

Abstract
Sliding clamps are toroidal proteins that encircle DNA and act as mobile platforms for DNA replication and repair machinery. To be loaded onto DNA, the eukaryotic sliding clamp Proliferating Cell Nuclear Antigen (PCNA) must be splayed open at one of the subunit-subunit interfaces by the ATP-dependent clamp loader, Replication Factor C, whose clamp-interacting sites form a right-handed spiral. Earlier molecular dynamics (MD) studies suggested that when PCNA opens, it preferentially adopts a right-handed spiral to match the spiral of the clamp loader. Here, analysis of considerably longer MD simulations shows that although the opened form of PCNA can achieve conformations matching the helical pitch of Replication Factor C, it is not biased toward a right-handed spiral structure. A coarse-grained elastic model was also built; its strong correspondence to the all-atom MD simulations of PCNA suggests that the behavior of the open clamp is primarily due to elastic deformation governed by the topology of the clamp domains. The elastic model was further used to construct the energy landscape of the opened PCNA clamp, including conformations that would allow binding to the clamp loader and loading onto double-stranded DNA. A picture of PCNA emerges of a rather flexible protein that, once opened, is mechanically compliant in the clamp opening process.
Introduction
Proliferating Cell Nuclear Antigen (PCNA) is a trimeric ring-shaped eukaryotic protein whose topology allows it to encircle double-stranded DNA. Its stability allows it to remain bound for extended periods of time by topologically tethering to the DNA. Its primary role is as a mobile platform on which the replicative DNA polymerases δ and ɛ bind (1,2). This confers upon the replisome the ability to rapidly and processively synthesize long stretches of DNA. Additionally, PCNA interacts with a myriad of other proteins, spatially organizing and temporally orchestrating them as required along the DNA strands (3,4).
PCNA is built from six structurally similar domains partitioned into three identical subunits. A series of six β-sheets, spanning domain and subunit interfaces, forms the outer diameter of the ring, against which 12 α-helices pack to form the lining of the ring's bore (Fig. 1) (5). The basic architecture of PCNA is shared by sliding clamps across all domains of life, although evolution has sown structural and organizational variation between species (5–9). In Escherichia coli, the homologous β-clamp is a homodimer, with each subunit containing three, rather than two, domains (7).
Figure 1.
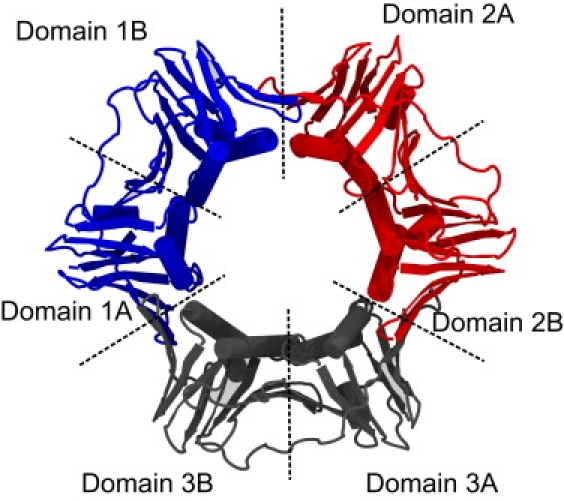
Overview of PCNA architecture. The crystal structure of PCNA from S. cerevisiae (PDB ID: 1PLQ) (5). PCNA contains three identical subunits assembled head-to-tail to form a ring. Each subunit contains two domains connected by a linker, giving the trimer pseudo sixfold symmetry.
PCNA is loaded onto DNA at a double-stranded/single-stranded junction by the ATP-dependent clamp loader, Replication Factor C (RFC) (10). RFC is a hetero-pentamer, whose five homologous subunits form a right-handed spiral, which orients the nucleic acid binding motifs to engage the DNA template (11). RFC facilitates loading by disrupting a single interface between PCNA subunits (12). Although the precise mechanism is unknown, several studies have illuminated key structural intermediates in the process. An electron microscopy reconstruction of the RFC-PCNA complex from the archae Pyrococcus furiosus shows a PCNA trimer that adopts an open lock-washer structure making extensive contacts with underside of RFC and matching its right-handed spiral (13). This structure is in stark contrast to the structure determined for the homologous RFC-PCNA complex from yeast (11), in which PCNA is closed and remains in the planar conformation observed for all isolated structures of PCNA to date (5,6,8,14,15). It is thought that these two structures of the RFC-PCNA complex might represent different intermediates along the loading pathway, although it has been suggested that the closed ring in the crystal structure might be the result of a synthetic sequence tag introduced into PCNA that stabilizes the subunit-subunit interface in that conformation (16). Nonetheless, it is clear that RFC potentiates a large conformational change in PCNA during the loading process, possibly in multiple steps (15).
