

Abstract
Formation of reactive oxygen and nitrogen species is a precipitating event in an array of neuropathological conditions. In response to excessive reactive oxygen species (ROS) levels, transcriptionally-dependent mechanisms drive the upregulation of ROS scavenging proteins which, in turn, limit the extent of brain damage. Here, we employed a transgenic approach in which CREB-mediated transcription is repressed (via A-CREB) to examine the contribution of the CREB/CRE pathway to neuroprotection and its potential role in limiting ROS toxicity. Using the pilocarpine-evoked repetitive seizure model, we detected a marked enhancement of cell death in A-CREB transgenic mice. Paralleling this, there was a dramatic increase in tyrosine nitration (a marker of reactive species formation) in A-CREB transgenic mice. In addition, inducible expression of PGC-1α (peroxisome proliferator-activated receptor gamma coactivator-1α) was diminished in A-CREB transgenic mice, as was activity of complex I of the mitochondrial electron transport chain. Finally, the neuroprotective effect of BDNF against ROS-mediated cell death was abrogated by disruption of CREB-mediated transcription. Together, these data both extend our understanding of CREB functionality and provide in vivo validation for a model in which CREB functions as a pivotal upstream integrator of neuroprotective signaling against ROS-mediated cell death.
Keywords: CREB, PGC-1α, ROS, seizure, striatum, cell death, oxidative stress
INTRODUCTION
Accumulating evidence has revealed a central role for oxidative and nitrosative stress in a wide range of congenital and acquired disorders of the central nervous system (CNS: Calabrese et al. 2000; Halliwell 2006; Sayre et al. 2008). The formation of reactive species is a by-product of mitochondrial respiration and also occurs as a result of second messenger signaling and metal ion redox chemistry (Calabrese et al. 2000; Chung et al. 2005; Aracena et al. 2006). Excessive ROS and reactive nitrogen species (RNS) production results in damage to proteins, lipids, DNA and RNA. Oxidative stress is associated with both the slow progression of neurodegenerative diseases such as ALS, and Parkinson’s (Albers and Beal 2000; Calabrese et al. 2006; Halliwell 2006) as well as the relatively rapid neuronal degeneration resulting from seizure activity and ischemic insults (Jarrett et al. 2008; Patel 2004; Love 1999; Warner et al. 2004). Importantly, in animal disease models, pharmacological approaches designed to reduce the oxidative load afford significant neuroprotection (Liang et al. 2007; Crow et al. 2005; Perluigi et al. 2006), thus attesting to the critical role that oxidative stress plays in a diverse array of neurological diseases and disorders.
To cope with the potential pathophysiological effects of ROS and RNS, CNS cells express an array of detoxifying enzymes. Some of the genes for these antioxidant enzymes are tonically expressed, whereas others, such as phase II detoxifying enzymes, can be induced in response to increased oxidative stress (Ishii et al. 2000). Thus, identification and functional characterization of the transcriptional pathways that couple potentially cytotoxic stimuli to the induction of neuroprotective gene expression is of great interest. Here, we examined the role of CREB as a regulator of ROS production and excitotoxic cell death. CREB is a member of the basic leucine zipper family of transcription factors that binds to the cAMP response element (CRE) and facilitates the expression of a large and diverse group of genes (Lonze and Ginty 2002). Within the CNS, CREB has been shown to play a critical role in activity-dependent enhancement of synaptic efficacy, which is thought to underlie adaptive processes such as learning and memory (Barco et al. 2003). In addition, CREB has been shown to be essential for normal CNS development, and for neuroprotection against an array of pathophysiological effectors (Bonni et al. 1999; Riccio et al. 1999; Walton et al. 1999; Rudolph et al. 1998; Lee et al. 2005; Mabuchi et al. 2001; Deak et al. 1998). Importantly, recent work has also indicated that CREB plays a role in the regulation of ROS detoxification. For example, several studies have shown that CREB is a direct regulator of antioxidant gene expression (Krönke et al. 2003; Bedogni et al. 2003), and induces the expression of PGC-1α, a key effector of ROS-detoxifying enzyme expression and mitochondrial biogenesis (St. Pierre et al. 2006; Lee and Wei 2005; Herzig et al. 2001). These findings raise the interesting prospect that CREB forms a central molecular building block of the cellular ROS defense mechanism.
In this study, we employed a transgenic mouse strain that expresses A-CREB, a dominant repressive form of CREB, to begin to explore the role of CREB as a regulator of neuronal responsiveness and vulnerability to oxidative stress. Using the pilocarpine model of SE, we report that A-CREB mice exhibit a profound increase in the vulnerability to repetitive seizure activity. Furthermore, there is an increase in both the basal and evoked ROS levels as well as a decrease in detoxifying enzyme expression, PGC-1α levels and decreased responsiveness to the neuroprotective effects of BDNF. Together, these data suggest that CREB functions as an essential upstream effector of neuroprotective signaling against ROS-mediated cell toxicity.
MATERIAL AND METHODS
Animals
Details regarding the generation of the A-CREB-eGFP transgenic mouse strain can be found in Lee et al. (2007). To drive A-CREB-eGFP transgene expression in the forebrain and striatum, mice were crossed with αCaMKII promoter-tTA transgenic mice (Mayford et al. 1996). Comparative analysis was performed between adult (6–10 weeks of age) A-CREB::αCaMKII-tTA (designated ‘A-CREB’ in rest of paper) mice and non-transgene littermates (denoted ‘WT’). The A-CREB strain was generated in a C57BL/6 background. To repress transgene expression, drinking water was supplemented with doxycycline (1 mg/mL: Sigma). CRE-β-galactosidase transgenic mice were provided by Dr. Daniel Storm (University of Washington) and genotyping was performed as described in Obrietan et al. (2002).
To induce status epilepticus, mice were first intraperitoneally (i.p.) injected with atropine methyl nitrate (1 mg/kg), and then, 30 min later with pilocarpine hydrochloride (325 mg/kg, i.p.). Motor seizure magnitude was defined using the Racine scale (Racine 1972). Thus, mice initially exhibited facial movements (stage 1) and then progressed to head nodding (stage 2), forelimb clonus (stage 3), rearing (stage 4), and rearing and falling (stage 5). After the initial stage 5 seizure, a subset of mice developed status epilepticus (SE), which was defined by persistent motor seizure activity with a duration of ≥ 3 hours; only animals that developed SE were used in this study. Control animals were injected with atropine methyl nitrate (1 mg/kg) and then with saline, instead of pilocarpine. The Ohio State University Lab Animal Use Committee approved all experimental procedures.
Cannulation and infusion
Mice were anesthetized with an i.p. injection of ketamine hydrochloride (91 mg/ml) and xylazine (9 mg/ml) and the placement of guide cannulae was performed as described in Butcher et al. (2002). The coordinates for striatal injection were: anterior 1.0 mm, lateral 1.8 mm and dorsoventral 2.2 mm from dura with head level. After surgery, animals were individually housed and allowed to recover at least 10 days. A stainless steel injector needle (30 gauge) extending 500 µm from the tip of the guide cannula was used to infuse saline or BDNF (50 ng/µl) at a rate of 0.40 µl/min.
Histology: Tissue collection
Initially, mice were anesthetized with ketamine hydrochloride and xylazine (as described above) and then transcardially perfused with 4% paraformaldehyde (PFA) in 0.1 M sodium phosphate buffer (pH 7.4). Brains were then removed and post-fixed (4 hours) in 4% PFA. Next, brains were cut into 600 µm coronal sections with a vibratome, and the sections were cryoprotected overnight in 30% sucrose. Thirty-µm thick coronal sections of the dorsal striatum (+1.1 mm to +0.5 mm from bregma) were cut on a freezing microtome and processed as described below.
Fluoro-jade B histochemistry
Tissue was processed as described in Choi et al. (2007). Briefly, fixed tissue was washed (3X) in distilled water and then incubated (10 min) in 0.06% potassium permanganate at room temperature with gentle agitation. Tissue was then washed (3X) in distilled water and incubated (20 min) in a solution of acetic acid (0.1%) and Fluorojade B (0.001%; Histo-Chem Inc., Jefferson, AR). Sections were then washed (3X) in distilled water, washed (2 min) in xylene and coverslipped with DPX (Electron Microscopy Sciences, Hatfield, PA).
