

Abstract
Single-molecule spectroscopy was employed to elucidate the fluorescence spectral heterogeneity and dynamics of individual, immobilized trimeric complexes of the main light-harvesting complex of plants in solution near room temperature. Rapid reversible spectral shifts between various emitting states, each of which was quasi-stable for seconds to tens of seconds, were observed for a fraction of the complexes. Most deviating states were characterized by the appearance of an additional, red-shifted emission band. Reversible shifts of up to 75 nm were detected. By combining modified Redfield theory with a disordered exciton model, fluorescence spectra with peaks between 670 nm and 705 nm could be explained by changes in the realization of the static disorder of the pigment-site energies. Spectral bands beyond this wavelength window suggest the presence of special protein conformations. We attribute the large red shifts to the mixing of an excitonic state with a charge-transfer state in two or more strongly coupled chlorophylls. Spectral bluing is explained by the formation of an energy trap before excitation energy equilibration is completed.
Introduction
Containing at least half of all the chlorophyll (Chl) pigments in chloroplasts, the main light-harvesting complex of plants and green algae, LHCII, serves as the principal collector of solar energy on earth (1,2). When a photon is captured, the absorbed energy is efficiently funneled via excitation energy transfer to the reaction center, where rapid charge separation temporally stabilizes the energy (3). The LHCII antenna is located on the periphery of photosystem II (PSII) and naturally assembles into a trimeric structure of three very similar pigment-protein subunits (4,5). The primary function of the protein is to bind pigments in an arrangement that ensures both efficient absorption of solar photons and protection against excess light and reactive oxygen species. The resolved structure of LHCII revealed eight Chl a's, six Chl b's, and four carotenoids (Cars), as well as their ensemble-average orientations and relative positions (2,6).
This detailed structural information allowed a decisive study of the spectroscopic and energy transfer properties of LHCII, which gave rise to a comprehensive picture of the energy equilibration in the trimer (7,8). The Chls in each monomeric subunit couple strongly into four excitonic clusters, resulting in ultrafast (<300 fs) relaxation within a cluster, followed by slower (subpicosecond to ∼10 ps) exciton hopping between them, and subsequent intermonomeric and eventually intertrimeric energy transfer. A robust disordered exciton model (7), based on modified Redfield theory, was developed to explain all of the spectroscopic features that were revealed by numerous experimental techniques. The model included 1), explicit estimates of the pigment-site energies; 2), intrinsic disorder of the pigment-site energies; 3), excitonic coupling between Chl pigments; and 4), an estimate of the interaction between the pigment electronic transitions and the protein's nuclear vibrations, known as exciton-phonon coupling. Nuclear perturbations (phonons) are often separated into two classes: those that relax on timescales much shorter than the characteristic fluorescence lifetime (denoted as dynamic disorder), and considerably slower collective motions (static disorder). The strong interpigment excitonic interactions of the Chls in LHCII delocalize the excitations over multiple pigments, whereas the disorder promotes localized states. The interplay between these couplings and the different contributions of disorder determines the energy equilibration in the system and the properties of the lowest exciton state.
The glass-like disorder of proteins (9) gives rise to heterogeneous exciton-phonon coupling in pigment-protein complexes and an essentially unique time-dependent realization of the static disorder for every complex. In the context of the protein energy landscape model (10), an excitation perturbation increases the probability of crossing energy barriers between different local minima, enabling the protein to traverse an extended region of its excited-state and ground-state landscapes. As a result of the strong exciton-phonon coupling and barriers of different heights, these conformational fluctuations give rise to spectral fluctuations of different extents and timescales (11). Various pigment-protein complexes exhibit considerable spectral diffusion, as revealed by single-molecule spectroscopy (SMS) (12–19). This technique provides insight into various fluctuation phenomena that are impossible to resolve with standard ensemble-averaging techniques.
SMS generally exploits fluorescence intensity fluctuations, allowing wavelength-integrated fluctuations on timescales down to 0.1–10 ms to be resolved (20–22). By investigating spectral diffusion on subsecond-to-second timescales, SMS is able to delicately elucidate the structure and function of various intrinsically fluorescent systems (12–19). The relatively robust light-harvesting complexes of purple bacteria are the most thoroughly studied natural multi-pigment-protein systems (see Cogdell and Köhler (23) for a recent review).
The first SMS studies of LHCII revealed intensity fluctuations at room temperature, as well as polarization and small spectral fluctuations at cryogenic temperatures, with a photobleaching behavior that was found to be independent of methods used to achieve complex immobilization (24,25). However, the two studies reported opposing outcomes for the energetic coupling between monomeric subunits, which was ascribed to small differences in the complex environment or sample treatment.
In this work, we investigated the fluorescence spectral dynamics of single LHCII trimers close to room temperature, for the first time to our knowledge. Many of the spectral properties are qualitatively related to simulated results to describe the energy delocalization of the various accessible lowest excitonic states.
Materials and Methods
Sample isolation
PSII antenna complexes were isolated from spinach thylakoids as described previously (26,27), with minor modifications. Additional gel filtration substantially reduced the fraction of monomers and free Chl pigments (see Fig. S1 in the Supporting Material). No observable increase in the nontrimeric contribution was found after a freeze-thaw cycle of a sample stored at −80°C. A high-performance liquid chromatography analysis (28) revealed that a third of the complexes contained Vio, and only trace amounts of contaminants were present.