Experimentally, it is difficult to determine the role of PCNA's mechanical properties in the mechanism of clamp loading, and also whether RFC is required to perform additional mechanical work to open PCNA after it disrupts one of the subunit-subunit interfaces. Specifically, is it necessary for PCNA, once an interface is broken, to preferentially adopt a spiral structure to avoid RFC having to pay a large energetic penalty on binding, or could spontaneous fluctuations result in binding to RFC? To investigate these questions, we performed all-atom explicit solvent molecular dynamics (MD) simulations on PCNA from yeast. The series of simulations presented here represents nearly an order-of-magnitude longer sampling than previous studies (17); the combination of significantly longer trajectories and the use of multiple replicas (18,19) allow for more exhaustive sampling of PCNA's conformational flexibility.
Our results indicate that PCNA does not display a preference for forming helical structures with a particular handedness, but instead diffuses over a large range of conformations, including those that come close to matching the helical pitch of RFC. We also develop a simple elastic network model (ENM) of PCNA that accurately reproduces its fluctuations and collective motions in the open state at equilibrium and allows us to directly calculate the energetic cost of mechanically deforming PCNA. The results of these studies indicate that PCNA appears to behave much like a simple elastic material, suggesting that once opened, RFC must direct the conformational transitions necessary for loading PCNA onto DNA.
Materials and Methods
Simulation details
Models of the DNA sliding clamp were generated using the crystal structure of PCNA from budding yeast Saccharomyces cerevisiae (Protein DataBank ID code 1PLQ) (5). Four identical dimeric systems were generated by removing one of the subunits and solvating the remaining two subunits in an explicit solvent box containing 150 mM NaCl. Independent trajectories were obtained by randomly initializing the velocities of both protein and solvent molecules for each replicate. All simulations were performed with the NAMD package (20) using the CHARMM22 protein force field (21) with CMAP corrections (22). Additional details can be found in the text of the Supporting Material.
Principal component analysis
We use principal component analysis (PCA) (23,24) to describe the collective motions of PCNA observed during the MD simulations. By diagonalizing the positional covariance matrix
| (1) |
where xi and xj are the atomic coordinates and x0i and x0j are the average position of atoms i and j after a least-squares superposition of all structures in the trajectory onto the average structure, we obtain a set of orthogonal modes describing the directions of maximal variation observed in the conformational ensemble. Note that the least-squares superposition removes the overall translational and rotational motions of the molecule before the calculation of Cij. Specifically, during the MD simulations a series of conformations was recorded every 4 ps. The first 5 ns of each trajectory were discarded as an equilibration period, and we analyzed the remaining conformations as follows. For each recorded protein conformation, a rigid fit to the first nonequilibration structure of the first trajectory was made such that the protein Cα atoms were fit to the coordinates of the same atoms in the starting structure by minimizing the root-mean-square deviation (RMSD) in the atomic positions of those atoms. This gives the average internal position of each Cα atom. The instantaneous structure from the MD simulations with the smallest RMSD (0.2 Å) from the average structure is used as the reference position in Eq. 1. This reference structure is also used as the reference structure for the anisotropic network model (ANM) in Eq. 2 and for PCA analysis of the ensemble of structures generated via the ANM. The GROMACS g_covar and g_anaeig tools (25) were used to calculate and analyze Cij.
Anisotropic network model
The ANM (26) is a coarse-grained elastic model of protein deformations. We calculate the normal modes of the system governed by the potential
| (2) |
where dij and dij0 are the instantaneous and reference distances between residues i and j, represented by their Cα positions, N is the number of atoms in the network, and H(x) is the Heaviside function, which excludes interactions between residues that are farther than the distance dcut apart in the reference structure. The force constant γ and dcut are both determined by fitting the root-mean squared fluctuations (RMSF) obtained from the all-atom MD simulations.
Reconstruction of a coarse-grained free energy surface
A coarse-grained free energy surface, W(X, Y), can be computed for the in- and out-of-plane order parameters (X and Y, respectively) by calculating
| (3) |
where P(X, Y) is a kernel density estimate (27,28) of the underlying probability density, computed using a Gaussian kernel with bandwidth σ
| (4) |
where kB is the Boltzmann factor, T is the temperature of the simulated system, σ is the bandwidth of the kernel density estimator, and N is the total number of conformations in the ensemble used to estimate the free energy. For each conformation in the ensemble, i, a kernel is placed at the corresponding point in the two-dimensional in- and out-of-plane space. Summing over all kernels gives an estimate of the conformational probability density. We have chosen an empirical value of σ = 2 Å for the surface shown later in Fig. 6.
Figure 6.
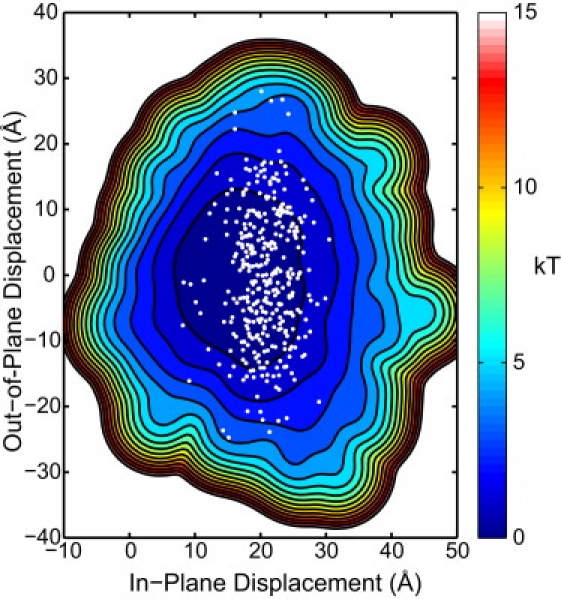
Coarse-grained free energy landscape for an open PCNA trimer. The conformational ensemble from the elastic network model is used to calculate the energetic cost of deforming an PCNA trimer with one disrupted subunit-subunit interface. Contour isolines are plotted in 1-kBT increments. The open points overlaid on the free energy landscape correspond to conformations in the ensemble that are consistent with the FRET distance measured in the presence of ATP and the clamp loader RFC (15).