Immunolabeling
For diaminobenzidine (DAB)-based immunohistochemical labeling, free-floating sections were initially treated with 0.3% H2O2 diluted in phosphate-buffered saline (PBS) to inhibit endogenous peroxidase activity, and then blocked (30 min) with 10% goat serum diluted in PBS with 0.1% Triton X-100 (PBST). Sections were then incubated overnight with an antibody against β-galactosidase (rabbit polyclonal, 1:10000 dilution; Cortex Biochem., San Leandro CA, USA), GFP (rabbit polyclonal, 1:5000 final dilution, University of Alberta, Edmonton, Canada) or Heme-oxygenase 1 (rabbit polyclonal, HO-1, 1:2000; Stressgen biotech, Victoria, BC, Canada). After sections were incubated a with biotylated secondary antibody, the tissue was processed using the ABC labeling technique (Vector Labs) and nickel-intensified DAB was used to visualize the signal. Sections were then mounted, dehydrated in xylene, and sealed with Permount (Sigma). Images were acquired using a Leica DMIRB inverted microscope connected to a 16 bit digital camera (Princeton Instruments; Monmouth Junction NJ) driven by Metamorph software (Universal Imaging, Downingtown PA,).
For fluorescence immunolabeling, tissue was blocked and permeabilized as described above, and then incubated (overnight at 4° C) with different combinations of the following antibodies against: NeuN (mouse monoclonal, 1:500; Chemicon, Temecula, CA), GFP (rabbit polyclonal, 1:5000 final dilution, Univ. Alberta), 3-NT (mouse monoclonal, 1:1000; Chemicon), and ND6 (mouse monoclonal, 1:1000; Molecular Probes, Eugene OR). Cells were then incubated (4 h at RT) with Alexa 488- and 594-conjugated secondary antibodies (1:250 final dilution, Molecular Probes) against the IgG domains of the primary antibodies. Tissue was then incubated (30 min) with the DNA stain DraQ5 (2 µM; Biostatus Limited, UK), washed 3X in PBST, and then mounted with cytoseal. Images were captured with a Zeiss 510 META confocal microscope (2 µm–thick z-axis) and data analysis was performed with Metamorph software.
Quantification and data analysis
To quantitate the number of Fluoro-Jade B positive cells in Fig. 2, fluorescent images (10× magnification) of the striatum were captured (~ +0.9 to +0.5 mm from bregma). One mm2 digital boxes (2 per hemisphere) were then placed over the medial striatum, and the total number of positive cells was counted within each box. Bilateral counting was performed from 2 sections (~ 300 µm apart) per animal (8 regions total) and mean values determined and used for averaging within a group. To quantitate the protective effects of BDNF infusion (Fig. 7), 2 boxes were places ~ 300 µm from the infusion site and FJB-positive cells were counted. Counts were performed from 2 sections (~ 300 µm apart) and the mean value for each animal was determined for the ipsilateral and contralateral striatum.
Fig. 2. Status Epilepticus-induced cell death in the striatum.
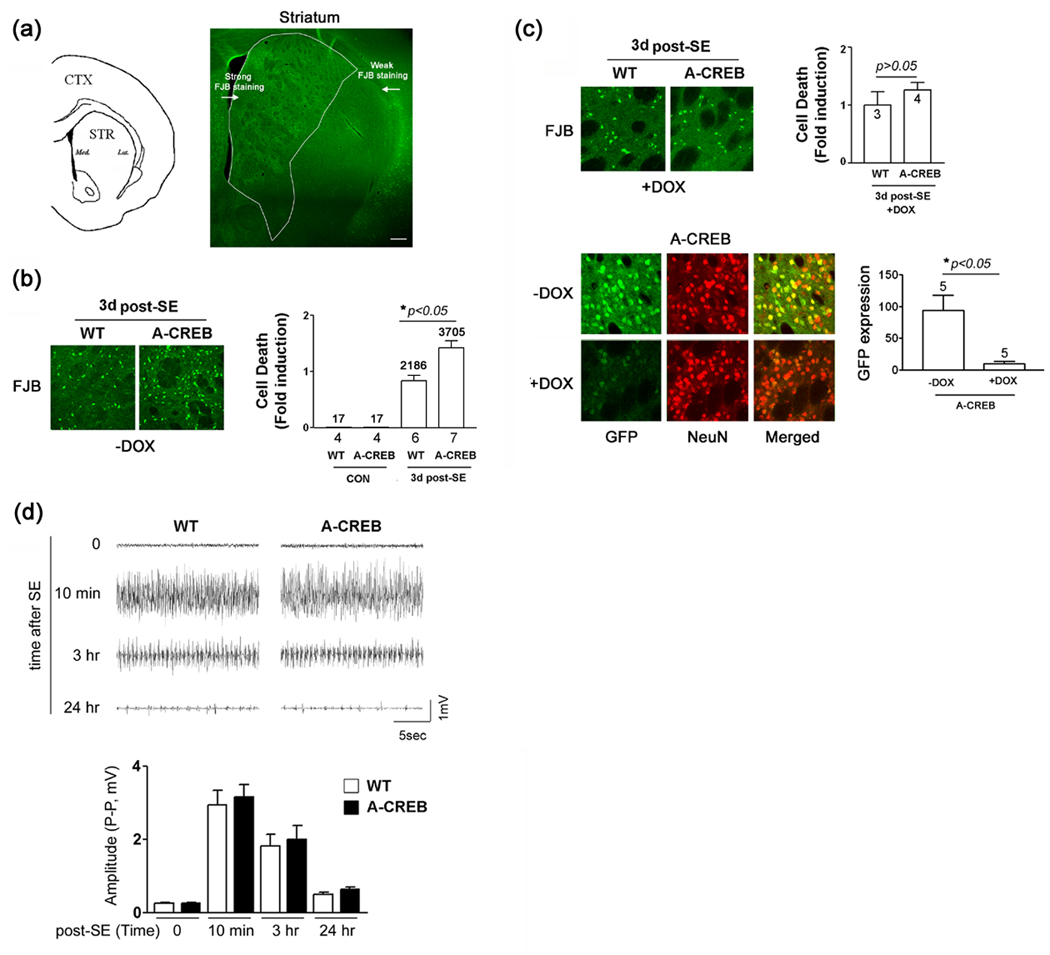
(a) Left panel) Line drawing denoting the approximate location (anterior/posterior: +1.10 from bregma) of striatal cell counting. Right panel) Representative low magnification image of FJB labeling in the striatum. Status epilepticus (SE) was induced with pilocarpine (325 mg/kg) and FJB labeling was used to detect dead and dying cells at 3 days post-SE. Bright green dots are FJB-positive cells. A higher level of cell death was consistently observed in the medial striatum. Bar = 250 µm. (b) Left panel) Representative images of striatal FJB labeling in WT and A-CREB transgenic mice. Tissue was collected 3 days post-SE. Note the higher relative number of dead cells in the A-CREB transgenic striatum. Right panel) Quantitation of FJB-positive cells in wild type and A-CREB transgenic mice. Under control conditions (no pilocarpine injection), little FJB labeling was detected in transgenic and wild type mice. Values below bars indicate the number of animals analyzed. Numbers above bars indicate the aggregate number of FJB-positive cells counted. SE-induced cell death in WT mice was normalized to a value of 1. Please see the Methods section for a detailed description of the counting procedure. (c) Doxycycline administration attenuates the effects of A-CREB on SE-induced cell death. Top left panel) Representative FJB labeling from WT and A-CREB transgenic mice treated for 2 weeks with doxycycline (1 mg/ml) in the drinking water. Tissue was collected 3 days post-SE. Top right panel) FJB cell death quantitation was performed as described in ‘b’. No significant difference in cellular vulnerability between WT and A-CREB transgenic mice was detected. Numbers in bars denote the number of animals analyzed. Bottom left panel) Representative double labeling of striatal tissue for GFP and NeuN in either the absence of, or following 2 weeks of doxycycline (1 mg/ml) treatment. Bottom right panel) Quantitation of GFP expression in A-CREB transgenic mice that were (Dox +) and were not (Dox −) treated with doxycycline. Values above the bar indicate the number of animals analyzed. GFP expression was analyzed via densitometry and normalized (a value of 100) to the – DOX condition (d) Top) Representative EEG recordings of pilocarpine-evoked seizure activity in a wild-type and A-CREB transgenic mice immediately prior to (time = 0), and at multiple time points following SE onset. Importantly, the EEG patterns of WT and transgenic mice were similar, as was the duration of SE, indicating that A-CREB does not markedly alter the electrophysiological characteristics of the SE episode. Bottom) Measurement of the EEG activity amplitude (peak to peak: P-P) for wild type and A-CREB transgenic mice. There was no significant difference in EEG activity between two lines. Significance was determined using the two-tailed Student's t test. Four wild type and 7 A-CREB mice were used for analysis.
Fig. 7. CREB couples BDNF to neuroprotection following SE.

