Type 0 cross-linker modification as variable. Validate results and export unmatched spectra for a second round search (Approx. 40% reduction).
Round 2: Search unmatched spectra from Round 1 for Type 1 cross-links on the same standard protein sequence database but using the Type 1|2 cross-linker modification as variable. Validate results and export unmatched spectra for a third round search (less than 5% reduction).
Round 3: Search unmatched spectra from Round 2 for Type 2 cross-links [intra-protein] against the xComb generated database for intra-protein cross-links only using again the Type 1|2 cross-linker modification as variable. Validate results and export unmatched spectra for a final round search (less than 5% reduction).
Round 4: Search unmatched spectra from Round 3 for Type 2 cross-links [inter-protein] against the xComb generated database from inter-protein cross-links only using again the Type 1|2 cross-linker modification as variable. Validate results.
Parallel comparison of cross-link identification in the CYP2E1-B5 complex by Phenyx, Sequest, OMSSA, X!Tandem and Mascot
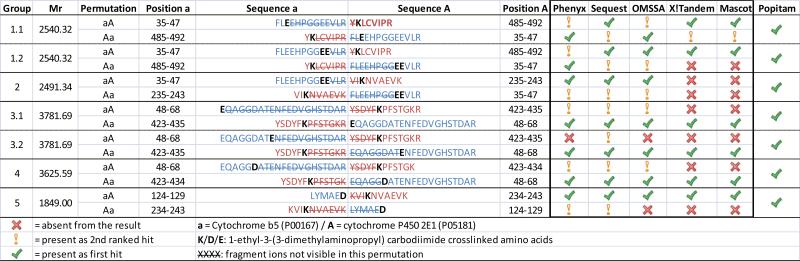
To demonstrate the potential of our xComb strategy to be generalizable across search engine platforms, we analyzed the protein complex formed by Cytochome P450 2E1 (cyp 2E1) and cytochrome b5 (b5) cross-linked with 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) as previously described 12,21 using five different search engines: Phenyx, Sequest, OMSSA, X!Tandem and Mascot. For each search engine, the same database built from the two respective protein sequences was uploaded in their respective database management systems. The database for Cyp 2E1 and b5 cross-linked with EDC consisted of 1'667'684 cross-linked peptide sequences. A peak list file was generated from the raw instrument file and used “as is” with Phenyx, OMSSA and Sequest because of their capability to interrogate high charge state precursor and fragment ions (z > 2). In the case of X!Tandem and Mascot, a deconvolution step was added to convert all high charge state spectra to a form consisting of a precursor of charge state z=2 and fragment ions of charge z=1. In all cases the four round searches described above was applied to generate the results for all five search engines shown in Table 1 and compared to the results previously described by our open- modification search strategy using Popitam 12,22. Six out of the seven cross-link sites previously characterized by our open- modification search pipeline were identified by all five search engines using the xComb strategy. However, one cross-linked peptide (FLEEHPGGEEVLR linked to VIKNVAEVK / cross-link 2 in Table 1) could not be identified by Mascot and X!Tandem. In order to rule out that this could be due to the deconvolution step which is only employed with Mascot and X!Tandem, we also searched the deconvoluted spectra with Phenyx, OMSSA and Sequest. The latter three search engines identified this cross-link with or without the addition of the deconvolution step. Thus, the lack of identification for cross-link 2 by Mascot and X!Tandem cannot be attributed to deconvolution and is most likely due to intrinsic differences in scoring routines. As previously mentioned, in the best case scenario, both permutations are identified for a given tandem mass spectrum of a cross-linked precursor. Unfortunately, most search engines, report only the best ranked hit for each tandem mass spectrum and access to other hits is not available in the final results. Sequest, however, provides a rank ordered list of the best hits in a table for each tandem mass spectrum that is easily accessible and modifiable to include as many possible secondary sequences matches in the list as the user desires. Interestingly to the performance of the xComb database concept, Sequest always identified both sequence permutations as the first and second ranked hit. Identically, Phenyx produced a similar result in that one can set the search not to resolve conflicts for the same tandem mass spectrum allowing both permutations to be detected. Thus, two peptides with a z-Score above the threshold are reported as a group, but in some cases Phenyx did not generate a sufficiently high z-Score to be considered in the report. This may require some further exploration on the part of Phenyx users who seek to optimize the search engine performance with xComb. Similarly, OMSSA reports more than one hit for the same tandem mass spectrum if they both score above the E-value threshold. In the case of X!Tandem and Mascot, both permutations were only reported for cross-link 1.1 (Table 1) and we could not set the search parameter such that it would report more of these pairs, but this should be possible with appropriate software permissions.