Results
We have adopted a strategy similar to that used by Kazmirski et al. (17) in their pioneering computational study of PCNA. Thus, in setting up our simulations, we omitted one of the three subunits from the crystal structure of the PCNA trimer (PDB entry 1PLQ) (5) in order to model the open state without knowing the details of how the trimer reorganizes after an interface is disrupted. The choice of which subunit to delete is arbitrary due to the threefold symmetry imposed during the crystallographic refinement. We have deemed this approach preferable to attempting to drive the full trimer into an open conformation by applying external forces, because little is known about the opening pathway, and application of a force along ad hoc reaction coordinates could bias the resulting behavior of the system. The dimer represents a minimal unit with which to examine the mechanical properties of PCNA, containing one intact subunit-subunit interface. To this end, we have performed and analyzed four independent equilibrium simulations of dimeric PCNA from yeast, as well as one equilibrium simulation of the full trimeric yeast PCNA. From the four simulations, we obtain nearly 240 ns of total simulation time, with individual simulations of 92, 46, 51, and 48 ns in length. (See Materials and Methods for details.)
Analysis of the stability of the protein reveals the effect of removing one of the subunits. At the onset of the simulations, the PCNA dimer rapidly relaxes away from the starting conformation of crystallized trimeric PCNA, as measured by the rapid growth in RMSD in the Cα positions of the entire protein (Fig. 2). The individual domains, however, are quite stable, with an average RMSD of <1.0 Å, indicating that this conformational change is mainly due to the rigid body motions of the individual domains relative to one another.
Figure 2.
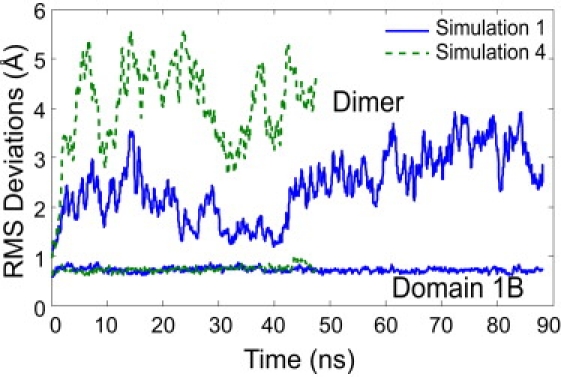
The RMSDs in Cα positions from the crystal structure for two of the four simulations. The dimer rapidly diverges from its starting conformation in both simulations; however, individual domains display small deviations when superimposed individually. The RMSDs of domain 1B are <1 Å over the entire simulation. The other three domains display similar RMSDs.
In-plane relaxation and out-of-plane motions of PCNA
To probe the effect these interdomain motions may have on the overall conformation of the dimer, we define two order parameters that monitor the conformational fluctuations observed during each simulation. Relative to the starting conformation and initial plane of the ring, these parameters decompose the fluctuations into in-plane and out-of-plane motions.
The degree to which PCNA fluctuates out-of-plane was measured by calculating the vertical displacement of the center-of-mass of domain 1A (Fig. 1) with respect to plane of the ring in the crystal structure. To accomplish this, a rectangular coordinate system was defined such that the z axis is the principal axis perpendicular to the plane of the ring of the PCNA crystal structure. Each instantaneous structure in a simulated trajectory was then aligned onto domain 2B of the crystal structure, and the z coordinate of the center-of-mass of domain 1A was recorded. Positive and negative values correspond to right- and left-handed out-of-plane deformations, respectively. In-plane opening was tracked by constructing a trimer of PCNA artificially using two copies of the instantaneous conformation. The first copy is aligned as described for the calculation of the out-of-plane coordinate. The second copy is then translated and rotated to superimpose domain 2A onto domain 1A of the first copy. This positions domain 1A and 1B of the second copy in the position of the third subunit in the trimer (domains 3A and 3B, respectively). Merging the coordinates of the first copy and domains 1A and 1B of the second creates an artificial trimer with a gap between subunits 2 and 3. Even though the motions of subunit 1 and 3 are artificially correlated, they should still capture the range of conformations of the open trimer that are thermally accessible. The conformational change in the in-plane direction is measured as d–d0, where d is the distance between the center-of-mass of domain 1A and the center-of-mass of 3B projected onto the xy plane, and d0 is the distance measured in the crystal structure. Conformations are sampled at 40-ps intervals after discarding the first 5 ns from each simulation.