Wild type and A-CREB transgenic mice were cannulated in the dorsal striatum and infused with BDNF (1 µl: 50 ng/ml) or vehicle 24 hours prior to SE induction: mice were killed 3 days after SE. Representative FJB images of the BDNF infused striatum (Ips-BDNF) and the contralateral striatum. Arrows in the low magnification images denote the sites of infusion. High magnification images: note the marked reduction in FJB-positive cells in the BDNF infused hemisphere of the WT animal. In contrast, the neuroprotective effects of BDNF were blunted in the A-CREB transgenic mice. Bottom panel) Quantitative analysis of FJB-positive cells. For each experimental group, the number of FJB-positive cells in the infused striatum was divided by the number of positive cells in the contralateral striatum. These values were averaged across all animals for each group and expressed as the ratio and normalized to a value of 1 for each group. In WT mice, BDNF infusion significantly attenuated cell death relative to the contralateral striatum. In contrast BDNF did not confer significant neuroprotection in A-CREB transgenic mice. A detailed description of the cell quantitation is found in the Methods section. Of note, the higher level of cell death detected in the infused striatum likely results from an additive effect of infusion-induced brain trauma and the SE insult.
To determine the effects of doxycycline on transgene expression (Fig. 2c), digital images (20× magnification: 4 per section) of GFP expression were captured from 2 coronal sections through the striatum. A 1000 pixel digital circle was then placed in the center of the image and GFP intensity was measured using a 0–256 scale. Intensity values were then background subtracted, averaged for each mouse.
To determine the percentage of GFP-positive neurons in the striatum, tissue was double labeled for GFP and NeuN. Next, fluorescent images (20× magnification) of the striatum were captured (~ +0.9 to +0.5 mm from bregma) and 200 µm2 digital boxes (2 per hemisphere) were placed over the medial striatum, and the total number of NeuN-positive cells were counted and digitally ‘marked’. The marks were then transferred to the GFP channel, and the number of ‘marked’ GFP positive cells was determined. Cells were counted from 2 sections for each animal. Of note, all GFP-positive cells were also NeuN positive. Data are presented as the mean ± SEM, and significance was assessed using a two-tailed Student’s t-test.
Western blotting
Mice were anesthetized as described above, decapitated, and brains were isolated and immediately immersed in chilled and oxygenated physiological saline and then cut into 600 µm sections with a vibratome and the dorsal striatum (~ +1.1 to +0.5 mm ~ mm from bregma) were placed on a microscope slide and frozen with dry ice. Striatal tissue was isolated and then sonicated in 100 µl of RIPA buffer (150 mM NaCl, 50 mM Tris pH 7.4, 1 mM EDTA, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 5 mM NaF, 25 µM sodium vanadate supplemented with protease inhibitor cocktail: complete mini tablet, Roche Diagnostics). Total protein levels were determined using a Bradford assay and 30 µg of protein were diluted in 3X sample buffer and loaded onto a 10% SDS-PAGE gel. After standard electrophoreses and transblotting procedures onto polyvinylidene fluoride (Immobilon P; Millipore), membranes were blocked (1 hr) with powdered milk (5% wt/vol), and then incubated (4°C overnight) with primary antibodies against 3-NT (1:1000; Chemicon), PGC-1α (1:500; Santa Cruz Biotech.), ND6 (1:1000; Molecular Probes), GFP (1:10,000; Univ. of Alberta) or ERK1/2 (1:1000; Cell signaling Technology, Beverly, MA). Samples were then incubated with horseradish peroxidase- or alkaline phosphatase-conjugated secondary antibodies (1:2500; Perkin-Elmer, Wellesley, MA) directed against the IgG domains of the primary antibodies. Signals were detected using the Western-CDP star (Perkin-Elmer) or Renaissance bioluminescent (New England Nuclear) detection systems. Between each antibody treatment, blots were washed 3X (5 min per wash) in PBST with 5% milk. Digital densitometric analysis of X-ray film band intensity was performed with Metamorph software. Band intensity was normalized to total Erk-2 for the corresponding lane. Each experiment was repeated a minimum of three times. Significance was determined via a two-tailed Student’s t test.
Real Time (RT)-PCR: PGC-1α and cyclophilin
Twenty-four hours after pilocarpine-induced SE, striatal tissue was resected as described above, and total RNA was isolated with Trizol Reagent (Invitrogen). RNA was reverse transcribed using SuperScript™ III First-Strand Synthesis kit (Invitrogen) and PCR amplification was performed using the Applied Biosystems Sybr Green Master Mix kit. All of steps were performed according to manufacturer’s instructions. The real time PCR reaction was performed using the Step One Plus Real Time PCR System (Applied Biosystems, Foster City, CA) and data analysis was performed using the comparative cycle time (Ct) method (Applied Biosystems, 2003; Bookout and Mangelsdorf, 2003). The following primer sets were used: PGC-1α : forward (AGCCGTGACCACTGACAACGAG), reverse (GCTGCATGGTTCTGAGTGCTAAG), cyclophilin: forward (CTCATCCTCAAATTTCTCTCCG), reverse (GACAGCAGAAAACTTTCGAGCT).
Complex I activity assay
Three days after SE, striatal tissue was isolated from whole brain and then homogenized in ice cold buffer containing 10 mM Tris-HCl (pH 7.4), 320 mM sucrose and 1 mM EDTA. Mitochondria were isolated via two centrifugation steps. Initially, the homogenate was centrifuged at 1000 ×g for 4 min and then the supernatant was collected and centrifuged at 15,000 ×g for 20 min. Pelleted mitochondria were resuspended in 25 mM PBS (pH 7.2) supplemented with 10 mM MgCl2 and treated to 3 freeze-thaw cycles. Complex I activity was estimated spectrophotometrically using the method described by Birch-Machin and Turnbull (2001). Initially, purified mitochondrial protein (25 µg) and decylubiquinone (50 µM) were added to the assay buffer (PBS [25 mM, pH 7.2] supplemented with MgCl2 [5 mM], KCN [1 mM], bovine serum albumin [BSA; 3.5 g/L], and antimycin A [2 µg/ml]). The total volume of reaction mixture was 0.2 ml. After preincubation for 2 min, the reaction was initiated by addition of NADH (0.25 M final concentration) and the decrease in the absorbance rate was monitored at 340 nm for 5 min. Rotenone (5 µM) was added to measure complex I-specific enzyme activity. Blank reactions were carried out in the absence of NADH and these values were subtracted from test samples.
Primary neuronal cell culture
Striatal tissue was dissected from embryonic day 18 Sprague Dawley rat pups. Tissue was then enzymatically digested into a single-cell suspension using the methods and reagents described by Lee et al. (2005). Cell were plated at a density of 2.2 × 105/cm2 onto poly-D-lysine-(>540 kDa; Sigma) coated 24 well dishes and maintained in Minimal Essential Media (Invitrogen) containing 2% B-27 (Invitrogen) and 100 U/ml penicillin/streptomycin at 37°C and 5% CO2 in an incubator.
Transfection and immunostaining of cultured neurons
Primary cultured striatal neurons were transfected (1.5 µg of DNA per well) after 7 days in vitro using a calcium phosphate transfection kit (Invitrogen). Transfections were performed using the modifications described by Xia et al. (1996). The following constructs were used: pcDNA3.1 empty vector (1 µg/well), VP16-CREB (1 µg/well), A-CREB (1 µg/well), and CMV-Venus (200 ng/well). For the A-CREB transfection assays (Fig. 6c), B27 was withdrawn from the media 24 hrs prior to H2O2 stimulation, and returned to the media after removal of the H2O2. This experimental design allowed for a more straightforward assessment of the neuroprotective effects of BDNF. For the VP16-CREB transfection assays (Fig. 6d), B27 was only withdrawn during H2O2 treatment. This longer withdrawal of B27 during the BDNF/A-CREB assays likely results in the enhanced toxicity of H2O2, relative to the VP16-CREB assays. After stimulation, the cells were fixed with 4% (w/v) PFA for 15 min, washed five times in PBST, and then blocked as described above. Cultures were immunolabeled (overnight at 4°C) with antibodies against the NeuN (mouse monoclonal, 1:500; Chemicon), and GFP (rabbit polyclonal, 1:5000 final dilution; Univ. Calgary). Cells were then incubated (4 h at RT) with Alexa 488- or 594-conjugated secondary antibodies (1:250 final dilution). To examine cell health, cultures were incubated (10 min) with the DNA stain Hoechst 33342 (Hoechst, 1 µg/ml; Molecular Probes) before mounting with gel mount (Biomedia, Foster City, CA). Photomicrographs were captured using the inverted epifluorescence microscope described above at 200× magnification. All tabulated data are expressed as the mean ± SEM. Significance was determined using the two-tailed Student's t test. Cell counts for each condition were performed from three slides.