Sample preparation and immobilization
The purified LHCII trimeric complexes were solubilized in a buffer (20 mM Tris, pH 8.0; 1 mM MgCl2; and 0.03% (w/v) n-dodecyl-β,D-maltoside) and diluted to a final concentration of ∼10 pM. A drop of a few microliters was deposited onto a substrate of poly-l-lysine (PLL; Sigma, Schnelldorf, Germany) and a few minutes were allowed for the complexes to settle onto the substrate, after which unbound complexes were removed by washing the sample cell with deoxygenated buffer. This ensured a monolayer of ∼10 complexes per 10 × 10 μm2 in an oxygen-free environment. Oxygen in the buffer was thoroughly scavenged by an enzymatic system of 200 μg/mL glucose oxidase, 7.5 mg/mL glucose, and 35 μg/mL catalase (all from Sigma), and by simultaneous flushing of gaseous nitrogen. This significantly reduced photooxidation of the complexes and consequently prolonged their survival times by more than one order of magnitude. The presence of MgCl2 in the buffer enhanced attachment of the complexes onto the PLL substrate.
The PLL layer was created by immersing a standard silica microscope coverslip in a 0.01% (v/v; pH 6.8) solution of PLL at room temperature, followed by copious rinsing in distilled water. Under these conditions, the PLL, being in a random coil conformation, is expected to adsorb into a dense, smooth, even layer on the silicon surface (29). Under acidic and neutral aqueous conditions, most of the terminal amines of the PLL side chains are protonated (30). When a silica surface is coated, the protruding strands are thus expected to form a layer of positively charged amino groups, which predominantly electrostatically adhere to the carboxyl groups of a protein. An LHCII trimer is expected to bind at multiple sites at the N- or C-terminal domains, and hence on an even surface, binding all three monomeric subunits simultaneously.
Experimental setup and data analysis
An elaborate description of the utilized setup is given in Fig. S2. Fluorescent particles were located by performing a raster scan of a specified area on the buffer-substrate interface and sending the resulting fluorescence to the avalanche photodiode. The matrix of detected intensities was then associated with the piezo stage coordinates and converted into an image, after which fluorescence spots with intermediate intensity were selected and investigated spectroscopically. The intensity of these spots corresponds well with the expected fluorescence intensity from trimers (see Supporting Material). The properties of the fluorescence spectral profiles were determined by skewed Gaussian approximations (Supporting Material).
Results
Continuous irradiation of a single trimer made it possible to follow the spectral dynamics over the course of tens of seconds. The majority of complexes exhibited spectrally stable fluorescence peaks (FLPs) that fluctuated within ∼1 nm of the intermediate peak position of ∼682 nm. The extent of diffusion decreased considerably with the frequency of occurrence, and was strongly complex-dependent and only occasionally time-dependent. Large shifts of the entire spectrum were rare; more often, an additional, red-shifted band appeared. Fig. 1 shows the spectral time traces of three selected trimers from which such double-band spectra were observed. The spectrally stable behavior illustrated in Fig. 1 B typifies most of the double-band states; however, significant spectral diffusion of the long-wavelength band occurred occasionally (Fig. 1 C) and with a much larger fractional frequency than observed for the ∼682-nm band. The appearance, disappearance, and shifts of the red band generally did not notably modify the ∼682-nm spectral profile. The relative intensity of the two bands was primarily complex-dependent, i.e., it varied substantially between different complexes, whereas the time-dependent intensity fluctuations of the two bands were generally correlated (Fig. 1 B and Fig. S4 A), thus denoting single quantum systems with coupled emitters. However, energetically fluctuating red bands often were not correlated with the bluer band (Fig. 1 C). The spectral peak distribution obtained from ∼2000 complexes is displayed in Fig. 2. Examples of reversibility were found for the whole distribution of spectral profiles and peak positions.
Figure 1.

Fluorescence (FL) spectral time traces from single LHCII trimeric complexes at 5°C, acquired during continuous irradiation corresponding to a power of 1 μW at 630 nm. FL was binned into 1-s integration times. The distinct spectroscopic states are denoted by I, II and III, respectively, where I/III indicates alternation between states I and III. Spectra on top are the time averages of the respective spectral states. FL is expressed in counts per second (cps).
Figure 2.
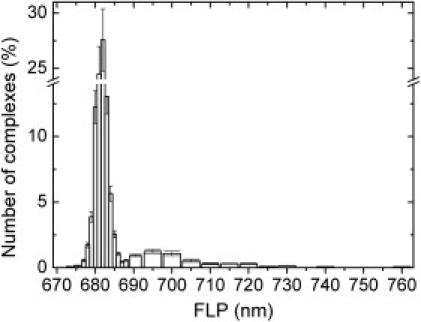
FLP distribution from ∼2000 individually measured LHCII trimers. On average, ∼100 consecutive spectra were acquired for every complex. Bins of 1 nm and 5 nm were used for the narrow and broad bars, respectively. Narrow bars denote the weighted time-dependent frequency of occurrence, whereas the broad bars describe the fraction of complexes that exhibited a peak in the specified wavelength window during the course of a spectral time trace. Broad bars comprise mainly the peak positions of the redder bands of double-band states (see Table 1). Standard errors of 10% and 20% were estimated on the narrow and broad bars, respectively.