Fig. 3 shows the projection of the longest simulation onto the above-defined parameters as a function of time, while Fig. 4 A shows the projections of all four simulations onto the two-dimensional space of the same parameters.
Figure 3.
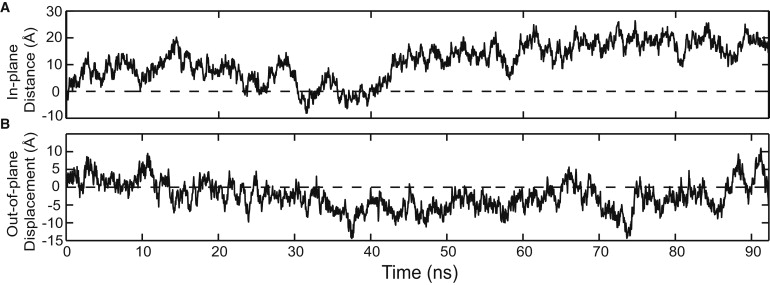
In-plane and out-of-plane motions of PCNA. (A) In-plane and (B) out-of-plane order parameters from dimer simulation 1 were calculated at 40-ps intervals. PCNA relaxes in the in-plane direction, while simultaneously diverging from the planar conformation found in the crystal structure of the closed trimer. In panel B, positive values of the order parameter correspond to conformations consistent with a right-handed spiral, while negative values correspond to left-handed spirals. Over the course of the 92-ns simulation, PCNA takes on conformations consistent with both right- and left-handed spirals.
Figure 4.
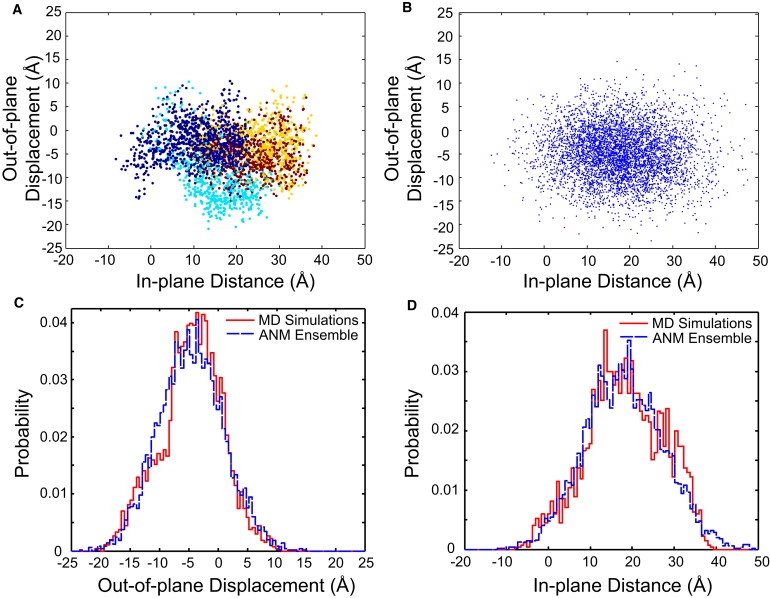
In-plane and out-of-plane conformations of PCNA. In-plane and out-of-plane order parameters from each of the four simulations were calculated at 40-ps intervals after removal of the first 5 ns of simulation. (A) Projection of in- and out-of-plane conformations. Each point represents a single conformation, colored by independent trajectory. (B) Conformational ensemble obtained using the anisotropic network model. (C) Histogram of out-of-plane displacements. (D) Histogram of in-plane distances. The data presented in panels C and D represent the aggregate of the MD simulations shown in panel A.
In all four simulations, PCNA relaxes in-plane to adopt a more open conformation. While there are oscillations in the in-plane distance, the peak in the distributions for the aggregate of all four simulations is ∼15 Å (Fig. 4 D). In-plane distances larger than 25 Å are observed in all four simulations. The in-plane distance can be related to the gap across the open interface (see the Supporting Material text); in all four simulations, gaps are observed which are sufficient to allow duplex DNA to be threaded through the opening.
In addition to the in-plane motions discussed above, the simulations also reveal divergences from the planar structure seen in the crystal structure of the full trimer. Fluctuations in the out-of-plane direction allow PCNA to realize conformations that are consistent with both right- and left-handed spirals. The distribution of out-of-plane displacements is strongly peaked with an average value of –4.8 ± 1.4 Å (± denotes standard error of the mean; see Supporting Material) (Fig. 4 C). Previously, Kazmirski et al. (17) observed primarily right-handed fluctuations in two out of three (10 ns) simulations of PCNA from yeast. In simulations that displayed predominantly conformations consistent with a left-handed spiral, they attributed that behavior to a rare energetically unfavorable breaking of two backbone hydrogen bonds in the intersubunit β-sheet. Our results, from >200 ns of simulation time, indicate no strong preference for spirals of a particular handedness, nor do they indicate a bistability in handedness induced by a reorganization of internal interactions. These results are robust to the choice of force field (see Supporting Material). Two 38-ns simulations of dimeric PCNA from P. furiosus (pfuPCNA) show a similar broad out-of-plane distribution, but with a slight bias in the opposite handedness and an average value of 5.5 ± 2.3 Å (Fig. S4).