Fig. 6. BDNF-mediated neuroprotection from oxidative injury: role of CREB.

(a) BDNF attenuates seizure-induced tyrosine nitration. Wild type mice were cannulated in the dorsal striatum and infused with vehicle or BDNF (1 µl: 50 ng/µl) 24 hours prior to pilocarpine injection. Mice were killed 6 h after pilocarpine injection and striatal tissue was collected and probed for 3-NT levels as described in Fig. 3. BDNF infusion reduced the seizure-induced increase in 3-NT levels. Each condition was run in duplicate. (b) Top panel) Striatal cells were isolated from embryonic day 18 rat pups, cultured for 7 days and then transfected with the marker gene Venus and either A-CREB or an empty expression vector (pcDNA3.1). Two days later, cell were treated with BDNF (100 ng/µl), followed 24 hrs later by exposed to H2O2 (200 µM, 30 min). Cells were fixed 4 hrs after H2O2 treatment, immunolabeled for Venus and NeuN and cell death was scored via Hoechst labeling. Relative to mock-treated samples, H2O2 led to a pronounced increase in cell death. Pretreatment with BDNF significantly attenuated H2O2 toxicity. Arrows denote dead transfected neurons (e.g., fragmented or condensed nuclei): arrowheads denote live cells. Bottom panel) Quantitative analysis revealed that the neuroprotective effects of BDNF were inhibited by A-CREB. Data are from triplicate determinations. Error bars denote SEM. Numbers in the bars indicate the number of neurons assayed. There was no significant difference (n.s., p>0.05) between the A-CREB transfected groups. (c) Top panel) Cultured striatal neurons were transfected with the marker gene Venus and either VP16-CREB or an empty expression vector (pcDNA 3.1) and stimulated as described above with H2O2. After 30 min stimulation with H2O2, cells were washed with culture media and maintained for eight more hours. Cells were then immunolabeled and scored for cell viability as described above. Representative images reveal that VP16-CREB conferred protection against H2O2-mediated cell death. Arrow indicates dead transfected neurons and arrowheads denote live neurons. Bottom panel) Quantitative analysis of cell death under the different stimulus and transfection conditions. Data are from triplicate determinations. Error bars denote SEM. Numbers in bars indicate the total number of neurons counted. Of note, the elevated level of H2O2-evoked cell death in panel ‘c’ relative to panel ‘d’ is the likely result of the extended withdrawal (24 hrs) of B27 prior to H2O2 treatment, which was required to effectively test the neuroprotective effects of BDNF. Please see the Methods section for additional details of the experimental approach.
Electroencephalogram (EEG) recording
For EEG recording, mice were anesthetized as described above, and surgically implanted with two bipolar recording electrodes (Plastics One, Roanoke, VA). The implantation procedure was the same as described above for cannulation. One electrode was positioned within area CA1 of the hippocampus (anterior −1.8 mm; lateral 1.1 mm and dorsoventral 1.2 mm from dura with the head level). The other electrode was positioned in the cortex (anterior −2.8mm; lateral 1.1 mm and dorsoventral 1.2 mm from dura with head level). Ten days after surgery, mice were injected with atropine (1 mg/kg), and then, 30 min later with pilocarpine (325 mg/kg). EEG recording started just prior to atropine injection and behavior was monitored continuously for the first three hours following SE. Polysomnographic signals were recorded on a personal computer (Pentium 4) using a computerized acquisition device (MP150, Biopac Systems, Santa Barbara, CA) and software (Acknowledge 3.9.0, Biopac Systems). EEG data were collected and compared at four time points: ~ 10 min after atropine injection and 10 min, 3 hr and 24 hr after SE onset. Average peak-to-peak (P-P) values were obtained from 20 sec EEG traces using Acknowledge 3.9.0 software and used for comparative analysis.
RESULTS
A-CREB transgenic mice
To investigate the role of CREB in cell stress and neuroprotection, we generated a tetracycline-regulated transgenic mouse strain which expresses A-CREB, a potent negative regulator of CREB/CRE-mediated transcription (Ahn et al. 1998). To identify transgenic cells, the A-CREB construct was designed to express green fluorescent protein (GFP) from an internal ribosomal entry site (IRES) (Fig. 1a). In several of our founder lines, crossing the A-CREB mice with the αCaMKII-tTA activator line (Mayford et al. 1996) drove robust transgene expression in the striatum (Fig. 1b). Double labeling for the transgene marker GFP and the neuronal marker NeuN revealed that ~ 61% of striatal neurons expressed the transgene construct (Fig. 1c). In a previous report (Lee et al. 2007), we found that CRE-mediated gene expression was potently repressed in A-CREB transgenic mice. Additional representative data presented here (Fig. 1d) show that SE-induced expression of the CREB-regulated gene Jun-B was attenuated specifically in the striatum, where high levels of the A-CREB transgene are expressed. Since a number of studies have shown that disruption of CRE-mediated transcription can affect cell viability (reviewed by Walton and Dragunow, 2000), we examined striatal neuron density in A-CREB transgenic mice. Quantitation of NeuN-expressing cells revealed that there was no significant difference in the average cell density between A-CREB transgenic and WT littermates (Fig. 1e). Similarly, histological examination with Fluoro-jade B (FJB), a marker of dead and dying cells, did not indicate an elevated level of striatal cell death in A-CREB transgenic mice (Fig. 2b). Together these findings reveal that expression of the transgene does not alter cell viability under normal physiological conditions.
Fig. 1. A-CREB transgenic mice.
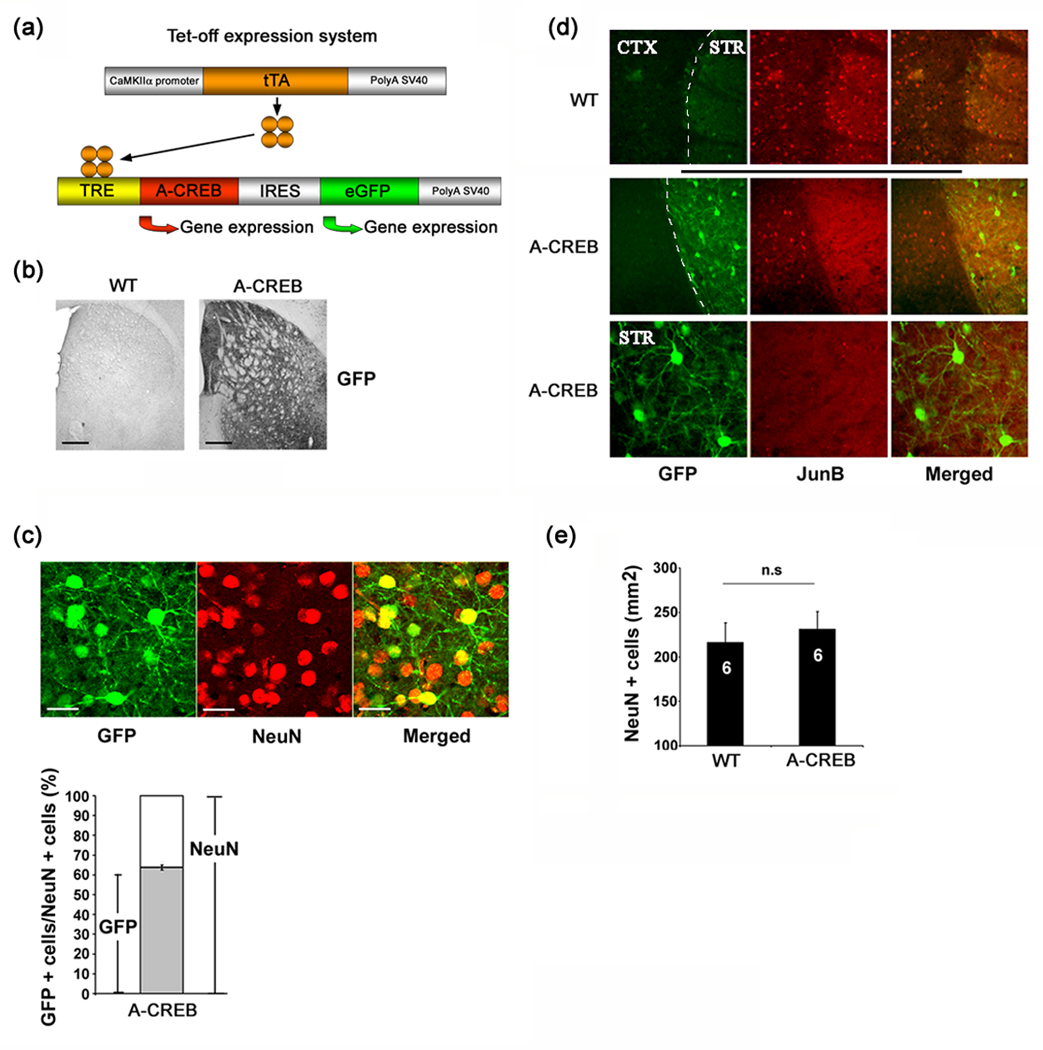
(a) Schematic of the bitransgenic tet-off system used to drive the expression of A-CREB and the marker GFP. (b) Tissue was immunohistochemically-processed for the transgene maker GFP. Note the robust staining in the striatum. As a test for antibody specificity, tissue from a WT littermate was processed for GFP expression. Bar = 250 µm. (c) Representative GFP (green) and NeuN (red) double labeling in the striatum. Bar = 25 µm. Quantitation of transgenic cell numbers is shown below. Approximately 64% of striatal neurons had detectable levels of GFP expression. Analysis was performed on ~ 1000 striatal cells from five animals. (d) Representative double labeling for the transgene marker GFP and JunB. Animals were killed 90 min after pilocarpine-evoked SE. WT tissue shows marked JunB expression in the striatum (STR) and cortex (CTX). In contrast, in A-CREB transgenic mice, JunB expression was not detected in the striatum, where high levels of the transgene are expressed. Magnified representative double labeling from A-CREB transgenic tissue is shown in the bottom set of panels. (e) Quantitation of NeuN positive cells in wild type (N=6) and A-CREB transgenic (N=6) mice. No significant difference (n.s) in cell density between transgenic and wild type mice was detected. Please see the Methods section for a detailed description of the counting process.
