

Abstract
For over a decade, scientists have been attempting to know more about the conformational dynamics of fatty acid binding proteins (FABPs), to answer the puzzling question of how ligands could access the internalized binding site(s). Conformational exchange of FABPs on the microsecond to millisecond timescales has been found in many FABPs and offers an important hypothesis for the ligand entry mechanism. Despite the potential significance, the validity of this hypothesis has not been verified yet. In this study, the slow dynamics of human liver fatty acid binding protein (hLFABP) that was shown previously to be highly flexible on millisecond timescales was quantitatively characterized in detail. In addition, the interaction between hLFABP and 1,8-ANS was studied using NMR spectroscopy, and the kinetic rate of ANS association to hLFABP was measured. We believe the current result excludes the possibility that the intrinsic millisecond dynamics of hLFABP represents a critical conformational reorganization process required for ligand entry, but implies that it may represent the exchange between the apo-state and a state resembling the singly-bound conformation. Furthermore, we suggest these results show that the ligand-entry related functional dynamics could occur on the microsecond/submicrosecond timescales, highly encouraging future computational studies on this topic.
Introduction
Fatty acid binding proteins (FABPs) form an important family of intracellular proteins, which have high affinity with the poorly soluble lipid molecules and facilitate their intracellular transportation (1,2). Structural biology studies have shown that all FABPs share a well-conserved β-barrel structure with at least one internalized ligand binding site (3–8). However, despite the high resolution structures of FABPs solved, the mechanisms of how the ligand molecules access the internalized binding site have not yet been fully understood. Considering that the crystal structures of FABPs do not show obvious openings for ligand entry/exit, transient conformational reorganizations would play a critical role in ligand association/dissociation.
The area delimited by α-helix II and the turns between βC-βD and βE-βF strands, based on inspection of the crystallographic model of intestinal FABP (IFABP), was proposed previously as a portal region for ligand entry/exit (9). This region has since then received much attention (10–16). Until now, one of the strongest experimental evidences supporting this hypothesis was the dynamics studies on rat IFABP by Hodsdon and Cistola (12), which suggested that the portal region was significantly more flexible over a wide range of timescales (as evidenced by the extremely low order parameters (S2), remarkable contribution of conformation exchange to transverse relaxation (Rex), and significant amide proton exchange) than other parts of the protein. The dynamics data supported the hypothesis of a conformational disorder-order transition (11) although the timescale of the functional dynamics was not determined. Recently, another independent work by Zhang et al. (17) showed that the portal region of human IFABP was much more rigid on ps-ns timescales than suggested previously by Hodsdon and Cistola (12). In addition, fatty acid binding was not found to significantly change the backbone mobility of human IFABP on the ps-ns timescale (17). Although the dynamical behavior of IFABP on ps-ns timescales remains controversial, the dynamics studies on ps-ns timescales for heart, epidermal, muscle, and liver FABPs have shown the relative backbone rigidity of the portal region for these proteins (18–21). Thus, it is reasonable to surmise that the functional dynamics might happen on a much slower timescale. The previous kinetics study on IFABP implies that the ligand binding might be regulated by the protein conformational isomerization on the millisecond timescale (22). In addition, protein conformational dynamics on μs-ms timescales would have significant contribution to the NMR transverse relaxation rate. Indeed, the existence of Rex was noticed for various FABPs in the model free analyses (12,18,20) and was hypothesized to represent a conformational exchange of portal residues between the close and open states (18), providing an important hypothesis for the ligand entry mechanism. For evaluating this hypothesis, detailed and quantitative characterization of the slow motions of FABPs is indispensable, which, unfortunately, is missing in the literature.
Liver FABP (LFABP) is a unique family member, which can accommodate two fatty acids as well as many bulky and rigid ligands such as fatty acid-CoA thioesters, lysophosphatidic acid, bile salts, heme, 1,8-ANS, lipophilic drugs, etc. (2,23,24). The x-ray structures of both apo-LFABP and holo-LFABP (6,25) do not show any obvious openings on the protein surface, thus, significant conformational dynamics is expected for this protein so as to allow ligand entry and exit. He et al. (26) proposed recently that the apo-rat LFABP adopted an open conformation and ligands binding to LFABP might occur through conformational fluctuations of the α-helix cap. This open conformation is not found in the dominant conformation of apo-human LFABP (25,27), however, we recently identified that human LFABP (hLFABP) is quite flexible on millisecond timescales, indicating the potential conformational exchange between the open and closed states (28). This assumption is, nonetheless, pending for more rigorous experimental test. This study presents what we believe to be a detailed analysis of the millisecond dynamics of hLFABP. In addition, the validity of the hypothesis about the potential correlation between the millisecond dynamics and the ligand entry process of hLFABP is examined by analyzing the kinetic rates of hLFABP-ligand interaction.
Materials and Methods
Protein purification and NMR sample preparation
The procedure for expression and purification of 15N-labeled apo-hLFABP were described previously (21). Delipidation of the purified hLFABP followed the widely adopted lipidex method (29). By thorough buffer exchange, protein samples (∼1 mM hLFABP, 50 mM NaCl, 1 mM EDTA, pH 5.5) were prepared for relaxation experiments. The protein concentrations were determined according to the UV absorbance at 280 nm. For the preparation of partially saturated hLFABP by 1,8-ANS, a concentrated ANS solution (pH 5.5) was added into the hLFABP protein solution to obtain three samples with respective final molar ratios ([ANS]/[hLFABP]) of ∼0.25:1, 0.5:1, and 2.5:1. Note that the concentration of ANS or hLFABP hereafter denotes the total concentration instead of the free form in the solution of hLFABP-ANS complex. The pH values of the complex solutions were verified using a pH meter maintained at 5.5.