Spectral profiles that deviate from the ensemble spectrum, which characteristically peaks at ∼682 nm, are classified into three groups in Table 1, with selected examples displayed in Fig. 3. In addition to the reversible single- and double-band spectra mentioned above, a third group (group 3) comprises irreversible blue-shifted spectra that characteristically peak below 670 nm. To date, we have not found conditions that establish the reversibility of these spectral profiles. These spectra were generally visible from the onset of illumination, but were in a few individual observations preceded by a nondeviating spectrum. Although the irreversibility strongly points to a degraded system, the environmental sensitivity of the complexes to exhibit extreme blue states prompted us to investigate this phenomenon. The emitting states of these complexes were frequently stable for several minutes before irreversible photobleaching occurred, indicating a stable underlying conformation. Indeed, single, denatured complexes of LHCII trimers were predominantly characterized by similar blue-shifted spectral profiles (data not shown). Moreover, a blue emission band in an ensemble spectrum of LHCII trimers was attributed to denatured complexes (31,32). Remarkably, we occasionally observed similar blue spectra when we performed SMS on small LHCII aggregates, minor antenna complexes (CP24, CP26 and CP29), and photosystem I antennae (data not shown).
Table 1.
Properties of deviating fluorescence spectral profiles, classified into three groups
| Peak 1 (nm) | Peak 2 (nm) | Fraction∗ (%) | Reversible | Proposed explanation | |
|---|---|---|---|---|---|
| Group 1 | 673–678, 686–695∼700–760 | – | 4–5†< 0.5 | YesSometimes | Static disorderCT mixing‡ |
| Group 2 | 680–685 | ∼690–755 | 4–5 | Yes | CT mixing‡ |
| Group 3 | 650–670 | 690–715 | Ill-defined§ | No | Denaturation |
A deviating profile contains at least one band that does not peak within 682 ± 3 nm. Reversibility denotes switching back to a state characterized by an ensemble spectral profile.
Fraction of complexes that exhibited at least one spectrum peaking within the specified wavelength windows during the course of a spectral time trace.
Value taken from Fig. 2, denoting a time-weighted percentage.
Mixing between an exciton and CT state.
Strongly varying between different sets of measurements performed under very similar conditions, but small for a typical experiment.
Figure 3.
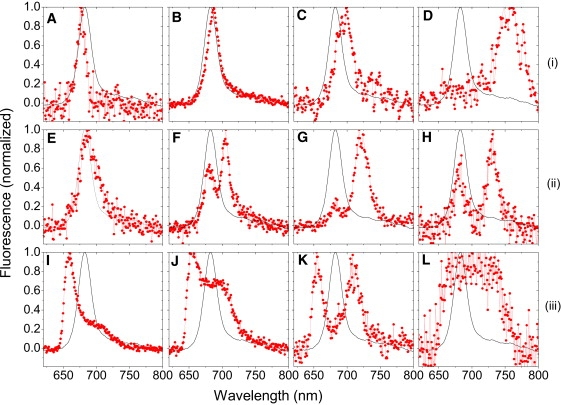
Selection of deviating spectral profiles from single LHCII complexes (points connected by lines, red online). The rows correspond to the three groups in Table 1, respectively. Spectra are time averages of similar spectral profiles, with each average corresponding to one complex. The reference spectrum (solid lines) is a time- and population-averaged single-molecule spectrum. The sharp decrease of the left wing below 640 nm in I–L stems from the fluorescence filter cutoff. All spectra are normalized to facilitate comparison of different profiles. See text and Table 1 for further details.
Group 3 states were observed to be interreversible among at least three emitting states with primary maxima at ∼655 nm, 660 nm, and 665 nm, respectively. In addition, these spectral profiles were characterized by a long red tail, which frequently enhanced into a defined peak with a maximum above 695 nm (Fig. 3, J and K). The red band generally displayed intensity fluctuations in correlation with the extreme blue band (Fig. S4 B), signifying coupled emitters in a single system. Although the extreme blue emission is reminiscent of Chl b fluorescence, its frequency of occurrence was found to be independent of the excitation wavelength. The frequency of occurrence was also independent of the excitation intensity, the time of illumination, and the number or duration of interspersed dark periods. Curiously, extreme blue spectra were rare when ensembles of >100 complexes were illuminated, which suggests that this phenomenon is enhanced under single-molecule conditions.
Discussion
The primary objective of this study was to investigate the rate and extent of spectral diffusion in single trimeric complexes of LHCII close to physiological conditions, and to establish a feasible connection to the properties of the corresponding pigment electronic states.
A large distribution of spectral inhomogeneity from LHCII trimers is reported here. The wealth of spectral dynamics can be well understood in the context of the protein energy landscape model (10). Such an energy landscape is a hypersurface in the high-dimensional space of all the atomic coordinates of the protein and therefore represents all possible protein conformations as a function of the protein's potential energy. The structural flexibility of a protein allows it to explore different regions of its landscape. The landscape includes a great number of local minima, each representing a quasi-stable conformational substate (CS). The height of the barriers between these substates determines the extent of the landscape that can be traversed by the protein under certain environmental conditions. The protein evidently traverses a more extended region at physiological temperatures than at reduced temperatures. Furthermore, charge displacement resulting from excitation of a pigment causes a rapid perturbation in the protein structure due to strong pigment-phonon coupling. After such a perturbation, the protein may relax into a different CS. In addition, the average dissipated energy of the LHCII pigments after photon absorption under the utilized conditions greatly exceeds the Boltzmann energy. This energy is released as heat into the local environment of the protein and adds a significant amount of disorder to the protein.