Although the simulations of the PCNA dimer show that the molecule is able to sample nonplanar conformations, it is important to examine to what extent the closure of the full trimeric ring restricts those motions. Therefore, we performed a simulation (32 ns) of the full trimer to measure to what extent the ring maintained a planar conformation. We measure deviations from planarity using the same out-of-plane displacement order parameter used to analyze the dimer simulations. The sign of the displacement, in this context, no longer indicates whether the fluctuations are right- or left-handed; the continuity of the closed ring dictates that for any displacement out-of-plane, part of the protein will form a left-handed spiral, while the rest of the ring must form a right-handed spiral with a matching pitch. Interestingly, even the closed PCNA ring moves in and out of planarity several times during the trajectory. The maximum vertical displacement of the center-of-mass of domain 1A is ∼6 Å above and 7 Å below the plane, compared to the maximum displacement of 10 Å and 20 Å above and below the plane, respectively, observed in the dimer simulations (see Supporting Material).
A common set of internal deformations drive large-scale conformational changes
By connecting the global deformations of the dimer to a set of internal collective motions, we aim to understand the role of local protein flexibility in the conformational changes we observe in the dimer simulations. To this end, we have analyzed the conformational ensemble of the four MD simulations using principal component analysis (PCA) (23,24). PCA extracts a set of nonredundant modes that are ordered by their contribution to the total RMSF observed over the entire MD trajectory. The first two modes correspond to a bending and twisting motion of the dimer, induced by a bending and twisting motion of the β-sheet embedded in the intersubunit interface (Fig. S6). Projecting the simulated dynamics back onto these two modes demonstrates that they are uncorrelated (linear correlation coefficient r = –0.004), echoing the results derived from PCA of the static structures of anti-parallel β-sheets found in different proteins (29). Modes 3–5 correspond to similar bending and twisting deformations within the individual subunits mediated by the intrasubunit β-sheets. The additional contacts between the interdomain linker and the β-sheets within a subunit appear to stiffen those sheets relative to the flexibility observed in the unhindered intersubunit sheet. Together, these five modes account for 82% of the total displacement observed in the simulations.
Brownian-driven elastic deformations dominate the motions of dimeric PCNA
To investigate to what degree the topology and material elasticity govern the conformational dynamics, we construct a simpler physical model of PCNA, and ask whether it can describe the conformational changes that we observe in the all-atom MD simulations. Instead of working with the complete set of atomic coordinates, we build a coarse-grained ANM (26), such that PCNA is represented by only the Cα atoms connected by a homogeneous network of harmonic springs. This model allows for dynamics governed only by topologically determined conformational elasticity, without allowing for internal reorganization of interactions that could give rise to metastable conformational states. ANMs have been used to examine the conformational fluctuations of proteins around their native state, and recent work has shown a close correspondence between the collective motions described by an ANM and those characterized by performing PCA on ensembles of structures determined by nuclear magnetic resonance, X-ray crystallography, and MD simulations for HIV-1 Protease (30).
Here an ANM, based on the average structure obtained from the MD simulations, was parameterized with a single homogenous spring constant and cutoff distance to best reproduce the pattern and magnitude of the RMSF calculated from the all-atom simulations (Fig. 5 A). By optimizing the least-squares difference between the residue fluctuations obtained by the two methods, we obtained a correlation coefficient of 0.93, resulting from a cutoff of 13 Å and a uniform spring constant of 0.4 kcal/(mol Å2).
Figure 5.
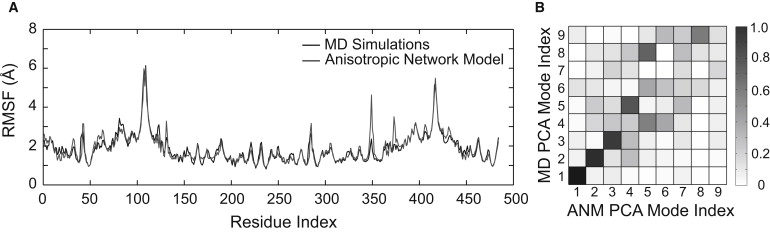
Comparison of all-atom MD simulations and anisotropic network model (ANM). (A) RMSF of Cα atoms from MD simulations (solid line) and ANM (shaded line). The ANM was fit to best reproduce the pattern and magnitude of the fluctuations. The linear correlation coefficient for the two sets of data is r = 0.93. (B) Grayscale plot showing the overlap between pairs of PCA modes calculated from the ANM (x axis) and MD simulations (y axis). The correspondence between modes is indicated by the shading of the block.
To make a direct comparison between the ANM and our all-atom simulations, we generated an ensemble of 10,000 independent structures based on the first 100 normal modes with nonzero eigenvalues, λi, by displacing the atomic coordinates along the eigenvector by a random normal variate with variance kBT/λi. We then performed PCA on the ensemble of conformations generated by the ANM using the same method described for the all-atom simulations.