A-CREB increases vulnerability to excitatory insults
Repetitive seizure activity (i.e. status epilepticus: SE) triggers marked cell death in the striatum of C57BL/6 mice (McLin and Steward, 2006). Consistent with this work, at 3 days post-SE, we observed robust and reproducible cell death in the striatum. The highest density of FJB-positive cells was detected in the medial striatum (Fig. 2a). To test the role of CREB as a regulator of SE-induced toxicity, A-CREB transgenic mice were challenged with SE and the level of cell death (assessed using FJB labeling), relative to WT littermates, was examined 3 days after pilocarpine injection. Expression of the A-CREB transgene significantly increased the excitotoxic effect of SE (Fig. 2b). Next, we tested whether augmentation of cell death could be reversed by repressing A-CREB expression. For these experiments, A-CREB transgenic and WT mice were treated with doxycycline (1 mg/ml) in their drinking water for 2 weeks, and then the effects of SE were assayed via FJB quantitation. Interestingly, A-CREB-mediated augmentation of cell death was repressed by doxycycline (Fig. 2c). These data reveal that the effects of A-CREB on cell vulnerability are largely reversible, and, by implication, that tonic transgene expression does not significantly affect neuronal maturation, or health. Rather, A-CREB appears to specifically enhance vulnerability to excitatory stimulation. To confirm the efficacy of doxycycline treatment, tissue from A-CREB transgenic mice was examined for GFP levels. Relative to the untreated transgenic mice, doxycycline administration led to an ~ 90% decrease in GFP transgene expression, as assessed by immunolabeling densitometry (Fig. 2c).
The increased vulnerability of the A-CREB mice could be the result of an enhanced SE phenotype. However, no distinguishable differences in the duration (Table 1) or the motor manifestations of the SE episode between the A-CREB and WT mice were noted. Additionally, EEG recordings detected a similar SE-evoked electrical signature in WT and A-CREB transgenic mice (Fig. 2d). High amplitude electrical discharges were observed 10 min after behavioral manifestation of SE onset. Recordings captured 3 hours after SE onset reveal repetitive high voltage discharges, which are stereotypical of repetitive, chronic, motor seizure activity (Honchar et al. 1983). Twenty-four hours after SE, EEG activity exhibited low level discharges. Importantly, the EEG patterns of WT and transgenic mice were similar, indicating that the expression of the transgene does not markedly affect key electrophysiological characteristics of the SE episode. Of note, though, A-CREB mice did exhibit a significantly shorter SE latency and a higher likelihood to exhibit SE than WT littermates (Table 1). Together, these data suggest that the cell death phenotype of A-CREB mice is not the result of enhanced seizure activity, but rather, results from an increased sensitivity to the excitatory insult.
Table 1.
Tabulation of behavioral observations following pilocarpine injection. Data are represented as mean ± SEM.
| Stage 5 seizure | Stage 6 seizure (SE) | Status Epilepticus | ||||||
|---|---|---|---|---|---|---|---|---|
| Animals | Number of mice |
Latency to onset (min) |
Percentage | Latency to onset (min) |
Percentage | Duration of SE (min) |
Percentage | Mortality (%) |
| WT | 18 | 41.5±8.1 | 83 | 45.1±9.1 | 50 | 286±6.2 | 28 | 22 |
| A-CREB | 16 | 18.5±1.5 | 100 | 27.7±3.0 | 100 | 259±8.0 | 50 | 31 |
| *p<0.05 | *p<0.05 | n.s. | ||||||
CREB, oxidative stress, and mitochondrial function after SE
These findings led us to examine the potential mechanism by which CREB regulates cell viability. A number of studies have shown that SE increases ROS production (Tejada et al. 2007; Yu et al. 2008; Kovács et al. 2002), which may enhance neuronal cell vulnerability. This, combined with recent work showing that CREB affects ROS detoxifying enzyme expression (Krönke et al. 2003; St. Pierre et al. 2006) raised the prospect that increased vulnerability in the A-CREB transgenics may result from a repressed antioxidant response mechanism. To begin to address this issue, we first tested whether SE triggers oxidative stress in the striatum. To this end, levels of peroxynitrite-mediated tyrosine nitration (a marker of oxidative protein damage) were detected with the use of an antibody to 3-nitrotyrosine (3-NT). Western analysis of striatal lysates revealed an increase in 3-NT levels from 6 hrs to 72 hrs after SE onset. Interestingly, relative to WT mice, the A-CREB transgenic mice exhibited a more robust increase in 3-NT levels at the early, 6 hr post-SE, time point (Fig. 3a). Probing blots for the transgene marker GFP validated the genotypes of the groups and total protein levels were assayed by blotting for total ERK 1/2. Of note, under control conditions, higher levels of 3-NT were detected in A-CREB transgenic than in WT mice, thus suggesting that CREB-mediated transcription regulates ROS/RNS generation and/or the antioxidant response profile of neurons under normal conditions. Lastly, to determine whether this increase in 3-NT levels occurred in A-CREB transgenic neurons, tissue from animals with SE was triple labeled for the transgene marker GFP, 3-NT and the DNA/nuclear marker DraQ5. Cellular analysis revealed that A-CREB expressing neurons exhibited elevated levels of 3-NT relative to adjacent non-transgenic cells (Fig. 3b). Together, these data indicate that disruption of CREB signaling elevates both basal and the SE-induced ROS/RNS levels in the striatum.
Fig. 3. SE-induced oxidative stress in A-CREB transgenic mice.

(a) Western blot showing increased 3-nitrotyrosine (3-NT) levels as a function of time following SE. Striatal tissue was collected at the noted times and lysates were probed using an antibody against 3-NT. In WT mice, an increase in 3-NT incorporation was noted from 6–72 hrs post-SE. Compared to WT mice, the level of 3-NT incorporation at the 6 hr time point was much higher in A-CREB transgenic mice. The blot was stripped and probed for expression of the GFP transgene, and, as a protein loading control, the blot was also probed ERK 1/2 levels. (b) Triple fluorescence labeling for GFP, 3-NT and DraQ5 in the striatum of A-CREB transgenic mice. The representative image shows that relatively high levels of 3-NT are detected in GFP positive (transgenic) cells (arrows). Tissue was collected at 24 hrs post-SE onset. Bar = 10 µm.
Next, to test whether CREB regulates detoxifying enzyme expression, WT and A-CREB transgenic tissue was immunolabeled for heme oxygenase I (HO-1). In a number of model systems, HO-1 has been shown to be under the influence of the CREB/CRE transcriptional pathway (Gong et al. 2002; Krönke et al. 2003). Representative labeling and quantitative analysis revealed that A-CREB mice had lower basal HO-1 levels than WT mice (Fig. 4a).
Fig. 4. CREB regulates HO-1 and PGC-1α expression.