Due to the strong exciton-phonon coupling in LHCII, all of these structural fluctuations that are manifested in the local environment of the embedded pigments result in variations of the pigment-site energies. This pigment-site disorder is mostly determined by proton interactions (33) and, at room temperature, by thermal fluctuations. In a system with sufficient structural flexibility, environmental or photo-induced structural changes of the chromophores give rise to additional site disorder (34,35). This pigment-site inhomogeneity is reflected in the absorption and fluorescence properties of the complex and accordingly results in spectral diffusion. Most of these spectral changes occur on timescales shorter than the experimental time resolution, giving rise to inhomogeneous broadening and single-molecule spectra with shapes and widths similar to those of the ensemble spectrum (Fig. S5) (16). However, assuming that all spectral profiles are connected to specific protein CSs, the observation that the deviating spectra are stable on timescales of seconds to tens of seconds suggests that large energy barriers surround the associated CSs (36,37). In the same context, the rapid transition between the different deviating spectral states indicates steep barriers. Furthermore, the histogram spectrum in Fig. 2 represents inhomogeneous disorder on a timescale of seconds and is therefore significantly narrower than the steady-state fluorescence spectrum. The infrequency of switching between states with distinctly different spectral properties adds to the height of the barriers between different CSs.
It remains to be determined whether the whole distribution of observed spectral profiles can be explained by a reasonable amount of disorder in the complex. For this purpose, we investigated the disorder-induced spectral flexibility of LHCII in solution at room temperature by using a modified Redfield disordered exciton model (7). In this model, an excitation perturbation is simulated by randomly varying the static disorder of all the pigment-site energies and calculating the subsequent energy equilibration and resulting fluorescence spectral shape. This site disorder is expected to account for potentially large thermally or charge-induced fluctuations in the local environment of the pigments. Different realizations of the static disorder indeed induce various spectral shape alterations (Fig. 4). By fitting these simulated profiles to the measured profiles, one can describe the related lowest exciton states in terms of a quantitative delocalization of the energy over all the pigments in the complex.
Figure 4.
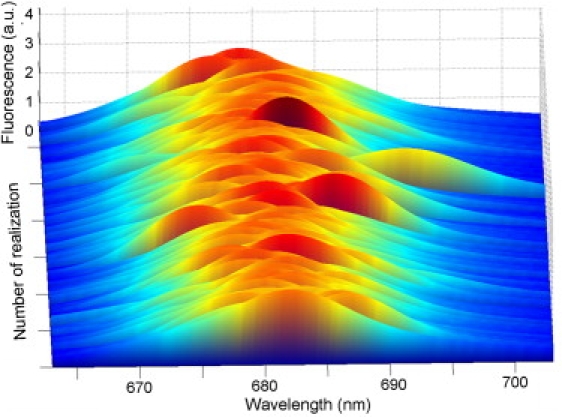
Fluorescence profiles calculated for different realizations of the static disorder for a single LHCII complex at room temperature. The realizations were obtained by using different sets of the site energies (Qy transition energies for 14 Chls), randomly taken from a Gaussian distribution with full width at half-maximum of 90 cm−1. This disorder width was the same as for the Gaussian fit of the bulk spectrum.
In the calculations, we used the structural data of Liu et al. (2) and took into account the interactions between 14 Chls of a monomeric subunit of the LHCII trimer while neglecting interactions between Chls from adjacent monomeric subunits. The latter make a minor contribution to the lowest exciton states that determine the steady-state fluorescence spectrum. Therefore, in our simplified model, 14 exciton components were used (see Fig. S6). To improve the fit of the room-temperature bulk spectra (Fig. S6), the parameters of the model were slightly adjusted. Generally, the emissive properties of chromophores are sensitive to their environment, indicating that spectra of immobilized LHCII complexes can differ from the spectra of the same complexes in solution. For example, to reproduce the single-molecule spectra from immobilized bacterial LH2 complexes, the exciton-phonon coupling should be 1.5–1.7 times the value of freely diffusing complexes in solution, and the site energies should be shifted uniformly (7,16). In contrast, the single-molecule spectra from LHCII can be reproduced with the same phonon couplings and disorder values as for the bulk spectrum. However, the typical disorder of 90 cm−1 gave rise to only rare occurrences of fluorescence spectral broadening (only 26 from 2000 realizations) and no double-band spectra. Increasing the disorder to 140 cm−1 led to an improved fit of the single-molecule profiles, including more occurrences of spectral broadening and rare realizations with double-band spectral shapes (see Fig. S7 and Fig. S8). To maintain the values of the reorganization shift, the coupling strengths to low- and high-frequency modes were decreased by a factor of 1.15.
Fig. 5 displays a selection of modeled profiles that correspond well with a number of selected measured profiles. The histograms show the thermally averaged participation ratio (PR) of the pigments from n = 1–14 (corresponding to Chl 601–614 in the notation of Liu et al. (2)), defined as , where pk is the steady-state population of the k-th exciton state, and is the wave-function amplitude corresponding to the participation of the n-th pigment in the k-th exciton state. PRn is therefore indicative of the degree of excitation localization at the n-th site.
Figure 5.
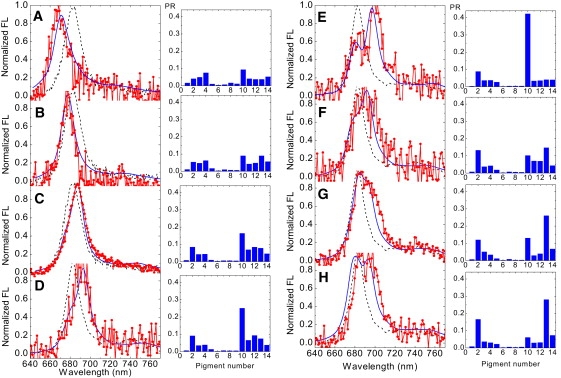
Comparison of eight selected modeled (solid lines, blue online) and measured (points connected by lines, red online) deviating fluorescence spectral profiles, along with the calculated PR of the different pigments to the lowest exciton state (histograms to the right of corresponding spectra). All spectra are normalized and compared with the nondeviating reference spectrum (dashed lines). Measured spectra are averages of spectral profiles with similar shapes, and calculated spectra are averaged over a number of realizations. For all realizations, the disorder was 140 cm−1. PRn is averaged over a number of realizations of the static disorder within each group. See text for details.