To quantify the similarity between the collective motions extracted from the MD simulations and those from the ANM, we calculate the overlap, Oi, j between modes i and j, defined as the normalized dot product of the eigenvectors vi and vj for different models (31). An overlap of 1 indicates exact co-linearity of the motion described by that pair of modes. A pairwise comparison of the first nine modes from each model is shown in Fig. 5 B. Such a plot confirms the strong correspondence between the modes derived from the all-atom and elastic network modes; this is shown by the tight clustering of overlaps with values approaching 1 along the ideal diagonal. In particular, the first three modes have an overlap of 0.96, 0.88, and 0.83, respectively. These modes account for 75% of the total variance in the all-atom simulations.
Further comparison can be made between the ANM and the all-atom MD by projecting the ensemble of independent conformations onto the in- and out-of-plane order parameters. Projection of the ensemble onto the two-dimensional order parameter space is shown in Fig. 4 B, and the histograms of the conformations for each is compared with the all-atom simulations in Fig. 4, C and D. The in-plane distances from the two methods are shown to be substantially similar using the Kolmogorov-Smirnov test (p = 0.06). Although qualitatively similar, the out-of-plane displacements observed in the two ensembles of conformations do not display matching distributions with statistical significance. It is likely that we have not obtained an equilibrium distribution from our all-atom simulations, resulting in the spurious shoulder at −10 Å, which accounts for most of the difference between the distributions. Having only fit the ANM using the scalar mean-squared fluctuation of individual residues, the degree to which this simple model fits the directionality of the collective motions as well as the approximate distributions of conformations projected onto the two order parameters is remarkable. This is especially true when compared to systematic studies of large numbers of protein motions using ANM (32) and direct comparisons between ANM and MD simulations for specific proteins (30,33). This suggests that the dynamics of the open ring may be dominated by the elastic properties imbued by the topology of PCNA, rather than infrequent transitions between discrete conformational states.
Coarse-grained free energy landscape of trimeric PCNA in the open state
To estimate the energetic cost of deforming PCNA during the ring opening process once one of the interfaces has been disrupted, we build an ANM of the full trimer based on the parameterization that we have validated with the dimeric system. The PCNA trimer was constructed artificially by using two copies of the average structure from the MD simulations as described above. Using the procedure described for the ANM of the dimer, we generate an equilibrium ensemble consisting of 100,000 independent open-clamp conformations which we call the ensemble of trimers. We then calculate the extent of in- and out-of-plane opening in this ensemble, using the same order parameters described for the dimer, with the modification that the out-of-plane deviations are based on the vertical displacement of domain 3A. We construct a coarse-grained free energy landscape by calculating the probability distribution in the two-dimensional space of these order parameters using a kernel density estimate; the free energy is then obtained using Eq. 3.
The resulting coarse-grained two-dimensional free energy landscape is shown in Fig. 6, and suggests that once the contacts at an interface are disrupted either spontaneously or by interactions with RFC, PCNA easily diffuses among the conformations that are relevant to its biological function. Specifically, the structure of the archael clamp loader bound to PCNA in an open conformation (13) requires a vertical displacement of domain 3A of ∼18 Å, coupled with a small in-plane opening distance of ∼5–10 Å. Our coarse-grained free energy landscape suggests that this conformation transition relative to the preferred open-state, in the absence of RFC, requires only 3–5 kBT from the external environment.
To date the only available structural information on the open structure of the PCNA clamp is from an electron microscopy image of the clamp and RFC clamp loader on DNA (13) and a measurement of the distance across the gap based on fluorescent resonance energy transfer (FRET) measurements done in the presence of RFC and ATP (15). Onto our calculated free energy landscape (Fig. 6), we overlay points corresponding to conformations in the ensemble of trimers that match the distance computed from the FRET efficiency (34 ± 0.5 Å) observed between the two fluorescently labeled residues (15). We note that the vast majority of the points consistent with that distance range fall within a few 1–2 kBT and fewer than 2% of conformations are >3 kBT in energy of the minimum of the surface. The potential of mean force, calculated along the coordinate defined by the FRET pair distance, yields a similar result (Fig. S7): the optimum FRET distance measured lies in the middle of a wide, flat potential-of-mean-force minimum that rises <1 kBT over a range of ∼20 Å.
Discussion
Although structural studies of PCNA and the RFC complex that loads PCNA onto DNA indicate that it must interconvert between a closed planar ring and an open right-handed spiral (11,13,15), our understanding of this process is limited because it is difficult to measure the material properties of PCNA experimentally. In particular, it is essential to understand the conformational flexibility and energetic cost of deforming PCNA in the absence of RFC in order to understand how the complicated pieces of the loading machinery work in concert.
Our simulations provide insight into the mechanical properties of PCNA in isolation by examining the conformational fluctuations of the molecule in two forms. In the first form, we mimic an open ring by removing a single subunit, which allows the molecule to fluctuate as if the ring were maintained in its open form. These simulations show that PCNA relaxes in the in-plane direction, while transiently sampling out-of-plane conformations consistent with both left- and right-handed spirals. We also simulated PCNA in a second form—the full trimer in its stable closed configuration—which allows us to measure the effect of the closure constraint on the conformational fluctuations at equilibrium. While the closed ring does not allow lateral relaxation of the ring in-plane as observed in the dimer, the continuity of the ring does not inhibit PCNA from fluctuating out-of-plane.