Table 1.
Search engine comparison of cytochrome P450 2E1/cytochrome b5 complex data.
|
As previously mentioned, for some cross-linked peptides a single permutation (either αβ or βα) may cover almost the full range of theoretical fragment ions, while in other cases both permutations are complimentary and thus required for correct identification. All following examples are from searches performed with the search engine Phenyx. Figure 3 shows the identification of cross-linked peptide 5 (Table 1) which is known to be a combination of the peptide with amino acid sequence LYMAED (peptide α) cross-linked to the peptide with amino acid sequence KVIKNVAEVK (peptide β). In this case, the C-terminal aspartic acid (D6) of peptide α is covalently linked to lysine at position 4 (K4) of peptide β (also shown as K10 in permutation αβ). Because of the position of the cross-linked amino acids, as illustrated by Figure 3A, almost all fragment ions can be matched to the permutation αβ (Figure 3B and 3C) with a high z-Score of 14 while permutation βα poorly matches to the spectrum with a z-Score of 3.56 (data not shown). In this example, while identification of both permutations is a strong indication of a hit to a tandem mass spectrum of a cross-linked peptide, only permutation αβ clearly identifies the cross-link and the position of the cross-linked amino acids. Supplementary Figure 5 and 6 illustrate the identification of cross-link 1.1 (Table 1) which is known to be a combination of the peptide with amino acid sequence YKLCVIPR (peptide α) cross-linked to the peptide with amino acid sequence FLEEHPGGEEVLR (peptide β). In this cross-linked peptide example, lysine K2 of peptide α has been cross-linked by EDC to Glutamic acid E3 of peptide β (i.e. E11 in permutation αβ or E3 in permutation βα). Unlike the previous example, here fragment ions are more equally distributed in both permutations as illustrated in Supplementary Figure 5A and 6A. Permutation αβ is identified with a z-Score of 11.0 (Supplementary Figure 5B and 5C) and a z-Score of 7.65 for permutation βα (Supplementary Figure 6B and 6C). Therefore, in this example, both permutations are useful in identifying the cross-linked peptide pair and the position of the cross-linked amino acid. In both examples, an important contribution to the validation of a particular cross-link is the presence of b- and y-ions that are past the two cross-linked amino acids (e.g. fragment ions b10-15 or y10-15 in permutation αβ in Figure 3C), be it from one permutation or both. These strongly validate that the identified tandem mass spectrum is from a cross-linked peptide pair as they represent molecular masses corresponding to one peptide and a cross-linked portion of the second one. Finally, in some cases, fragment ions corresponding to a fragmentation in the cross-linker itself can occur as illustrated in Figure 3A by the dashed b- and y-ion pair or by the matched b6 and y10 fragment ion in Figure 3C. These are again good indicators that the tandem mass spectrum originates from a cross-linked peptide pair, but these observations are rare and most likely only occur via CID in cross-linkers, such as EDC, where a CID-susceptible amide bond links the two peptides.
Figure 3. Example of an identification based on only one permutation in the cytochrome P450 2E1/cytochrome b5 complex.

A) Aspartic acid D6 of peptide α is cross-linked to lysine K4 of peptide β. The linearization αβ depicted here covers 80% of the fragment ions. B) and C) Spectrum matching to the linearization αβ. Almost all fragment ions are assigned to this spectrum with a high z-Score using Phenyx. The extent of fragment assignment allows validation of the position of the cross-linked amino acid. Unlike linearization αβ, βα poorly matches to the spectrum with a low z-Score of 3.56 (data-not shown).
Experimental section
Mass spectrometric data
Mass spectrometric data acquisition and processing were done according to our previously published protocol 12. Briefly, all data were acquired in an LTQ-Orbitrap mass spectrometer 23 using data-dependent initiated acquisition of tandem mass spectra by collision induced dissociation (CID) of ions [M+4H]4+ and higher. Both precursor ion and product ion spectra were acquired at high mass accuracy in the Orbitrap. Peak list files were searched with Phenyx, OMSSA and Sequest without further modifications. In the case of Mascot and X!Tandem, a deconvolution step was performed using Hardklor 24 and then peaklist files written with lower charge state precursor (+2) and fragment ions (+1) subjected to searches.