Fig. 5 reveals that different spectral shapes can be related to different corresponding excitation energy localization patterns. In general, blue-shifted spectra are connected to delocalized states, whereas red shifts and double-band states are characterized by localization of the energy on specific low-energy sites, resulting in a more nonuniform distribution of the PR. The different spectral features will be considered individually.
Blue shifts of the whole spectrum, with simultaneous spectral shape alterations, occur for realizations with nearly uniform distribution within the three Chl a clusters, i.e., delocalization within the a602-a603, a610-a611-a612, and a613-a614 clusters (Fig. 5, A and B). Localization at a613 in most cases produces significant broadening on the red side, whereas the blue wing still corresponds to the position of the bulk profile (Fig. 5, G and H). Shifting of the whole spectrum to the red, without significant broadening, corresponds to an increasingly more nonuniform PR with predominant localization at a610 and a smaller localization at a602 (Fig. 5, C and D). A disorder-induced red shift of the a602 or a610 sites breaks the delocalization within the Chl a clusters on the stromal side (i.e., within the a610-a611-a612 and a602-a603 clusters), thus increasing the reorganization shift with further red shifting of the whole spectrum. However, increasingly more localization at a610 and delocalization within the a613-a614 cluster give rise to significant broadening and the appearance of two bands, with the red contribution being more intense than the blue part and the position of the blue band corresponding to that of the bulk spectrum (Fig. 5 E). Alternatively, broadening of the red wing without shifting of the blue wing is determined by localization within the a602-a603 and a613-a614 clusters while delocalization is preserved within the a610-a611-a612 cluster (Fig. 5 F). In this case, the localized states are shifted to the red, whereas the delocalized states still contribute to the blue part of the spectrum. The two components in such spectra correspond to two emitters within a monomeric subunit of LHCII. Alternatively, the two emitters may be located in different monomeric units of a trimer.
Although the excitation energy density was substantially higher than under in vivo conditions, local heating was negligible (Supporting Material) (38,39). However, the high excitation rate may drive the protein into conformational states that occur less frequently under natural conditions but are still intrinsic to the system (16). Considering the very similar spectral shape of the single-molecule time and population average compared to the ensemble spectrum (Fig. S5), as well as the reversibility of the deviating states, we suggest that repetitive laser excitation at most increases the probability of slight conformational changes into states that are accessible in vivo.
Large red shifts
The pure exciton model of LHCII with standard disorder can explain spectral peaks up to ∼695 nm. The inclusion of substantially increased disorder gave rise to peaks up to ∼705 nm on rare occasions. However, SMS demonstrated the capability of LHCII to emit photons with even lower energy, i.e., emission peaks beyond this wavelength and up to ∼760 nm were observed. This indicates the occasional appearance of large changes in the electronic structure of some of the pigments. Given the inability of a pure exciton model to explain such conditions, we propose the existence of a protein conformational change to invoke these special low-energy states. Such a structural change may allow substantial wave-function overlap of some of the Chls to induce charge transfer (CT). Furthermore, such a CT state may strongly overlap spatially and energetically with one of the lowest exciton states, giving rise to coupling between these two states and subsequently a substantial red shift of the fluorescence (40,41). Indeed, the peripheral light-harvesting complex of photosystem I of plants (LHCI) characteristically exhibits an excessively red-shifted emission, which has been attributed to mixing of the excited state with a CT state of the excitonically coupled Chl ab dimer in the L2 site of this complex (42,43). Considering the homology between LHCI and LHCII, we suggest that the large red shifts in the latter may result from a similar mixing in the corresponding Chl a603-b609-L2 site (in the notation of Liu et al. (2)). Furthermore, fluctuation between different red-shifted states may arise from differing interaction strengths between the CT and excitonic state. Such large spectral shifts to the red have not yet been reported for LHCII trimers in solution.
The observation that the appearance and shifts of the red band did not notably modify the spectral shape of the 682-nm band can be interpreted as the appearance of an additional emitting site, i.e., a trimer with a red site. The characteristic double-band spectra from LHCI at room temperature have been explained by the existence of similar so-called Chls (44). Furthermore, the coupled intensity fluctuations of these two bands and the reversibility of the red state suggest that the double-banded spectra are not related to dissociated trimers.
Spectral red shifts exhibited by semiconductor quantum dots were also attributed to the Stark effect (denoted a quantum-confined Stark effect) and suggested to occur as the result of charge accumulation under slowly ionizing radiation (45). In particular, charge-carrier trapping induces an electric field at or near the surface of the quantum dot and accordingly perturbs the electron-hole energy levels. Fluctuations in the field due to charge hopping between trap sites leads to a redistribution of electron density in the quantum dot core that manifests as a diffusion of the exciton transition energy.