(a) Left) Representative photomicrographs of heme oxygenase 1 (HO-1) expression in WT and A-CREB mice under control conditions (no pilocarpine). Right) Quantitation of HO-1 expression. The value for WT mice was normalized to 1; numbers in bars denote the number of animals examined. (b) Western blotting of striatal samples for PGC-1α. Tissue was harvested at 0 hour (control), 24 hours and 3 day after SE onset from both A-CREB and wild type (WT) mice. Left) Representative blot showing that in wild type mice, SE induced a marked increase in PGC-1α expression at 24 hrs and 3 days post-SE. In contrast SE did not evoke PGC-1α in A-CREB transgenic mice. The blot was stripped and probed for expression of the GFP transgene, and the protein loading control, ERK 1/2. Right) Quantitative densitometric analysis of PGC-1α expression. Levels of PGC-1α were normalized to ERK 2 levels for each lane, and averaged across all triplicate determinations; control (no SE) values were normalized to a value of 1. Values in the bar indicate the number of blots analyzed. (c) Sybr Green-based quantitative real time PCR analysis of PGC-1α transcript expression. Animals were sacrificed 24 hrs after SE onset. Relative to WT mice, SE-induced PGC-1α expression was blunted in A-CREB transgenic mice. Data are presented as the relative PGC-1α expression amd averaged using quadruplicate determinations from 4–5 animals for each condition. (d) SE evoked CRE-mediated reporter gene expression in the striatum. Representative photomicrographs of striatal β-galactosidase expression under control conditions and 8 hrs after SE onset. Tissue was processed using DAB labeling with nickel intensification.
Recent work has shown that CREB functions as a signaling intermediate that couples ROS production to the induction of PGC-1α, and, in turn, enhanced mitochondrial biogenesis, free radical scavenging and neuroprotection (St. Pierre et al. 2006). Given these data, and our findings showing that repression of CREB function enhances protein nitration, we examined the potential connection between CREB and PGC-1α expression. Western analysis of striatal lysates showed that SE stimulated a significant increase in PGC-1α expression. This induction appears to be dependent on CREB-mediated transcription, since the SE-induced expression of PGC-1α was abrogated in A-CREB transgenic mice (Fig. 4b). The elevated level of PGC-1α in the A-CREB control lane results from a relatively high level of protein loading (note the increased relative ERK1/2 expression in this lane). We also employed Sybr Green-based quantitative RT PCR to profile PGC-1α transcript levels (Fig. 4c). In a parallel with the Western data, we found that SE triggered a marked increase in PGC-1α transcript expression in WT mice, whereas in A-CREB mice, this increase was inhibited. Finally, to validate the physiological relevance of the A-CREB data, we used a CRE-β-galactosidase reporter mouse strain to validate that SE stimulates CRE-mediated gene expression in the striatum. As expected, at 8 hrs post-SE onset, robust induction of β-galactosidase was detected in the striatum (Fig. 4d). A more rigorous examination of SE-induced, CRE-mediated transcription can be found in Lee et al. (2007). Together these data reveal that the CREB/CRE signaling pathway is both activated by SE and augments inducible PGC-1α expression.
Since A-CREB mice had a blunted response to SE-induced upregulation of PGC-1α we next determined whether CREB disruption affected mitochondrial gene expression and function. Initially, we examined the expression of the mitochondrial-encoded gene ND6, which is a subunit of complex I of the mitochondrial respiratory chain, and thus subject to regulation through PGC-1α co-activator pathways. Compared to WT littermates, basal expression of ND6 was lower in the striatal lysates of A-CREB mice (Fig. 5a). Furthermore, SE induced a significant increase in ND6 expression in WT mice which was not detected in A-CREB transgenic animals (Fig. 5a), indicating that CREB functions as an upstream regulator of ND6 expression. As an aside, as part of our standard analysis of transgene expression, we consistently noted a marked upregulation of GFP expression 2–3 days after the SE episode. One potential explanation for this is that the SE-insult stimulates expression of the transgene activator tTA.
Fig. 5. Mitochondrial complex I expression and activity in A-CREB and WT mice following SE.

(a) Left) Western blotting for ND6, a subunit of complex I. In wild type mice, SE induced a modest increase in ND6 expression at 3 days post-SE. In A-CREB transgenic mice, SE did not elevate ND6 levels. In addition, relative to WT mice, a lower level of ND6 expression was detected in A-CREB transgenics. Blots were probed for the transgene marker (GFP) and for total protein levels (ERK 1/2). Two samples were run for each condition. Right) ND6 band intensity was normalized to ERK 2 intensity for each lane; control (no SE) values were normalized to a value of 1. (b) Representative triple labeling for GFP, DraQ5 and ND6 (complex I). The images reveal that transgenic cells have a lower level of ND6 expression than non-transgenic cells. Arrows denote GFP-positive cells. Arrowheads denote GFP-negative cells. (c) Complex I activity. Striatal mitochondria were isolated from A-CREB and WT mice under control conditions and three days after SE. Complex I activity (assayed in the crude mitochondrial preparation) was normalized to a value of 1 for WT mice. Values are presented as the mean ± SEM. The numbers in the bars denotes the number of animals (samples) used for the analysis.
Importantly, these data were complemented by triple-labeling immunofluorescence experiments showing that complex I (ND6) expression was selectively reduced in A-CREB-positive cells (Fig. 5b). Since the ND6 subunit is a key component of complex I, we also examined the activity of complex I. Consistent with the Western blot findings (Fig 5a), a significant reduction in complex I activity was detected in A-CREB transgenic mice (Fig. 5c). This relative decrease was detected under both control conditions and following SE. Together, these data indicate that CREB regulates inducible complex I protein levels and activity after SE. These findings, combined with data showing elevations in 3-NT levels and decreased induction of PGC-1α, indicate that protective responses to oxidative stimuli are suppressed in A-CREB mice, with likely detrimental consequences to cell viability.
BDNF protects striatal neurons from cell toxicity via a CREB-dependent mechanism
Recent work suggests that BDNF has therapeutic value as a treatment for neurodegenerative disorders such as Alzheimer's disease (Forero et al. 2006), Parkinson's disease (Fernandez-Espejo, 2004) and Huntington's disease (Davies and Ramsden, 2001). At a mechanistic level, oxidative stress appears to contribute to the pathophysiology of the three aforementioned diseases, thus raising the possibility that BDNF confers protection via the attenuation of oxidative stress. To test the effects of BDNF in vivo, wild type mice were cannulated in the dorsal striatum and infused with vehicle or BDNF (1 µl of 50 ng/µl) 24 hours prior to pilocarpine injection. Six hours after pilocarpine-evoked stage 3–4 seizure activity, mice were killed and striatal tissue was processed via Western blotting for 3-NT. Interestingly, the seizure-induced increase in tyrosine nitration was markedly attenuated by BDNF, thus revealing a potent antioxidant effect of BDNF (Fig. 6a).
Given that the CREB/CRE transcriptional pathway is tightly regulated by BDNF/TrkA receptor signaling, we wanted to know whether the neuroprotective effects of BDNF were dependent on CREB-mediated transcription. To address this question, cultured striatal neurons were transfected with A-CREB, pretreated with BDNF (24 hours, 100 ng/mL) and then challenged with H2O2 (200 µM) for 30 min. Eight hours after treatment, the cells were fixed and labeled for expression of the GFP derivative Venus (a transfection marker) and NeuN and nuclear condensation (a marker of cell death) was analyzed using Hoechst staining. Representative images and quantitative analysis revealed that A-CREB blocked the neuroprotective effects of BDNF (Fig. 6b). As an important validation of the functional role of CREB in protection against oxidative stress, transient transfection of a constitutively active form of CREB (VP16-CREB: Barco et al. 2002) conferred significant protection against H2O2-mediated cell death (Fig. 6c).
Finally, we examined whether BDNF-mediated neuroprotection is altered in A-CREB transgenic mice. To this end, BDNF (50 ng/µl) was unilaterally infused into the striatum 24 hrs prior to SE induction and the level of cell death was examined via FJB labeling. Representative FJB staining from mice sacrificed 3 days after SE, reveal that BDNF injection (ipsilateral) dramatically decreased the number of dead/dying cells compared to the contralateral hemisphere (Fig. 7). In contrast, the capacity of BDNF to attenuate cell death was markedly diminished in A-CREB transgenic mice; hence the level of cell death between the ipsilateral (BDNF-infused) and contralateral striatum was not significantly different in A-CREB transgenic mice. Collectively, these data strongly suggest a BDNF-actuated, CREB-mediated, signaling mechanism that potently attenuates oxidative stress and cell death.