Generation of databases with xComb
xComb is written in Perl. It is composed of two programs, Protein2digest.pl and Digest2cxdb.pl, available through a Common Gateway Interface (CGI) (http://phenyx.proteomics.washington.edu/CXDB/index.cgi) (see Supplementary Figure 7). Protein2digest.pl is used for the generation of a protein digestion file with extension ‘.digest’ for each protein. It currently supports several enzymes (Trypsin, Arg-C, Lys-C, Glu-C and Asp-N), allows users to specify the number of missed cleavages and reads both FASTA or DAT format (the latter format can be used by the program to retrieve protein processing information - e.g. signal peptide - before performing the digestion). Digest2cxdb.pl is the concatenated peptide sequence cross-link database generation tool. This program reads all “.digest” files and computes every possible cross-link combination by linearizing each pair of peptide. Because of the linearization process and in order to maximize the fragment ion coverage during the search (see Figure 2 for more details), each pair of peptides is assembled in two permutations (i.e. peptide A followed by peptide B or vice-versa). In order to increase the specificity of the database search process and reduce the database size, several parameters have to be set: i) type of database to be generated (intra-protein cross-link, inter-protein cross-links or both); ii) type of cross-linker used during the experiment allowing unwanted combinations to be discarded; iii) specificity for amine cross-linkers (at least one missed cleavage at a Lysine residue has to be present in the sequence, e.g. xxxKxxxxK or xxxKxxxxR); and iv) minimum peptide length for each peptide in the pair. A special formatting of the FASTA header has been added for use of the database with Phenyx. We have also added a test mode that adds a ‘|’ in between each peptide of the pair for easier proof reading of the database before final compilation. The final output is a FASTA formatted database where each entry is a single peptide pair with a header describing both peptide sequences used with protein information and peptide position. Following is an example of an xComb generated FASTA file:
>P05181_P00167_aA_152_18 a=YSDYFKPFSTGKR (423-435) Cytochrome P450 2E1
cx A=EQAGGDATENFEDVGHSTDAR (53-73) Cytochrome b5
YSDYFKPFSTGKREQAGGDATENFEDVGHSTDA
For the cytochrome P450 2E1/cytochrome B5 complex analysis, the following parameters were used to generate the database: i) digestion with trypsin, ii) up to two missed cleavages, iii) inter-protein cross-link database, iv) amine/carboxyl cross-linker, and v) amine cross-linkers missed cleavage set to “ON”. For Phenyx searches, the Phenyx header output was used.
For the statistical analysis of xComb, all cross-linkers were tested with various numbers of proteins as input (2,5,10,15,20,25,30,35,40,45,50). All other parameters were set as previously described. The 48 other proteins were selected from Uniprot human based on their similar molecular weight to cytochrome b5 and 2e1 (24 each).
Database search using Phenyx
Cross-link databases were uploaded using the database management system in Phenyx 2.6. A special scoring model for the Orbitrap was added that allowed use of high charge state fragment ions in the search. The following parameters were used to search the raw spectra: i) “do_not_cleave” enzyme was selected, ii) 10 ppm precursor ion tolerance, iii) ltq_orbitrap_0.1Da_xcomb scoring model, iv) turbo set to 5% b- and y-ions at 20 ppm mass accuracy, and v) modifications: Cys_CAM [fixed, all], Oxidation_M [variable, none]. In this case, no modification was added for the cross-linker. EDC (1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide) has a modification of -18 Da, thus counter balancing the linearization water loss of +18 Da. In the case where FDR were used, data were filtered at less than 1%.
Database search using Sequest
The database was uploaded on the computer running Bioworks Browser 3.3.1 SP1. The following parameters were used to search the raw spectra: i) a “do_not_cleave” enzyme mode was simulated by adding an enzyme that cleaves after a non-existing amino acid “J” thus not cleaving at all, ii) 10 ppm precursor ion tolerance, iii) precursor tolerance set to 0.01 Da, and iv) modifications: +57.0215 for cysteine residues [fixed], +15.9949 for methionine [variable]. Again, no modification was added for the cross-linker. EDC (1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide). Sequest search result were stored in SRF files and then filtered using an Xcorr higher than 0.5.