Spectral bluing
Although model calculations provide an explanation for spectra exhibiting a peak down to 670 nm (Fig. 5 A), SMS revealed that reversible spectral shifts were limited to at most 5 nm to the blue, with peaks beyond 675 nm being extremely rare. This indicates a possible overestimation of the disorder-induced spectral diffusion to the blue. However, the occurrence of extreme blue states characteristically peaking below 670 nm suggests the presence of a phenomenon that either obstructs efficient energy transfer from Chl b to Chl a, or exhibits large changes in the electronic structure of the lowest exciton state. In either case, sizeable spatial rearrangements of the pigment-protein structure are required. A possible mechanism is the uncoupling of the tetramer Chl a604-b605-b606-b607 from the rest of the trimeric complex and subsequent emission from the Chl b's in this cluster. This tetramer is the only loosely coupled cluster in a monomeric subunit (8,46). Moreover, Chl a604, which absorbs at relatively blue wavelengths, is a bottleneck site (8,43). In addition, the long red tail, which often appeared as a prominent band, may signify the presence of CT states, in particular involving Chl a604. The intensity fluctuation correlation of this red band with the extreme blue peak may accordingly indicate a strong interaction between the two emitters, and in particular a strong coupling of Chl a604 to the rest of the tetramer. The stability of this denatured state is possibly enhanced by stronger interaction with the substrate.
The phenomenon of extreme blue emission from single-molecule systems is generally known as spectral bluing (47). Reversible spectral bluing was observed in a number of bacterial LH2 antennae and attributed to a similar mechanism as explained above, i.e., the appearance of an energy trap before equilibration to the lowest exciton state (17). Intriguingly, irreversible and partially reversible spectral bluing is a well-known phenomenon of semiconductor quantum dots (QDs) (for a recent review, see Lee and Osborne (47)). In contrast to the similar phenomenon appearing for LHCII, spectral bluing from QDs is strongly intensity-dependent. This is generally believed to result from photooxidation of parts of a QD. However, under the essentially oxygen-free conditions we employed to investigate LHCII, it is highly unlikely that up to 100% of the complexes photooxidized. This suggests that different mechanisms are responsible for spectral bluing in QDs and pigment-protein complexes.
Comparison with bacterial antennae
The spectral behavior of LHCII is significantly different from that reported for LH2 antennae of bacterial systems (16,17), as evidenced by the following results: 1) LHCII exhibited a smaller spectral switching rate and reversibility under continuous irradiation. 2) The deviating emission states of LHCII showed a larger photostability. 3) The majority of deviating spectral profiles comprised double maxima, whereas only a small fraction of double maxima were observed for the bacterial systems. 4) Under similar experimental conditions, LHCII had a smaller fluorescence intensity and significantly shorter survival time before irreversible photobleaching occurred. 5) Spectral bluing was irreversible for LHCII, but reversible for LH2. 6) LHCII showed a preference for switching to lower energy values, whereas the jump size distribution of LH2 was symmetric about zero. 7) In LH2, all spectra apart from spectral bluing could be well explained by the disordered-exciton model, but this model proved to be inadequate to explain all the spectra originating from LHCII. 8) The PR of the red-shifted realizations of LHCII yielded a nonuniform distribution with predominant energy localization on one to three pigments; however, the LH2 antenna is highly symmetric, and thus localization can occur at any arbitrary pigment, depending on the disorder pattern. The first two characteristics described above may indicate that LHCII possesses larger potential energy barriers between its different CSs than LH2. The spectral behavioral differences between LHCII and LH2 can primarily be ascribed to the substantially different structures of these two systems.
Conclusions
LHCII samples its disorder, and the resulting, relatively small spectral fluctuations are well described by the disordered exciton-Redfield model. The variations of the associated site energies are related to small conformational changes in the pigment-protein scaffold. The pure exciton model with increased disorder can reproduce most typical experimental profiles with peaks lying between 670 nm and 705 nm, and with only rare occurrences of large spectral shifts. This signifies that these spectra may be related to a specific localization pattern that is controlled predominantly by the conformation-induced disorder of the pigment-site energies. Occasionally, LHCII switches to a new state, emitting primarily above 705 nm. These realizations may be connected to some special conformations that are not included in the normal exciton model, which we attribute to states with a substantial CT character. On a timescale of seconds to tens of seconds, single LHCII systems exhibit significantly different spectral behavior compared to bacterial systems, a phenomenon that should be expected in the light of the substantial dissimilarity of the respective structural arrangements.
Supporting Material
Additional methods, calculations, results, and nine figures are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(10)00360-7.
Supporting Material
Acknowledgments
The authors thank Henny van Roon for the sample isolation and pigment analysis.
This work was supported by the EU FP6 Marie Curie Early Stage Training Network via the Advanced Training in Laser Sciences project (T.P.J.K), the Netherlands Organization for Scientific Research via the Foundation of Earth and Life Sciences (C.I. and R.v.G.), the EU FP7 Marie Curie Reintegration Grants (C.I.), the Netherlands Organization for Scientific Research, and the Russian Foundation for Basic Research (grant 09-04-00605 to V.I.N.).
Contributor Information
Tjaart P.J. Krüger, Email: tpj.krueger@few.vu.nl.
Rienk van Grondelle, Email: rienk@few.vu.nl.