For verifying the Rex values detected in apo-hLFABP (delipidated by lipidex chromatography), an alternative delipidation method was also attempted to prepare apo-hLFABP. In this method, the purified hLFABP from Escherichia coli cells was denatured in 8 M urea to abort the protein's ability of binding lipids. The denatured protein was then applied to HPLC (hydrophobic column) to separate the unfolded LFABP and lipid molecules. After lyophilization of hLFABP (eluted from HPLC), the protein powder was redissolved in water and then changed to the desired buffer. In this way, hLFABP was found to refold quite successfully, producing identical 15N HSQC spectrum with the one delipidated by the lipidex method (without denaturation treatment). The hLFABP sample prepared by this unfolding-refolding method is believed to be lipid-free. However, the stability of the refolded hLFABP was found to be decreased, which resulted in slow precipitation. This sample, thus, was only used for verifying the Rex values of apo-hLFABP.
ANS titration
15N-labeled hLFABP (∼0.46 mM protein, 50 mM NaCl, pH 5.5, 90% H2O, and 10% D2O) was used for titration experiments at 20°C. Concentrated ANS solution (pH 5.5) was titrated into the protein solution at final concentrations of 0.11, 0.23, 0.46, 0.92, 1.8, and 3.57 mM, respectively. The HSQC spectrum at each ANS concentration was recorded on a 500 MHz spectrometer. Combined chemical shift perturbations (CCSP) (30,31) were calculated as follows:
| (1) |
where ΔδHN and ΔδN are the respective differences of 1H and 15N chemical shifts in the absence of ANS and in the presence of 0.46 mM ANS; αN is a scaling factor with a value of 0.17.
NMR relaxation experiments
Relaxation dispersion spectra for apo-hLFABP and partially saturated hLFABP ([ANS]/[protein] = 0.25:1 and 0.5:1) were recorded on Bruker 800 MHz and 500 MHz spectrometers at 20°C using the pulse scheme described previously (28). For the hLFABP sample partially saturated at the low affinity site ([ANS]/[protein] = 2.5:1), the relaxation dispersion profiles were recorded only at an 800 MHz field. A constant time delay (TCP = 50 ms) was used with a series of CPMG frequencies, ranging from 40 Hz to 960 Hz. For the correction of the apparent relaxation rates (28), the 15N relaxation times T1 and T1ρ, and 1H-15N NOEs of apo-hLFABP were measured at a 500 MHz field using inverse-detected two-dimensional NMR methods at 20°C (32,33). The relaxation delays of 5, 60, 150, 250, 360, and 505 ms were used for determination of T1 values. T1ρ values were determined by collecting seven points with delays of 5, 20, 40, 60, 80, 100, and 125 ms using a spin-lock field strength of 1600 Hz. Proton saturation time for the NOE measurement was 2.5 s, whereas the recycle delay was also 2.5 s. T2 values were calculated from T1 and T1ρ values. The corresponding T1 and T2 values at an 800 MHz field were calculated from the T1, T2, and NOE values at a 500 MHz field using the model free formalism (34). The correction values for the effective transverse relaxation rate () were generally <0.7 s−1. In the correction for partially saturated samples, minor changes of intrinsic relaxation rates due to ANS binding were neglected. The corrected dispersion profiles were subsequently fitted with the two-state exchange model to extract kinetics parameters. For the curve fitting of relaxation dispersion profiles of the apo-protein, the experimental data were fitted individually for each residue. For the curve fitting of relaxation dispersion profiles of the partially saturated hLFABP samples (of the concentration ratios 0.25:1 and 0.5:1), the experimental data of selected representative residues were fitted globally, assuming the same exchange rate and populations. In addition, the ΔωN values (between the apo-state and singly bound state) for individual residues were determined from the NMR titration result and were fixed in the curve fitting. The standard errors of the extracted parameters were estimated using the jackknife method.
Use of a temperature (20°C) lower than the physiological temperature is due to the better stability of protein-ANS complex when ANS is in excess ([ANS]/[hLFABP] = 2.5:1). At 20°C, the protein's function (interaction with fatty acids, ANS, and various other ligands) is well maintained. Thus, in vitro experiment at this temperature should be meaningful.
Results and Discussion
An assumptive model
To describe the hypothetical relationship between the intrinsic conformational exchange of hLFABP and the entry and exit of ligands, an assumptive model is presented as follows, which is based on the idea that protein conformational isomerization regulates the ligand binding process (22).
| (I) |
| (II) |
| (III) |
| (IV) |
| (V) |
In this model, the intrinsic conformational exchange is assumed to represent a dynamical equilibrium between open and closed states of the portal region(s), whereas the ligand molecules can only bind the protein through the open state that is invisible due to the low population. As a sequential multistep reaction, the overall exchange rates should be limited by the kinetic rates of individual steps. For binding of the first ligand at the high affinity site, the overall exchange can be represented as:
| (VI) |
that should satisfy:
| (2) |
where kon′ is the apparent on-rate, which depends on the concentration of the free ligand ([L]) in solution. This inequality condition (Eq. 2) will be rigorously tested in the following sections.