Database search using OMSSA
The cross-link fasta database was converted with formatdb (http://pubchem.ncbi.nlm.nih.gov/omssa/blast.htm) and uploaded to the OMSSA Browser software (http://pubchem.ncbi.nlm.nih.gov/omssa/browser.htm). The following parameters were used to search the raw spectra: i) enzyme set to “whole protein”, ii) maximum peptide length set to 10, iii) precursor m/z tolerance 0.01 Da, iv) charge state allowed 1 to 10 and multiple charge product start at 3, v) product m/z tolerance 0.01 Da, vi) maximum charge state for product set to 6, vii) charge dependency at m/z tolerance set to linear correction, and viii) modifications: carbamidomethyl C [fixed], oxidation of M [variable]. No modification was added for the cross-linker EDC.
Database search using X!Tandem
The database was uploaded on the computer cluster running X!Tandem. The following parameters were used to search the de-convoluted spectra: i) enzyme specificity was set to [J]|[X] thus not cleaving at all, ii) 10 ppm precursor tolerance, iii) 20 ppm product ion tolerance, iv) spectrum dynamic range and total peaks set to 100, v) maximum parent charge state set to 10, vi) noise suppression set to off and minimum peaks to 5, vii) no refinement, and viii) modifications: +57.021464@C [fixed], +15.994915@M [variable]. No modification was added for the cross-linker EDC.
Database search using Mascot
The cross-link database was uploaded into Mascot server version 2.2. The following parameters were used to search the de-convoluted spectra: i) a “do_not_cleave” enzyme mode was used by adding enzyme specificity of the amino acid “J” to simulate no cleavage, ii) 10 ppm precursor tolerance, iii) product ion tolerance set to 0.05 Da, iv) instrument set to ESI-FTICR, and v) modifications: carbamidomethyl (C) [fixed], oxidation (M) [variable]. No modification was added for the cross-linker EDC.
Conclusions
We have demonstrated that any of the most common database search engines used in proteomics laboratories can be used to select tandem mass spectra of cross-linked peptide pairs from those of linear peptides using a simplified version of the Rappsilber concept. Our xComb strategy is straightforward, easy to implement and requires no additional software for most of the search engines to perform well. The tools to generate this simplified concatenated cross-linked peptide sequence database are available as a web application on our website (http://phenyx.proteomics.washington.edu/CXDB/index.cgi) or a Perl script for those concerned with a lack of privacy. The xComb process leverages the capability of existing protein sequence database search software for which well developed scoring schemes exist to allow users to immediately identify tandem mass spectra of cross-linked peptide pairs using their preferred search engine. Because of the design of the cross-linked peptides database and the “do not cleave” enzyme used, the search is constrained to only cross-linked sequences and therefore is highly specific, especially with high mass accuracy data. Positional information – i.e. origin and position of peptide - is immediately available because each entry in the FASTA file contains this information. The strategy allows a quick assessment and validation of which amino acids are cross-linked via the user's search engine of choice. Finally, while the strategy may not yet be applied to a whole proteome, due to combinatorial limitations, experiments like in vivo cross-linking of a whole cell may be conducted in a targeted or iterative fashion. In this case the xComb strategy may be used as a very specific binocular to zoom into different regions of the proteome one biochemical pathway or functional group at a time by creating several cross-linked databases for each region. This would allow for a particular protein network/complex of interest to be interrogated selectively rather than as the whole cell network simultaneously, a practice which would necessarily increase false discovery rate to an unacceptable level. However, such strategy would be successful only for well-characterized pathways where composition is known and can therefore be predicted. In addition, monitoring dynamic changes between different states would be difficult unless hypothesized protein candidates are added to the database. Finally, the linearized, concatenated peptide-specific database concept of xComb may also be used in parallel with other traditional protein-specific approaches, e.g. with Popitam as we previously published, as a means to cross-validate results by an orthogonal method.
Supplementary Material
Acknowledgments
We thank Alexandre Masselot and Pierre-Alain Binz from GenBio for their thoughtful discussion and help on implementing our xComb strategy with Phenyx. DRG thanks the National Institutes of Health (NIH) for support: R33CA099139-01, 1S10RR023044-01 and 1U54 AI57141-01. AP acknowledge the Swiss National Science Foundation (SNF) for support (PBLAA—119623 and PA00P3_126252).