DISCUSSION
Dysregulation of ROS production and elimination plays a major role in the pathology of a number of CNS diseases (Halliwell, 2006; Calabrese et al. 2006; Albers and Beal, 2000). Given this, there is a clear need to gain deeper insight into ROS regulatory pathways that may be altered as a part of the disease process. To this end, we focused on the CREB/CRE pathway. Interest in this transcriptional pathway was heightened by a series of studies showing that CREB is a key regulator of neuroprotection and that repression of CREB-mediated signaling can contribute to neuronal pathology. Along these lines, CREB has been shown to function as a survival factor in a number of model systems (Riccio et al. 1999; Lee et al. 2005) and CREB null mice exhibit marked progressive degeneration of hippocampal and striatal neurons (Mantamadiotis et al. 2002). Likewise, disruption of CREB-phosphorylation at Ser-133 has been shown to increase the vulnerability of neurons to cytotoxic stimuli (Hardingham et al. 2002). Until recently, the principal neuroprotective signaling mechanism by which CREB regulates neuronal survival was thought to occur via upregulation of neurotrophin and antiapoptotic gene expression (Lonze and Ginty, 2002). However, data collected over the past few years has revealed a role for CREB in the regulation of ROS toxicity (Zou and Crews, 2006). Central to this is work by St. Pierre (2006) in which PGC-1α, a key effector of ROS detoxifying enzyme expression, as well as mitochondrial biogenesis, was shown to be induced in a CREB-dependent manner.
The goal of this study was to provide an in vivo, and context-specific assessment of CREB as a regulator of cell death and ROS signaling. To this end, we used a pilocarpine-evoked SE model to elicit excitotoxic cell death. Pilocarpine is a muscarinic receptor agonist that has been shown to initiate neuronal death in diverse brain regions, including the cortex, hippocampus and striatum (Fujikawa 1996). Importantly, pilocarpine-induced SE triggers a rapid and robust increase in ROS production throughout the CNS (Tejada et al. 2007). Consistent with this, we detected an SE-induced increase in oxidative stress in WT mice. In an important in vivo validation of the role of CREB as a regulator of oxidative stress, we detected a marked increase in the level of SE-induced protein nitration in A-CREB mice. This increase in oxidative stress was followed by a significant increase in the level of SE-induced cell death. As noted above, seizure severity and duration were not dramatically altered by A-CREB, indicating that vulnerability was dependent on the inherent capacity of the cell to effectively respond to the excitatory stimulus. In line with this, previous work using cultured neurons found that cell death evoked by exogenous glutamate application was markedly enhanced by repression of CREB-mediated transcription (Lee et al. 2005; Glover et al. 2004).
It is important to note that under basal conditions, neither a decrease in cell number nor an active cell death program was detected in A-CREB-transgenic mice, indicating that the SE stimulus was not simply accelerating an ongoing neurodegenerative process. In some respects, this contrasts with data from Long et al. (2001) who reported that A-CREB transgenic mice die minutes after birth. This difference is the likely result of distinct transgene targeting. Hence, Long and colleagues used the cartilage-specific collagen type II promoter/enhancer which induces gene expression in tibial chondrocytes from early developmental stage, whereas we used the CaMK II promoter (Mayford et al. 1996) to drive robust transgene expression within the forebrain.
The increase in oxidative stress observed in A-CREB transgenic mice led us to examine possible alterations in the expression and functionality of ROS producing and regulatory processes. To this end, we profiled the expression of PGC-1α. In WT mice, SE elicited a marked increase in PGC-1 α expression. However, in A-CREB transgenic mice, SE-evoked PGC-1α expression was blunted, indicating a critical upstream role for CREB in inducible PGC-1α expression. Given that PGC-1α drives the expression of many ROS-detoxifying enzymes, including GPx1 and SOD2 (St. Pierre et al. 2006), these data support a model wherein a CREB/ PGC-1α signaling cassette regulates oxidative stress.
Interestingly, CREB has also been shown to directly influence the expression of several cytoprotective antioxidant enzymes (Bedogni et al. 2003; Gong et al. 2002; Krönke et al. 2003). In accordance with this, A-CREB mice exhibited a marked decrease in the expression of the CREB regulated gene for HO-I. In addition, repression of CREB may also indirectly affect gene expression via the modulation of genetic regulatory networks. Along these lines, CREB repression of HO-I could result from repressed expression of the CREB target genes Jun and Fos. These AP-1 binding proteins have been shown to form a transcriptional complex with Nrf2 (Wild et al., 1998; Wild et al., 1999; Dhakshinamoorthy et al., 2000), a potent activator of stress evoked HO-1 expression (Alam and Cook, 2003). Thus, abrogation of CREB-dependent transcription could inhibit HO-1 via an indirect mechanism that reduces the expression of the Nrf2 binding partners. Additional studies will be required to address these issues.
We also found a significant A-CREB-dependent decrease in the expression of the complex I subunit ND6, both under basal and SE conditions. ND6 is encoded from mitochondria DNA (mtDNA) and is essential for complex I respiratory function (Bai and Attardi, 1998). This decrease in expression was paralleled by a decrease in complex I activity under both control conditions and following SE. Interestingly, deficient complex I activity has been shown to elevate ROS levels (Tretter et al. 2004; Votyakova and Reynolds, 2005) and is correlated with significant oxidative damage (Sherer et al. 2003; Beretta et al. 2006). Together, these findings raise the possibility that disruption of CREB enhances oxidative stress, in part, by compromising mitochondrial respiration efficiency.
An extensive literature has provided significant insight into the role of CREB as a signaling intermediate that couples preconditioning stimuli to neuroprotection. For example, Hara et al. (2003) found that CRE-mediated transcription is required for the induction of ischemic tolerance and Mabuchi et al. (2001) reported that the neuroprotective effects of ischemic preconditioning involve CREB-dependent transcription. Although much of the work examining CREB and neuroprotection has focused on its regulation of neurotrophins and antiapoptotic genes, the data presented here indicate that CREB also stimulates a potentially neuroprotective preconditioning response as well. Along these lines, our cell culture data revealed that pretreatment with BDNF conferred protection against H2O2-mediated cell death via a CREB-dependent mechanism. Furthermore, the in vivo neuroprotective effects of BDNF were dependent on CREB and were correlated with a decrease in oxidative load. Additional assays that employ a combination of mRNA microarray and proteomic interrogation approaches will be required to identify both the CREB-regulated genetic networks that give rise to antioxidant neuroprotection and how these networks regulate the oxidative state of specific proteins.
The data presented here highlight the central role of CREB as a regulator of oxidative stress and, in turn, suggest the possibility that disruption of CREB signaling may be a key event in disease-dependent increases in oxidative load. This, coupled with work revealing a central role for CREB in neurotrophin and antiapoptotic gene expression, raises the prospect that strategies focusing on augmentation of CREB-mediated transcription may prove clinically beneficial against an array of acquired and congenital neurological disorders.
Acknowledgements
We would like to thank Heather Dziema and Thong Ma for technical assistance. This work was supported by the National Institutes of Health (Grant number: MH62335 and NS47176).