Intrinsic millisecond dynamics of apo-hLFABP
The relaxation dispersion spectra recorded at an 800 MHz field revealed that a large number of residues underwent conformational exchange at 20°C. The Rex value of each residue was approximated as
| (3) |
The Rex values for the apo-hLFABP prepared using the lipidex column were consistent with those for the refolded apo-hLFABP (data not shown). Thus, the Rex should be attributed to the intrinsic slow motions of apo-hLFABP instead of the interference with a trace amount of lipids that remained bound to hLFABP after delipidation using the lipidex column. Using a cutoff of Rex ≥ 1.5 s−1, 36 residues were identified as exchanging residues, which are highlighted on the three-dimensional structure of hLFABP, as shown in Fig. 1.Disappointedly, these exchanging residues, instead of locating around certain portal regions, are quite spread on the protein structure. This distribution of exchanging residues indicated that the conformational exchange of hLFABP might not represent the closed to open exchange of portal region(s).
Figure 1.
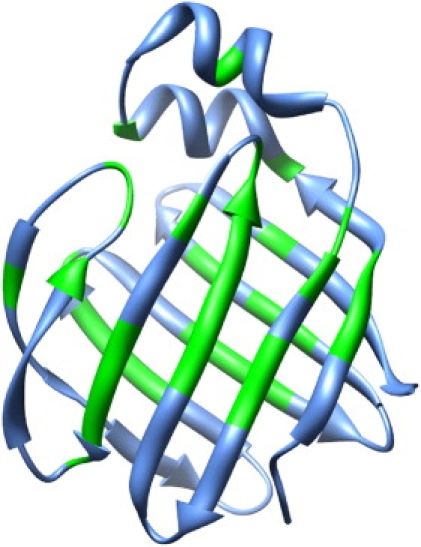
hLFABP residues undergoing conformational exchange (green). Based on the backbone assignment of hLFABP (27), these residues are identified as Y9, Q10, L11, N16, I24, I31, G39, V40, E42, I43, F52, T53, I54, T55, A56, G57, V60, N63, E64, F65, V67, E74, M76, T77, V81, V94, T95, T96, K98, V103, T104, E105, T112, N113, M115, K123. The protein structure is displayed using UCSF Chimera (39).
For quantitative analysis of the conformational exchange, the relaxation dispersion profiles were subsequently used for extracting exchange parameters (k1 and k−1). For reliable extraction of kinetics parameters through curve fitting, the Rex values of a given residue at both fields (500 MHz and 800 MHz) should be sufficiently large. Thus, in the current studies, only the residues that displayed Rex > 1.5 s−1 at both fields were fit into the theoretical model, which were E42, F52, T53, I54, T55, E64, F65, M76, and V94, as shown in Fig. 2. It is worth noting the extracted k1 values range from 4.2 s−1 to 12 s−1 (Fig. 2), which are considerably smaller than expected. According to the inequality condition (Eq. 2), kon′ should be limited to ≤∼10 s−1, provided that the conformational exchange observed on this timescale regulates the ligand binding.
Figure 2.
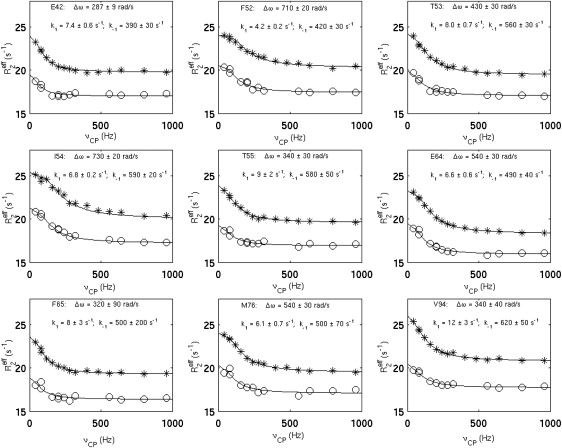
Relaxation dispersion profiles of the residues with intrinsic conformational exchange. The residues that displayed Rex ≥ 1.5 s−1 at both 500 MHz and 800 MHz fields were fitted with the two-state exchange model, and the kinetics parameters (k1 and k−1) and chemical shift difference (Δω) were extracted. Δω values are given in angular frequencies at 500 MHz field. The experimental data collected at 500 MHz and 800 MHz fields are represented as o and ∗, respectively. The solid lines are theoretical curves. The errors in k1, k−1, and Δω were estimated using the jackknife method, and they were similar to those estimated using the Monte Carlo method.