References
- 1.Back JW, de Jong L, Muijsers AO, et al. J Mol Biol. 2003;331(2):303. doi: 10.1016/s0022-2836(03)00721-6. [DOI] [PubMed] [Google Scholar]
- 2.Sinz A. Mass Spectrom Rev. 2006;25(4):663. doi: 10.1002/mas.20082. [DOI] [PubMed] [Google Scholar]
- 3.Singh P. Anal Chem. 2010 DOI:10.1021/ac1000724. [Google Scholar]
- 4. http://creativemolecules.com/
- 5. http://www.piercenet.com/products/browse.cfm?fldID=0203.
- 6.Rinner O, Seebacher J, Walzthoeni T, et al. Nat Methods. 2008;5(4):315. doi: 10.1038/nmeth.1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alley Stephen C., Ishmael Faoud T., Daniel Jones A, et al. Journal of the American Chemical Society. 2000;122(25):6126. [Google Scholar]
- 8.Fujii N, Jacobsen RB, Wood NL, et al. Bioorg Med Chem Lett. 2004;14(2):427. doi: 10.1016/j.bmcl.2003.10.043. [DOI] [PubMed] [Google Scholar]
- 9.Hurst GB, Lankford TK, Kennel SJ. J Am Soc Mass Spectrom. 2004;15(6):832. doi: 10.1016/j.jasms.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 10.Sinz A, Kalkhof S, Ihling C. J Am Soc Mass Spectrom. 2005;16(12):1921. doi: 10.1016/j.jasms.2005.07.020. [DOI] [PubMed] [Google Scholar]
- 11.Chu F, Mahrus S, Craik CS, et al. J Am Chem Soc. 2006;128(32):10362. doi: 10.1021/ja0614159. [DOI] [PubMed] [Google Scholar]
- 12.Singh P, Shaffer SA, Scherl A, et al. Anal Chem. 2008;80(22):8799. doi: 10.1021/ac801646f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maiolica A, Cittaro D, Borsotti D, et al. Mol Cell Proteomics. 2007;6(12):2200. doi: 10.1074/mcp.M700274-MCP200. [DOI] [PubMed] [Google Scholar]
- 14.Colinge J, Masselot A, Giron M, et al. Proteomics. 2003;3(8):1454. doi: 10.1002/pmic.200300485. [DOI] [PubMed] [Google Scholar]
- 15.Craig R, Beavis RC. Bioinformatics. 2004;20(9):1466. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- 16.Eng Jimmy K., McCormack Ashley L., Yates Iii John R. Journal of the American Society for Mass Spectrometry. 1994;5(11):976. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 17.Geer LY, Markey SP, Kowalak JA, et al. J Proteome Res. 2004;3(5):958. doi: 10.1021/pr0499491. [DOI] [PubMed] [Google Scholar]
- 18.Perkins DN, Pappin DJ, Creasy DM, et al. Electrophoresis. 1999;20(18):3551. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 19.Scherl A, Tsai YS, Shaffer SA, et al. Proteomics. 2008;8(14):2791. doi: 10.1002/pmic.200800045. [DOI] [PubMed] [Google Scholar]
- 20.Schilling B, Row RH, Gibson BW, et al. J Am Soc Mass Spectrom. 2003;14(8):834. doi: 10.1016/S1044-0305(03)00327-1. [DOI] [PubMed] [Google Scholar]
- 21.Gao Q, Doneanu CE, Shaffer SA, et al. J Biol Chem. 2006;281(29):20404. doi: 10.1074/jbc.M601785200. [DOI] [PubMed] [Google Scholar]
- 22.Hernandez P, Gras R, Frey J, et al. Proteomics. 2003;3(6):870. doi: 10.1002/pmic.200300402. [DOI] [PubMed] [Google Scholar]
- 23.Makarov A. Anal Chem. 2000;72(6):1156. doi: 10.1021/ac991131p. [DOI] [PubMed] [Google Scholar]
- 24.Hoopmann MR, Finney GL, MacCoss MJ. Anal Chem. 2007;79(15):5620. doi: 10.1021/ac0700833. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.