References
- 1.Kühlbrandt W., Wang D.N., Fujiyoshi Y. Atomic model of plant light-harvesting complex by electron crystallography. Nature. 1994;367:614–621. doi: 10.1038/367614a0. [DOI] [PubMed] [Google Scholar]
- 2.Liu Z.F., Yan H.C., Chang W. Crystal structure of spinach major light-harvesting complex at 2.72 A resolution. Nature. 2004;428:287–292. doi: 10.1038/nature02373. [DOI] [PubMed] [Google Scholar]
- 3.Van Grondelle R., Dekker J.P., Sundström V. Energy transfer and trapping in photosynthesis. Biochim. Biophys. Acta. 1994;1187:1–65. [Google Scholar]
- 4.Dekker J.P., Boekema E.J. Supramolecular organization of thylakoid membrane proteins in green plants. Biochim. Biophys. Acta. 2005;1706:12–39. doi: 10.1016/j.bbabio.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 5.Van Amerongen H., van Grondelle R. Understanding the energy transfer function of LHCII, the major light-harvesting complex of green plants. J. Phys. Chem. B. 2001;105:604–617. [Google Scholar]
- 6.Standfuss J., Terwisscha van Scheltinga A.C., Kühlbrandt W. Mechanisms of photoprotection and nonphotochemical quenching in pea light-harvesting complex at 2.5 A resolution. EMBO J. 2005;24:919–928. doi: 10.1038/sj.emboj.7600585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Novoderezhkin V.I., Palacios M.A., van Grondelle R. Excitation dynamics in the LHCII complex of higher plants: modeling based on the 2.72 Angstrom crystal structure. J. Phys. Chem. B. 2005;109:10493–10504. doi: 10.1021/jp044082f. [DOI] [PubMed] [Google Scholar]
- 8.van Grondelle R., Novoderezhkin V.I. Energy transfer in photosynthesis: experimental insights and quantitative models. Phys. Chem. Chem. Phys. 2006;8:793–807. doi: 10.1039/b514032c. [DOI] [PubMed] [Google Scholar]
- 9.Weber G. Energetics of ligand binding to proteins. Adv. Protein Chem. 1975;29:1–83. doi: 10.1016/s0065-3233(08)60410-6. [DOI] [PubMed] [Google Scholar]
- 10.Frauenfelder H., Sligar S.G., Wolynes P.G. The energy landscapes and motions of proteins. Science. 1991;254:1598–1603. doi: 10.1126/science.1749933. [DOI] [PubMed] [Google Scholar]
- 11.Hofmann C., Aartsma T.J., Köhler J. Direct observation of tiers in the energy landscape of a chromoprotein: a single-molecule study. Proc. Natl. Acad. Sci. USA. 2003;100:15534–15538. doi: 10.1073/pnas.2533896100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trautman J.K., Macklin J.J., Betzig E. Near-field spectroscopy of single molecules at room temperature. Nature. 1994;369:40–42. [Google Scholar]
- 13.Lu H.P., Xie X.S. Single-molecule spectral fluctuations at room temperature. Nature. 1997;385:143–146. [Google Scholar]
- 14.Hofkens J., Maus M., De Schryver F. Probing photophysical processes in individual multichromophoric dendrimers by single-molecule spectroscopy. J. Am. Chem. Soc. 2000;122:9278–9288. [Google Scholar]
- 15.Jelezko F., Tietz C., Bittl R. Single-molecule spectroscopy on photosystem I pigment-protein complexes. J. Phys. Chem. B. 2000;104:8093–8096. [Google Scholar]
- 16.Rutkauskas D., Novoderezhkin V.I., van Grondelle R. Fluorescence spectroscopy of conformational changes of single LH2 complexes. Biophys. J. 2005;88:422–435. doi: 10.1529/biophysj.104.048629. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17.Rutkauskas D., Olsen J., van Grondelle R. Comparative study of spectral flexibilities of bacterial light-harvesting complexes: structural implications. Biophys. J. 2006;90:2463–2474. doi: 10.1529/biophysj.105.075895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brecht M., Radics V., Bittl R. Protein dynamics-induced variation of excitation energy transfer pathways. Proc. Natl. Acad. Sci. USA. 2009;29:11857–11861. doi: 10.1073/pnas.0903586106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nieder J.B., Brecht M., Bittl R. Dynamic intracomplex heterogeneity of phytochrome. J. Am. Chem. Soc. 2009;131:69–71. doi: 10.1021/ja8058292. [DOI] [PubMed] [Google Scholar]
- 20.Basché T., Kummer S., Bräuchle C. Direct spectroscopic observation of quantum jumps of a single molecule. Nature. 1995;373:132–134. [Google Scholar]
- 21.Ha T., Enderle T., Weiss S. Quantum jumps of single molecules at room temperature. Chem. Phys. Lett. 1997;271:1–5. [Google Scholar]
- 22.García-Parajo M.F., Segers-Nolten G.M.J., van Hulst N.F. Real-time light-driven dynamics of the fluorescence emission in single green fluorescent protein molecules. Proc. Natl. Acad. Sci. USA. 2000;97:7237–7242. doi: 10.1073/pnas.97.13.7237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cogdell R.J., Köhler J. Use of single-molecule spectroscopy to tackle fundamental problems in biochemistry: using studies on purple bacterial antenna complexes as an example. Biochem. J. 2009;422:193–205. doi: 10.1042/BJ20090674. [DOI] [PubMed] [Google Scholar]
- 24.Tietz C., Jelezko F., Wrachtrup J. Single molecule spectroscopy on the light-harvesting complex II of higher plants. Biophys. J. 2001;81:556–562. doi: 10.1016/S0006-3495(01)75722-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gerken U., Wolf-Klein H., Paulsen H. Single molecule spectroscopy of oriented recombinant trimeric light harvesting complexes of higher plants. Single Mol. 2002;4:183–188. [Google Scholar]