ANS binding studied by NMR titration
1,8-ANS is a widely known fluorescent analog of fatty acids, which interacts with FABPs (24,35,36). Previous fluorescence measurements showed that ANS can bind to two different sites of rat LFABP with binding affinities differing by an order (24). In this study, a large number of hLFABP residues were perturbed on titration of ANS, due to both direct binding and allosteric effects, as shown in Fig. 3. Chemical shift walking in the titration indicates that the overall exchange rates are fast/intermediate on the chemical shift time regime, and that most perturbed peaks moved sequentially at two different directions shows clearly that LFABP can bind two molecules of ANS (Fig. 3), which agrees with the previous result in the literature (24). It is worth noting that in the presence of excessive ANS molecules, ANS might form additional weak association on the surface of the hLFABP, which is evidenced by the minor change of chemical shift walking directions at high concentrations of ANS (e.g., G34, I31, and K59 in Fig. 3). This weak association could be hardly told from the titration profiles at low ANS concentrations ([ANS] ≤ 1.8 mM, [ANS]/[hLFABP] ≤ 4:1), showing that the affinity of this nonspecific association is much lower than the specific binding inside the β-cavity. Further titration of ANS ([ANS]/[hLFABP] > 8:1) had negligible changes on protein chemical shifts, but the resonance intensity was significantly weakened, which would be attributed to protein random aggregation induced by the nonspecific binding.
Figure 3.
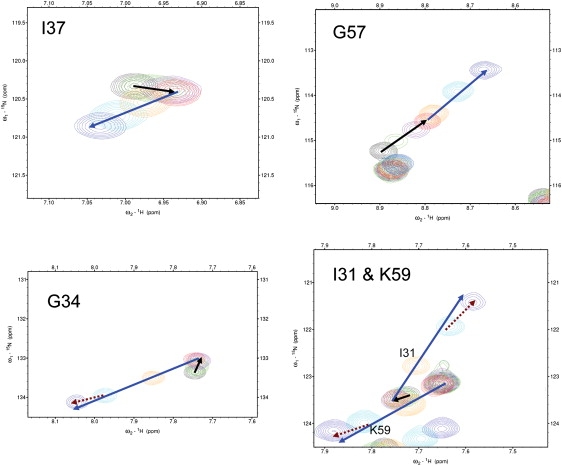
Representative residues in NMR titration experiments. The protein concentration was estimated to be ∼0.46 mM; the concentrations of ANS were: 0 (black), 0.11 (green), 0.23 (purple), 0.46 (red), 0.92 (orange), 1.8 (cyan), and 3.57 (light purple) mM. The direction of chemical shift walking due to ligand binding at the high (low) affinity site is indicated by the black (blue) arrow, (K59 did not show resonance shift on binding at the high affinity site); the minor change of the direction due to additional weak association is indicated by the dashed brown arrow.
Kinetic rates of ANS binding at the high affinity site
At low concentrations of ANS ([ANS]/[hLFABP] = 1/4 or 1/2), ANS primarily binds at the high affinity site, as shown in formula (Eq. VI). Due to the dominant populations of apo-LFABP and holo-LFABP(S), the exchange between apo-LFABP and holo-LFABP(S) would dominate the contribution to the Rex, although the exchange with other minor states of hLFABP might also exist.
To quantitatively compare kon′ and k1, the relaxation dispersion spectra of partially saturated proteins were recorded. When hLFABP was ∼1/4 or 1/2 saturated by ANS, the signal intensities for the residues showing chemical shift perturbation were significantly weakened, accompanied by the broadening of line-widths, indicating that the exchange rate of ANS for the high affinity binding site is close to the intermediate rate on the chemical shift time regime. To quantitate the exchange rates, selected residues, which displayed large Rex values and good signal/noise ratios, were fitted with the theoretical model. As shown in Fig. 4 A and B, global fitting of the dispersion profiles of four selected representative residues successfully extracted the apparent on-rates (kon′) and off-rate (koff), which agreed with the expected population ratios (pfree/pbound = koff/kon′) and confirmed that the exchange rate is comparable with the chemical shift differences (Δω). In addition, the off-rates extracted at two different ANS concentrations agreed with each other within experimental errors, which is expected as koff is independent of [L]. The fact that the on-rates (kon′, 85 and 216 s−1) are much (one order) larger than k1 (∼10 s−1) violates the basic condition (Eq. 2) for the initial assumptive model. Based on that, we conclude that the intrinsic millisecond timescale dynamics of hLFABP should not represent a conformational switch allowing ligand entry. The protein conformational rearrangement, which creates ligand exchange pathways, should occur on a much faster timescale.
Figure 4.
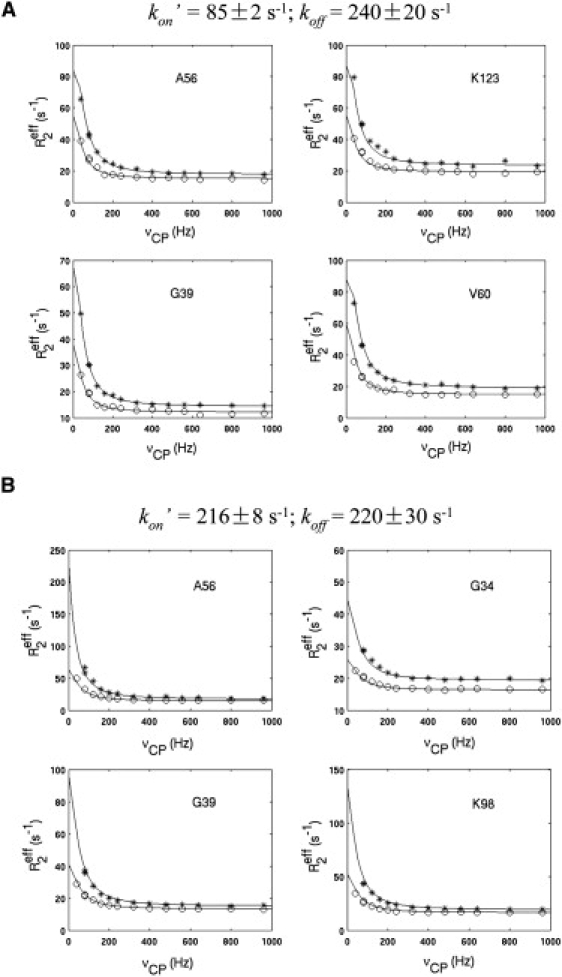
Kinetics of hLFABP-ANS interaction. The hLFABP-ANS samples were prepared by adding ANS into the solution of hLFABP with final molar ratios (ANS/hLFABP) of (A) ∼1:4 and (B) ∼1:2, respectively. The experimental data of representative residues recorded at 500 MHz (o) and 800 MHz (∗) fields were fitted with the two-state exchange model (solid lines) globally as described in the Materials and Methods.