The simulations suggest that the flexibility of PCNA appears sufficient for it to pattern itself onto the right-handed spiral of RFC once a ring-opening event has occurred, although PCNA itself does not have a strong preference to transiently adopt conformations of a particular handedness. In fact, PCNA from yeast and a thermophilic archaeon display small but opposite biases for fluctuations of a particular handedness. Although these biases may arise from incomplete sampling, our analysis nevertheless suggests that protein flexibility rather than equilibrium bias allows PCNA to deform to bind RFC.
Therefore, PCNA does not appear to be pretensioned to fit the binding surface of the clamp loader RFC (17), although our simulations confirm the observation that PCNA, like the E. coli β-clamp, relaxes in-plane when opened (17,34). Furthermore, in- and out-of-plane motions are uncorrelated, suggesting that if these motions are ordered along the opening pathway, the temporal arrangement of such intermediaries is dictated by RFC and not by energetic barrier imposed by PCNA's mechanical properties. In our simulations, PCNA appears to behave in a manner consistent with an elastic material undergoing stochastic deformations driven by Brownian forces from the solvent, suggesting a minimal role for long-range interactions or other structural elements within PCNA that would trap it in metastable conformations for long times. The small energetic cost of deforming PCNA over a wide range of conformations relative to kBT indicates that PCNA is likely compliant rather than complicit in the loading process.
Supporting Material
Further information containing details on the simulations and analysis, including two equations and seven figures, are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(10)00422-4.
Supporting Material
Acknowledgments
We thank Česlovas Venclovas and George Oster for critical reading of the manuscript. We thank the Livermore Computing Center at the Lawrence Livermore National Laboratory for simulation resources.
This work was supported by a U.S. Department of Energy Computational Science Graduate Fellowship (No. DE-FG02-97ER25308 to J.L.A), a distinguished postdoctoral fellowship from the California Institute for Quantitative Biosciences at the University of California, Berkeley (to J.D.C), funding from the Biotechnology and Biological Sciences Research Council through the University of Cambridge as well as sabbatical funding from Lawrence Livermore National Laboratory (to D.B.), and a Dreyfus New Faculty Award (to T.F.M). Part of this work was performed under the auspices of the U.S. Department of Energy by Lawrence Livermore National Laboratory under contract No. DE-AC52-07NA27344.
Footnotes
Joshua L. Adelman's present address is Department of Biosciences, University of Pittsburgh, Pittsburgh, PA.
References
- 1.Garg P., Burgers P.M.J. DNA polymerases that propagate the eukaryotic DNA replication fork. Crit. Rev. Biochem. Mol. Biol. 2005;40:115–128. doi: 10.1080/10409230590935433. [DOI] [PubMed] [Google Scholar]
- 2.Hubscher U., Maga G., Spadari S. Eukaryotic DNA polymerases. Annu. Rev. Biochem. 2002;71:133–163. doi: 10.1146/annurev.biochem.71.090501.150041. [DOI] [PubMed] [Google Scholar]
- 3.Maga G., Hubscher U. Proliferating cell nuclear antigen (PCNA): a dancer with many partners. J. Cell Sci. 2003;116:3051–3060. doi: 10.1242/jcs.00653. [DOI] [PubMed] [Google Scholar]
- 4.Moldovan G.-L., Pfander B., Jentsch S. PCNA, the maestro of the replication fork. Cell. 2007;129:665–679. doi: 10.1016/j.cell.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 5.Krishna T.S., Kong X.P., Kuriyan J. Crystal structure of the eukaryotic DNA polymerase processivity factor PCNA. Cell. 1994;79:1233–1243. doi: 10.1016/0092-8674(94)90014-0. [DOI] [PubMed] [Google Scholar]
- 6.Gulbis J.M., Kelman Z., Kuriyan J. Structure of the C-terminal region of p21(WAF1/CIP1) complexed with human PCNA. Cell. 1996;87:297–306. doi: 10.1016/s0092-8674(00)81347-1. [DOI] [PubMed] [Google Scholar]
- 7.Kong X.P., Onrust R., Kuriyan J. Three-dimensional structure of the β-subunit of E. coli DNA polymerase III holoenzyme: a sliding DNA clamp. Cell. 1992;69:425–437. doi: 10.1016/0092-8674(92)90445-i. [DOI] [PubMed] [Google Scholar]
- 8.Matsumiya S., Ishino Y., Morikawa K. Crystal structure of an archaeal DNA sliding clamp: proliferating cell nuclear antigen from Pyrococcus furiosus. Protein Sci. 2001;10:17–23. doi: 10.1110/ps.36401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moarefi I., Jeruzalmi D., Kuriyan J. Crystal structure of the DNA polymerase processivity factor of T4 bacteriophage. J. Mol. Biol. 2000;296:1215–1223. doi: 10.1006/jmbi.1999.3511. [DOI] [PubMed] [Google Scholar]
- 10.Yao N.Y., Johnson A., O'Donnell M. Mechanism of proliferating cell nuclear antigen clamp opening by replication factor C. J. Biol. Chem. 2006;281:17528–17539. doi: 10.1074/jbc.M601273200. [DOI] [PubMed] [Google Scholar]