- 26.Van Leeuwen P.J., Nieveen M.C., van Gorkom H.J. Rapid and simple isolation of pure photosystem II core and reaction center particles from spinach. Photosynth. Res. 1991;28:149–153. doi: 10.1007/BF00054128. [DOI] [PubMed] [Google Scholar]
- 27.van Roon H., van Breemen J.F.L., Boekema E.J. Solubilization of green plant thylakoid membranes with n-dodecyl-α,D-maltoside. Implications for the structural organization of the photosystem II, photosystem I, ATP synthase and cytochrome b6 f complexes. Photosynth. Res. 2000;64:155–166. doi: 10.1023/A:1006476213540. [DOI] [PubMed] [Google Scholar]
- 28.Gilmore A.N., Yamamoto H.Y. Resolution of lutein and zeaxanthin using a non-endcapped, lightly carbon-loaded C18 high-performance liquid chromatographic column. J. Chromatogr. A. 1991;543:137–145. [Google Scholar]
- 29.Subramanian A., Kennel S.J., Doktycz M.J. Comparison of techniques for enzyme immobilization on silicon supports. Enzyme Microb. Technol. 1999;24:26–34. [Google Scholar]
- 30.Latour R.A. Molecular modeling of biomaterial surfaces. Curr. Opin. Solid State Mater. Sci. 1999;4:413–417. [Google Scholar]
- 31.Ilioaia C., Johnson M.P., Ruban A.V. Induction of efficient energy dissipation in the isolated light-harvesting complex of photosystem II in the absence of protein aggregation. J. Biol. Chem. 2008;283:29505–29512. doi: 10.1074/jbc.M802438200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Oort B., van Hoek A., van Amerongen H. Equilibrium between quenched and nonquenched conformations of the major plant light-harvesting complex studied with high-pressure time-resolved fluorescence. J. Phys. Chem. B. 2007;111:7631–7637. doi: 10.1021/jp070573z. [DOI] [PubMed] [Google Scholar]
- 33.Brecht M., Studier H., Bittl R. Spectral diffusion induced by proton dynamics in pigment-protein complexes. J. Am. Chem. Soc. 2008;130:17487–17493. doi: 10.1021/ja806216p. [DOI] [PubMed] [Google Scholar]
- 34.Cogdell R.J., Isaacs N.W., Prince S. The structure and function of the LH2 (B800-850) complex from the purple photosynthetic bacterium Rhodopseudomonas acidophila strain 10050. Prog. Biophys. Mol. Biol. 1997;68:1–27. doi: 10.1016/s0079-6107(97)00010-2. [DOI] [PubMed] [Google Scholar]
- 35.McLuskey K., Prince S.M., Isaacs N.W. The crystallographic structure of the B800–820 LH3 light-harvesting complex from the purple bacteria Rhodopseudomonas acidophila strain 7050. Biochemistry. 2001;40:8783–8789. doi: 10.1021/bi010309a. [DOI] [PubMed] [Google Scholar]
- 36.Rutkauskas D., Novoderezkhin V., van Grondelle R. Fluorescence spectral fluctuations of single LH2 complexes from Rhodopseudomonas acidophila strain 10050. Biochemistry. 2004;43:4431–4438. doi: 10.1021/bi0497648. [DOI] [PubMed] [Google Scholar]
- 37.Jankowiak R., Hayes J.M., Small G.J. Spectral hole-burning spectroscopy in amorphous molecular solids and proteins. Chem. Rev. 1993;93:1471–1502. [Google Scholar]
- 38.Schönle A., Hell S.W. Heating by absorption in the focus of an objective lens. Opt. Lett. 1998;23:325–327. doi: 10.1364/ol.23.000325. [DOI] [PubMed] [Google Scholar]
- 39.Gulbinas V., Karpicz R., Valkunas L. Nonequilibrium heating in LHCII complexes monitored by ultrafast absorbance transients. Biochemistry. 2006;45:9559–9565. doi: 10.1021/bi060048a. [DOI] [PubMed] [Google Scholar]
- 40.Lathrop E.J.P., Friesner R.A. Vibronic mixing in the strong electronic coupling limit. Spectroscopic effects of forbidden transitions. J. Phys. Chem. 1994;98:3050–3055. [Google Scholar]
- 41.Renger T. Theory of optical spectra involving charge transfer states: dynamic localization predicts a temperature dependent optical band shift. Phys. Rev. Lett. 2004;93:188101. doi: 10.1103/PhysRevLett.93.188101. [DOI] [PubMed] [Google Scholar]
- 42.Croce R., Chojnicka A., van Grondelle R. The low-energy forms of photosystem I light-harvesting complexes: spectroscopic properties and pigment-pigment interaction characteristics. Biophys. J. 2007;93:2418–2428. doi: 10.1529/biophysj.107.106955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Romero E., Mozzo M., Croce R. The origin of the low-energy form of photosystem I light-harvesting complex Lhca4: mixing of the lowest exciton with a charge-transfer state. Biophys. J. 2009;96:L35–L37. doi: 10.1016/j.bpj.2008.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jennings R.C., Garlaschi F.M., Zucchelli G. The room temperature emission band shape of the lowest energy chlorophyll spectral form of LHCI. FEBS Lett. 2003;547:107–110. doi: 10.1016/s0014-5793(03)00687-2. [DOI] [PubMed] [Google Scholar]
- 45.Empedocles S.A., Bawendi M.G. Quantum-confined stark effect in single CdSe nanocrystallite quantum dots. Science. 1997;278:2114–2117. doi: 10.1126/science.278.5346.2114. [DOI] [PubMed] [Google Scholar]
- 46.Gradinaru C.C., Özdemir S., van Amerongen H. The flow of excitation energy in LHCII monomers: implications for the structural model of the major plant antenna. Biophys. J. 1998;75:3064–3077. doi: 10.1016/S0006-3495(98)77747-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee S.F., Osborne M.A. Brightening, blinking, bluing and bleaching in the life of a quantum dot: friend or foe? ChemPhysChem. 2009;10:2174–2191. doi: 10.1002/cphc.200900200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.