Kinetic rates of ANS binding at the low affinity site
Binding/unbinding of the second ANS molecule seems even faster than that of the first ANS, because no obvious reduction of signal intensities was observed when the low affinity binding site was partially saturated. To estimate the kex of the second binding site, the dispersion profile of hLFABP, in which the second binding site was ∼1/2 saturated (when [ANS]/[hLFABP] ≈ 2.5:1), was recorded. However, due to the fast exchange rate, a CPMG frequency of 960 Hz is not sufficient to suppress all the exchange contribution to transverse relaxation. In the fast exchange limit, the residual Rex at a given CPMG frequency can be written as (37):
| (4) |
in which is the relaxation rate free of the exchange contribution. was not directly measured, but could be estimated as the of the apo-hLFABP measured at νCP of 960 Hz because the exchange contribution to at this CPMG field strength was negligible for the apo-protein (as shown in the Supporting Material). Thus,
| (5) |
Relaxation dispersion profiles of four representative residues are shown in Fig. 5 A–D. It should be noted that in the presence of excessive ANS molecules, multiple exchange processes would contribute to the Rex values. The effects of millisecond exchange processes (e.g., protein conformational exchange and ligand association/dissociation at the high affinity site) can be effectively suppressed at the CPMG field strength of 960 Hz. However, fast exchange processes other than ligand association at the low affinity site (e.g., nonspecific association between ANS and hLFABP and weak self-aggregation of hLFABP) may also contribute to Rex, the effects of which may not be suppressed completely at νCP of 960 Hz. In addition, the protein aggregation would increase the intrinsic transverse relaxation rates. Therefore, the estimation (Eq. 5) actually gives only an upper limit of the Rex(νCP = 960 Hz) that is contributed by the fast exchange of ligand association at the low affinity site, i.e.,
| (6) |
Figure 5.
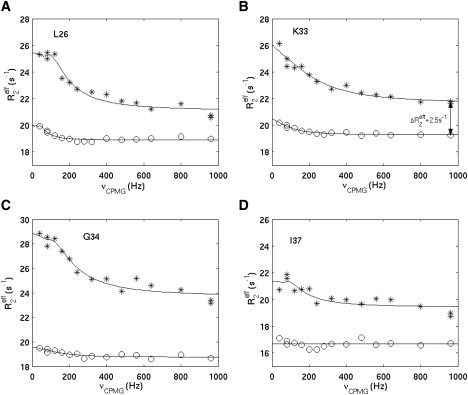
The relaxation dispersion profiles of representative residues. The ∗ represents the effective relaxation rates measured using the hLFABP sample, in which the low affinity site was ∼1/2 saturated. The o represents the effective relaxation rates measured using the apo-hLFABP sample. All the experiments were conducted using an 800 MHz Bruker machine at 20°C. The relaxation rates (∗) were fitted with the theoretical curve for demonstration purpose, which should represent the millisecond exchange that can be effectively suppressed at νCP of 960 Hz. It should not be confused with the fast exchange of ligand association/dissociation at the low affinity site.
Based on the upper limit of residual Rex at νCP of 960 Hz, the lower limit of kex can be calculated using Eq. 4. The residue K33 (Fig. 5 B), which shows the relatively high value, was used for calculation here. The ΔωN of K33 was 1.04 ppm, corresponding to 529 rad/s at an 800 MHz field. of K33 was 2.5 s−1, as shown in Fig. 5 B. The populations (pA, pB) were estimated from the population-weighted average chemical shift of K33, which was 56% and 44% respectively. Therefore,
and
Despite the complicated contributions to the transverse relaxation rates of hLFABP when the low affinity site was partially saturated, a rough estimation of the lower limit of the apparent on-rate for the low affinity site shows that it is one to two orders larger than that for the high affinity site. The very fast exchange rate for the low affinity binding site could also be qualitatively verified from the chemical shift walking over long distances (up to ∼0.3 ppm at the hydrogen dimension, corresponding to 942 rad/s at a 500 MHz field) without obvious weakening of the signal intensities in the middle way of the walking. This result also confirms that the formation of ligand entry/exit pathways should take place on a fast timescale, approaching the microsecond or submicrosecond timescale. The most recent advances in the algorithms, software, and computer hardware for molecular dynamics simulations have already made this timescale accessible (38), thus, the current result would highly encourage such long-timescale molecular dynamics simulations on FABPs, which could provide many mechanistic details of the ligand entry process.