- 11.Bowman G.D., O'Donnell M., Kuriyan J. Structural analysis of a eukaryotic sliding DNA clamp-clamp loader complex. Nature. 2004;429:724–730. doi: 10.1038/nature02585. [DOI] [PubMed] [Google Scholar]
- 12.Dionne I., Brown N.J., Bell S.D. On the mechanism of loading the PCNA sliding clamp by RFC. Mol. Microbiol. 2008;68:216–222. doi: 10.1111/j.1365-2958.2008.06150.x. [DOI] [PubMed] [Google Scholar]
- 13.Miyata T., Suzuki H., Morikawa K. Open clamp structure in the clamp-loading complex visualized by electron microscopic image analysis. Proc. Natl. Acad. Sci. USA. 2005;102:13795–13800. doi: 10.1073/pnas.0506447102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schurtenberger P., Egelhaaf S.U., Hübscher U. The solution structure of functionally active human proliferating cell nuclear antigen determined by small-angle neutron scattering. J. Mol. Biol. 1998;275:123–132. doi: 10.1006/jmbi.1997.1435. [DOI] [PubMed] [Google Scholar]
- 15.Zhuang Z., Yoder B.L., Benkovic S.J. The structure of a ring-opened proliferating cell nuclear antigen-replication factor C complex revealed by fluorescence energy transfer. Proc. Natl. Acad. Sci. USA. 2006;103:2546–2551. doi: 10.1073/pnas.0511263103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barsky D., Venclovas C. DNA sliding clamps: just the right twist to load onto DNA. Curr. Biol. 2005;15:R989–R992. doi: 10.1016/j.cub.2005.11.047. [DOI] [PubMed] [Google Scholar]
- 17.Kazmirski S.L., Zhao Y., Kuriyan J. Out-of-plane motions in open sliding clamps: molecular dynamics simulations of eukaryotic and archaeal proliferating cell nuclear antigen. Proc. Natl. Acad. Sci. USA. 2005;102:13801–13806. doi: 10.1073/pnas.0506430102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caves L.S., Evanseck J.D., Karplus M. Locally accessible conformations of proteins: multiple molecular dynamics simulations of crambin. Protein Sci. 1998;7:649–666. doi: 10.1002/pro.5560070314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ivetac A., McCammon J.A. Elucidating the inhibition mechanism of HIV-1 non-nucleoside reverse transcriptase inhibitors through multicopy molecular dynamics simulations. J. Mol. Biol. 2009;388:644–658. doi: 10.1016/j.jmb.2009.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.MacKerell A.D., Bashford D., Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 22.MacKerell A.D., Feig M., Brooks C.L. Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 23.Ichiye T., Karplus M. Collective motions in proteins: a covariance analysis of atomic fluctuations in molecular dynamics and normal mode simulations. Proteins. 1991;11:205–217. doi: 10.1002/prot.340110305. [DOI] [PubMed] [Google Scholar]
- 24.Kitao A., Hirata F., Go N. The effects of solvent on the conformation and the collective motions of protein: normal mode analysis and molecular dynamics simulations of melittin in water and in vacuum. Chem. Phys. 1991;158:447–472. [Google Scholar]
- 25.Hess B., Kutzner C., Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 26.Atilgan A.R., Durell S.R., Bahar I. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J. 2001;80:505–515. doi: 10.1016/S0006-3495(01)76033-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Silveman B.W. Monographs on Statistics and Applied Probability. Chapman and Hall; London: 1986. Density estimation for statistics and data analysis. [Google Scholar]
- 28.Yang S., Banavali N., Roux B. Mapping the conformational transition in Src activation by cumulating the information from multiple molecular dynamics trajectories. Proc. Natl. Acad. Sci. USA. 2009 doi: 10.1073/pnas.0808261106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Emberly E.G., Mukhopadhyay R., Wingreen N.S. Flexibility of β-sheets: principal component analysis of database protein structures. Proteins. 2004;55:91–98. doi: 10.1002/prot.10618. [DOI] [PubMed] [Google Scholar]
- 30.Yang L., Song G., Jernigan R.L. Close correspondence between the motions from principal component analysis of multiple HIV-1 protease structures and elastic network modes. Structure. 2008;16:321–330. doi: 10.1016/j.str.2007.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tama F., Sanejouand Y.H. Conformational change of proteins arising from normal mode calculations. Protein Eng. 2001;14:1–6. doi: 10.1093/protein/14.1.1. [DOI] [PubMed] [Google Scholar]
- 32.Yang L., Song G., Jernigan R.L. Protein elastic network models and the ranges of cooperativity. Proc. Natl. Acad. Sci. USA. 2009 doi: 10.1073/pnas.0902159106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lyman E., Pfaendtner J., Voth G.A. Systematic multiscale parameterization of heterogeneous elastic network models of proteins. Biophys. J. 2008;95:4183–4192. doi: 10.1529/biophysj.108.139733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jeruzalmi D., Yurieva O., Kuriyan J. Mechanism of processivity clamp opening by the δ-subunit wrench of the clamp loader complex of E. coli DNA polymerase III. Cell. 2001;106:417–428. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.