Chemical shift perturbation pattern—an implication for the nature of the minor state
From the titration result, it was noticed that the chemical shifts of a large number of residues were perturbed on ligand binding. When both binding sites were saturated, almost all the residues displayed obvious resonance shift; however, when only the high affinity binding site was saturated, the resonance shift was observed for only part of the protein residues. The combined CCSP was calculated for the high affinity binding site, as shown in Fig. 6, to highlight regions with obvious changes. Surprisingly, plotted on the structure of hLFABP, these residues are found to roughly overlap with those showing intrinsic conformational exchange in the absence of ligands. As shown in Fig. 7, there is a large common region (yellow) that shows both Rex in the apo-protein and significant CCSP in the current titration experiment. This result implicates that the minor state of apo-hLFABP might represent the bound conformation of hLFABP, in which the high affinity site is occupied whereas the low affinity site is free.
Figure 6.
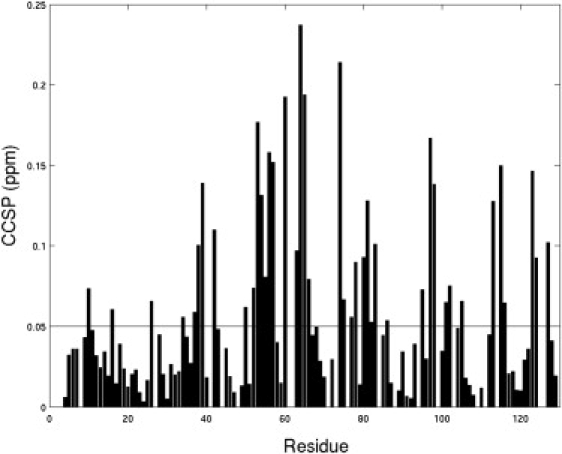
Histogram of CCSP of hLFABP on addition of ANS (at ∼1:1 ratio).
Figure 7.
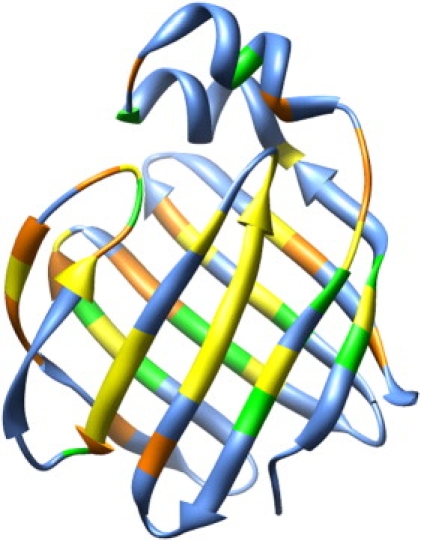
Comparison of protein regions showing resonance shift and those showing conformational exchange in the absence of ligands. Residues displaying significant resonance shift (CCSP ≥ 0.05 ppm) are shown in yellow and orange. Residues displaying significant Rex (≥1.5 s−1) are shown in yellow and green. The yellow region displayed both characteristics.
Distinct regions (Fig. 7, orange and green), which showed only CCSP (>0.05 ppm) or Rex (>1.5 s−1) in the apo-form, are also highlighted on the structure. Such a difference is not unexpected because the CCSP is contributed by 1H and 15N chemical shift changes caused by both conformational changes and direct protein-ligand contact. On the other hand, the Rex detected for the apo-form relies on the nitrogen chemical shift difference (ΔωN) between the minor and major apo-protein conformations. It is noteworthy that similar (but not completely identical) conformations could produce different backbone chemical shift patterns in practice. The minor state of apo-hLFABP may resemble the bound conformation with a single ANS, but they are not necessarily identical.
Conclusions
In this study, detailed characterization of the slow motions of hLFABP was conducted, and the potential correlation between slow dynamics and the ligand entry/exit processes was modeled and then tested by experiments. Although the initial purpose of this study was to establish a dynamics-function relationship, our experimental results, unfortunately, reject the assumed model due to the contradiction of timescales. Future investigation of the functional dynamics of hLFABP relevant to ligand entry, which may be due to the intrinsic dynamical property or be induced by the ligand molecules, should be focused on the microsecond or submicrosecond timescale. Because the current study is based entirely on hLFABP, these results do not exclude the possibility that other FABPs may adopt different mechanisms. Comparative studies between hLFABP and various other FABPs, including IFABP and its helix-less variant (22), would be an interesting future direction.
Regarding the nature of the conformational exchange of hLFABP on the millisecond timescales, our current experimental results are not yet sufficient for drawing any conclusion. However, qualitative comparison of the patterns of the intrinsic conformational exchange and the chemical shift perturbation implies that the invisible state might represent the singly bound state of hLFABP. In other words, this protein may have the intrinsic trend to access one bound-state conformation even in the absence of the ligands. This assumption, however, should be subjected to more rigorous experimental test in the future.
Supporting Material
Additional text and one figure are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(10)00413-3.
Supporting Material
Acknowledgments
The authors thank Mr. W. Song for sharing the initial titration results of hLFABP with various ligands.
This work was supported by the Biomedical Research Council, Agency for Science, Technology and Research of Singapore (R154000373305).
References
- 1.Storch J., Corsico B. The emerging functions and mechanisms of mammalian fatty acid-binding proteins. Annu. Rev. Nutr. 2008;28:73–95. doi: 10.1146/annurev.nutr.27.061406.093710. [DOI] [PubMed] [Google Scholar]
- 2.Thompson J., Reese-Wagoner A., Banaszak L. Liver fatty acid binding protein: species variation and the accommodation of different ligands. Biochim. Biophys. Acta. 1999;1441:117–130. doi: 10.1016/s1388-1981(99)00146-8. [DOI] [PubMed] [Google Scholar]
- 3.Xu Z., Bernlohr D.A., Banaszak L.J. The adipocyte lipid-binding protein at 1.6-A resolution. Crystal structures of the apoprotein and with bound saturated and unsaturated fatty acids. J. Biol. Chem. 1993;268:7874–7884. [PubMed] [Google Scholar]
- 4.Young A.C., Scapin G., Sacchettini J.C. Structural studies on human muscle fatty acid binding protein at 1.4 A resolution: binding interactions with three C18 fatty acids. Structure. 1994;2:523–534. doi: 10.1016/s0969-2126(00)00052-6. [DOI] [PubMed] [Google Scholar]
- 5.Lassen D., Lücke C., Rüterjans H. Three-dimensional structure of bovine heart fatty-acid-binding protein with bound palmitic acid, determined by multidimensional NMR spectroscopy. Eur. J. Biochem. 1995;230:266–280. doi: 10.1111/j.1432-1033.1995.tb20560.x. [DOI] [PubMed] [Google Scholar]
- 6.Thompson J., Winter N., Banaszak L. The crystal structure of the liver fatty acid-binding protein. A complex with two bound oleates. J. Biol. Chem. 1997;272:7140–7150. doi: 10.1074/jbc.272.11.7140. [DOI] [PubMed] [Google Scholar]
- 7.Hohoff C., Börchers T., van Tilbeurgh H. Expression, purification, and crystal structure determination of recombinant human epidermal-type fatty acid binding protein. Biochemistry. 1999;38:12229–12239. doi: 10.1021/bi990305u. [DOI] [PubMed] [Google Scholar]
- 8.Balendiran G.K., Schnutgen F., Sacchettini J.C. Crystal structure and thermodynamic analysis of human brain fatty acid-binding protein. J. Biol. Chem. 2000;275:27045–27054. doi: 10.1074/jbc.M003001200. [DOI] [PubMed] [Google Scholar]
- 9.Sacchettini J.C., Gordon J.I., Banaszak L.J. Crystal structure of rat intestinal fatty-acid-binding protein. Refinement and analysis of the Escherichia coli-derived protein with bound palmitate. J. Mol. Biol. 1989;208:327–339. doi: 10.1016/0022-2836(89)90392-6. [DOI] [PubMed] [Google Scholar]
- 10.Long D., Mu Y., Yang D. Molecular dynamics simulation of ligand dissociation from liver fatty acid binding protein. PLoS One. 2009;4:e6081. doi: 10.1371/journal.pone.0006081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hodsdon M.E., Cistola D.P. Discrete backbone disorder in the nuclear magnetic resonance structure of apo intestinal fatty acid-binding protein: implications for the mechanism of ligand entry. Biochemistry. 1997;36:1450–1460. doi: 10.1021/bi961890r. [DOI] [PubMed] [Google Scholar]
- 12.Hodsdon M.E., Cistola D.P. Ligand binding alters the backbone mobility of intestinal fatty acid-binding protein as monitored by 15N NMR relaxation and 1H exchange. Biochemistry. 1997;36:2278–2290. doi: 10.1021/bi962018l. [DOI] [PubMed] [Google Scholar]
- 13.Ory J., Kane C.D., Bernlohr D.A. Biochemical and crystallographic analyses of a portal mutant of the adipocyte lipid-binding protein. J. Biol. Chem. 1997;272:9793–9801. doi: 10.1074/jbc.272.15.9793. [DOI] [PubMed] [Google Scholar]
- 14.Richieri G.V., Low P.J., Kleinfeld A.M. Binding kinetics of engineered mutants provide insight about the pathway for entering and exiting the intestinal fatty acid binding protein. Biochemistry. 1999;38:5888–5895. doi: 10.1021/bi982703n. [DOI] [PubMed] [Google Scholar]
- 15.Jenkins A.E., Hockenberry J.A., Bernlohr D.A. Testing of the portal hypothesis: analysis of a V32G, F57G, K58G mutant of the fatty acid binding protein of the murine adipocyte. Biochemistry. 2002;41:2022–2027. doi: 10.1021/bi015769i. [DOI] [PubMed] [Google Scholar]
- 16.Mihajlovic M., Lazaridis T. Modeling fatty acid delivery from intestinal fatty acid binding protein to a membrane. Protein Sci. 2007;16:2042–2055. doi: 10.1110/ps.072875307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang X., Sui X., Yang D. Probing methyl dynamics from 13C autocorrelated and cross-correlated relaxation. J. Am. Chem. Soc. 2006;128:5073–5081. doi: 10.1021/ja057579r. [DOI] [PubMed] [Google Scholar]
- 18.Constantine K.L., Friedrichs M.S., Farmer B.T. Backbone and side chain dynamics of uncomplexed human adipocyte and muscle fatty acid-binding proteins. Biochemistry. 1998;37:7965–7980. doi: 10.1021/bi980203o. [DOI] [PubMed] [Google Scholar]
- 19.Lücke C., Fushman D., Rüterjans H. A comparative study of the backbone dynamics of two closely related lipid binding proteins: bovine heart fatty acid binding protein and porcine ileal lipid binding protein. Mol. Cell. Biochem. 1999;192:109–121. [PubMed] [Google Scholar]
- 20.Gutiérrez-González L.H., Ludwig C., Lücke C. Solution structure and backbone dynamics of human epidermal-type fatty acid-binding protein (E-FABP) Biochem. J. 2002;364:725–737. doi: 10.1042/BJ20020039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Long D., Yang D. Buffer interference with protein dynamics: a case study on human liver fatty acid binding protein. Biophys. J. 2009;96:1482–1488. doi: 10.1016/j.bpj.2008.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cistola D.P., Kim K., Frieden C. Fatty acid interactions with a helix-less variant of intestinal fatty acid-binding protein. Biochemistry. 1996;35:7559–7565. doi: 10.1021/bi952912x. [DOI] [PubMed] [Google Scholar]
- 23.Thompson J., Ory J., Banaszak L. The liver fatty acid binding protein—comparison of cavity properties of intracellular lipid-binding proteins. Mol. Cell. Biochem. 1999;192:9–16. [PubMed] [Google Scholar]
- 24.Chuang S., Velkov T., Scanlon M.J. Characterization of the drug binding specificity of rat liver fatty acid binding protein. J. Med. Chem. 2008;51:3755–3764. doi: 10.1021/jm701192w. [DOI] [PubMed] [Google Scholar]
- 25.Kursula, P., A. G. Thorsell, C. Arrowsmith, H. Berglund, A. Edwards, et al. 2005. Crystal structure of human FABP1. PDB entry: 2F73.
- 26.He Y., Yang X., Stark R.E. Solution-state molecular structure of apo and oleate-liganded liver fatty acid-binding protein. Biochemistry. 2007;46:12543–12556. doi: 10.1021/bi701092r. [DOI] [PubMed] [Google Scholar]
- 27.Xu Y., Long D., Yang D. Rapid data collection for protein structure determination by NMR spectroscopy. J. Am. Chem. Soc. 2007;129:7722–7723. doi: 10.1021/ja071442e. [DOI] [PubMed] [Google Scholar]
- 28.Long D., Liu M., Yang D. Accurately probing slow motions on millisecond timescales with a robust NMR relaxation experiment. J. Am. Chem. Soc. 2008;130:2432–2433. doi: 10.1021/ja710477h. [DOI] [PubMed] [Google Scholar]
- 29.Glatz J.F., Veerkamp J.H. Removal of fatty acids from serum albumin by Lipidex 1000 chromatography. J. Biochem. Biophys. Methods. 1983;8:57–61. doi: 10.1016/0165-022x(83)90021-0. [DOI] [PubMed] [Google Scholar]
- 30.Tochio H., Hung F., Zhang M. Solution structure and backbone dynamics of the second PDZ domain of postsynaptic density-95. J. Mol. Biol. 2000;295:225–237. doi: 10.1006/jmbi.1999.3350. [DOI] [PubMed] [Google Scholar]
- 31.Fan J.S., Cheng Z., Yang D. Solution and crystal structures of mRNA exporter Dbp5p and its interaction with nucleotides. J. Mol. Biol. 2009;388:1–10. doi: 10.1016/j.jmb.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 32.Farrow N.A., Muhandiram R., Kay L.E. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry. 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- 33.Ran X., Miao H.H., Yang D. Structural and dynamic characterization of a neuron-specific protein kinase C substrate, neurogranin. Biochemistry. 2003;42:5143–5150. doi: 10.1021/bi0271751. [DOI] [PubMed] [Google Scholar]
- 34.Yang D., Mok Y.K., Kay L.E. Contributions to protein entropy and heat capacity from bond vector motions measured by NMR spin relaxation. J. Mol. Biol. 1997;272:790–804. doi: 10.1006/jmbi.1997.1285. [DOI] [PubMed] [Google Scholar]
- 35.Kirk W.R., Kurian E., Prendergast F.G. Characterization of the sources of protein-ligand affinity: 1-sulfonato-8-(1′)anilinonaphthalene binding to intestinal fatty acid binding protein. Biophys. J. 1996;70:69–83. doi: 10.1016/S0006-3495(96)79592-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Velkov T., Chuang S., Scanlon M.J. The interaction of lipophilic drugs with intestinal fatty acid-binding protein. J. Biol. Chem. 2005;280:17769–17776. doi: 10.1074/jbc.M410193200. [DOI] [PubMed] [Google Scholar]
- 37.Luz Z., Meiboom S. Nuclear magnetic resonance study of the proteolysis of trimethylammonium ion in aqueous solution-order of the reaction with respect to solvent. J. Chem. Phys. 1963;39:366–370. [Google Scholar]
- 38.Klepeis J.L., Lindorff-Larsen K., Shaw D.E. Long-timescale molecular dynamics simulations of protein structure and function. Curr. Opin. Struct. Biol. 2009;19:120–127. doi: 10.1016/j.sbi.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 39.Pettersen E.F., Goddard T.D., Ferrin T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.