REFERENCES
- Ahn S, Olive M, Aggarwal S, Krylov D, Ginty DD, Vinson C. A dominant-negative inhibitor of CREB reveals that it is a general mediator of stimulus-dependent transcription of c-fos. Mol. Cell. Biol. 1998;18:967–977. doi: 10.1128/mcb.18.2.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam J, Cook JL. Transcriptional regulation of the heme oxygenase-1 gene via the stress response element pathway. Curr Pharm Des. 2003;9:2499–2511. doi: 10.2174/1381612033453730. [DOI] [PubMed] [Google Scholar]
- Albers DS, Beal MF. Mitochondrial dysfunction and oxidative stress in aging and neurodegenerative disease. J. Neural. Transm. Suppl. 2000;59:133–154. doi: 10.1007/978-3-7091-6781-6_16. [DOI] [PubMed] [Google Scholar]
- Applied Biosystems. Essentials of Real Time PCR. 2003 [Google Scholar]
- Aracena P, Aguirre P, Muñoz P, Núñez MT. Iron and glutathione at the crossroad of redox metabolism in neurons. Biol. Res. 2006;39:157–165. doi: 10.4067/s0716-97602006000100017. [DOI] [PubMed] [Google Scholar]
- Bai Y, Attardi G. The mtDNA-encoded ND6 subunit of mitochondrial NADH dehydrogenase is essential for the assembly of the membrane arm and the respiratory function of the enzyme. EMBO J. 1998;17:4848–4858. doi: 10.1093/emboj/17.16.4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barco A, Alarcon JM, Kandel ER. Expression of constitutively active CREB protein facilitates the late phase of long-term potentiation by enhancing synaptic capture. Cell. 2002;108:689–703. doi: 10.1016/s0092-8674(02)00657-8. [DOI] [PubMed] [Google Scholar]
- Barco A, Pittenger C, Kandel ER. CREB, memory enhancement and the treatment of memory disorders: promises, pitfalls and prospects. Expert Opin. Ther. Targets. 2003;7:101–114. doi: 10.1517/14728222.7.1.101. [DOI] [PubMed] [Google Scholar]
- Bedogni B, Pani G, Colavitti R, Riccio A, Borrello S, Murphy M, Smith R, Eboli ML, Galeotti T. Redox regulation of cAMP-responsive element-binding protein and induction of manganous superoxide dismutase in nerve growth factor-dependent cell survival. J. Biol. Chem. 2003;278:16510–16519. doi: 10.1074/jbc.M301089200. [DOI] [PubMed] [Google Scholar]
- Beretta S, Wood JP, Derham B, Sala G, Tremolizzo L, Ferrarese C, Osborne NN. Partial mitochondrial complex I inhibition induces oxidative damage and perturbs glutamate transport in primary retinal cultures. Relevance to Leber Hereditary Optic Neuropathy (LHON) Neurobiol. Dis. 2006;24:308–317. doi: 10.1016/j.nbd.2006.07.016. [DOI] [PubMed] [Google Scholar]
- Birch-Machin MA, Turnbull DM. Assaying mitochondrial respiratory complex activity in mitochondria isolated from human cells and tissues. Methods Cell. Biol. 2001;65:97–117. doi: 10.1016/s0091-679x(01)65006-4. [DOI] [PubMed] [Google Scholar]
- Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and - independent mechanisms. Science. 1999;286:1358–1362. doi: 10.1126/science.286.5443.1358. [DOI] [PubMed] [Google Scholar]
- Bookout AL, Mangelsdorf DJ. Quantitative real-time PCR protocol for analysis of nuclear receptor signaling pathways. Nucl. Recept. Signal. 2003;1:e012. doi: 10.1621/nrs.01012. Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher GQ, Dziema H, Collamore M, Burgoon PW, Obrietan K. The p42/44 mitogen-activated protein kinase pathway couples photic input to circadian clock entrainment. J. Biol. Chem. 2002;277:29519–29525. doi: 10.1074/jbc.M203301200. [DOI] [PubMed] [Google Scholar]
- Calabrese V, Bates TE, Stella AM. NO synthase and NO-dependent signal pathways in brain aging and neurodegenerative disorders: the role of oxidant/antioxidant balance. Neurochem. Res. 2000;25:1315–1341. doi: 10.1023/a:1007604414773. [DOI] [PubMed] [Google Scholar]
- Calabrese V, Guagliano E, Sapienza M, Mancuso C, Butterfield DA, Stella AM. Redox regulation of cellular stress response in neurodegenerative disorders. Ital. J. Biochem. 2006;55:263–282. [PubMed] [Google Scholar]
- Choi YS, Lin SL, Lee B, Kurup P, Cho HY, Naegele JR, Lombroso PJ, Obrietan K. Status epilepticus-induced somatostatinergic hilar interneuron degeneration is regulated by striatal enriched protein tyrosine phosphatase. J. Neurosci. 2007;27:2999–3009. doi: 10.1523/JNEUROSCI.4913-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung KK, Dawson TM, Dawson VL. Nitric oxide, S-nitrosylation and neurodegeneration. Cell. Mol. Biol. 2005;51:247–254. [PubMed] [Google Scholar]
- Crow JP, Calingasan NY, Chen J, Hill JL, Beal MF. Manganese porphyrin given at symptom onset markedly extends survival of ALS mice. Ann. Neurol. 2005;58:258–265. doi: 10.1002/ana.20552. [DOI] [PubMed] [Google Scholar]
- Davies S, Ramsden DB. Huntington's disease. Mol. Pathol. 2001;54:409–413. doi: 10.1136/mp.54.6.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deak M, Clifton AD, Lucocq LM, Alessi DR. Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J. 1998;17:4426–4441. doi: 10.1093/emboj/17.15.4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhakshinamoorthy S, Long DJ, 2nd, Jaiswal AK. Antioxidant regulation of genes encoding enzymes that detoxify xenobiotics and carcinogens. Curr. Top. Cell. Regul. 2000;36:201–216. doi: 10.1016/s0070-2137(01)80009-1. [DOI] [PubMed] [Google Scholar]
- Fernandez-Espejo E. Pathogenesis of Parkinson's disease: prospects of neuroprotective and restorative therapies. Mol. Neurobiol. 2004;29:15–30. doi: 10.1385/MN:29:1:15. [DOI] [PubMed] [Google Scholar]
- Forero DA, Casadesus G, Perry G, Arboleda H. Synaptic dysfunction and oxidative stress in Alzheimer's disease: emerging mechanisms. J. Cell. Mol. Med. 2006;10:796–805. doi: 10.1111/j.1582-4934.2006.tb00439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujikawa DG. The temporal evolution of neuronal damage from pilocarpine-induced status epilepticus. Brain Research. 1996;725:11–22. doi: 10.1016/0006-8993(96)00203-x. [DOI] [PubMed] [Google Scholar]
- Glover CP, Heywood DJ, Bienemann AS, Deuschle U, Kew JN, Uney JB. Adenoviral expression of CREB protects neurons from apoptotic and excitotoxic stress. Neuroreport. 2004;15:1171–1175. doi: 10.1097/00001756-200405190-00018. [DOI] [PubMed] [Google Scholar]
- Gong P, Stewart D, Hu B, Vinson C, Alam J. Multiple basic-leucine zipper proteins regulate induction of the mouse heme oxygenase-1 gene by arsenite. Arch. Biochem. Biophys. 2002;405:265–274. doi: 10.1016/s0003-9861(02)00404-6. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Oxidative stress and neurodegeneration, where are we now? J. Neurochem. 2006;97:1634–1658. doi: 10.1111/j.1471-4159.2006.03907.x. [DOI] [PubMed] [Google Scholar]
- Hara T, Hamada J, Yano S, Morioka M, Kai Y, Ushio Y. CREB is required for acquisition of ischemic tolerance in gerbil hippocampal CA1 region. J. Neurochem. 2003;86:805–814. doi: 10.1046/j.1471-4159.2003.01847.x. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Herzig S, Long F, Jhala US, Hedrick S, Quinn R, Bauer A, Rudolph D, Schutz G, Yoon C, Puigserver P, Spiegelman B, Montminy M. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature. 2001;413:179–183. doi: 10.1038/35093131. [DOI] [PubMed] [Google Scholar]
- Honchar MP, Olney JW, Sherman WR. Systemic cholinergic agents induce seizures and brain damage in lithium-treated rats. Science. 1983;220:323–325. doi: 10.1126/science.6301005. [DOI] [PubMed] [Google Scholar]
- Ishii T, Itoh K, Takahashi S, Sato H, Yanagawa T, Katoh Y, Bannai S, Yamamoto M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J. Biol. Chem. 2000;275:16023–16029. doi: 10.1074/jbc.275.21.16023. [DOI] [PubMed] [Google Scholar]
- Jarrett SG, Liang LP, Hellier JL, Staley KJ, Patel M. Mitochondrial DNA damage and impaired base excision repair during epileptogenesis. Neurobiol. Dis. 2008;30:130–138. doi: 10.1016/j.nbd.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovács R, Schuchmann S, Gabriel S, Kann O, Kardos J, Heinemann U. Free radical-mediated cell damage after experimental status epilepticus in hippocampal slice cultures. J. Neurophysiol. 2002;88:2909–2918. doi: 10.1152/jn.00149.2002. [DOI] [PubMed] [Google Scholar]
- Krönke G, Bochkov VN, Huber J, Gruber F, Blüml S, Fürnkranz A, Kadl A, Binder BR, Leitinger N. Oxidized phospholipids induce expression of human heme oxygenase-1 involving activation of cAMP-responsive element-binding protein. J. Biol. Chem. 2003;278:51006–51014. doi: 10.1074/jbc.M304103200. [DOI] [PubMed] [Google Scholar]
- Lee B, Butcher GQ, Hoyt KR, Impey S, Obrietan K. Activity-dependent neuroprotection and cAMP response element-binding protein (CREB): kinase coupling, stimulus intensity, and temporal regulation of CREB phosphorylation at serine 133. J. Neurosci. 2005;25:1137–1148. doi: 10.1523/JNEUROSCI.4288-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B, Dziema H, Lee KH, Choi YS, Obrietan K. CRE-mediated transcription and COX-2 expression in the pilocarpine model of status epilepticus. Neurobiol. Dis. 2007;25:80–91. doi: 10.1016/j.nbd.2006.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HC, Wei YH. Mitochondrial biogenesis and mitochondrial DNA maintenance of mammalian cells under oxidative stress. Int. J. Biochem. Cell Biol. 2005;37:822–834. doi: 10.1016/j.biocel.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Liang LP, Huang J, Fulton R, Day BJ, Patel M. An orally active catalytic metalloporphyrin protects against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity in vivo. J. Neurosci. 2007;27:4326–4333. doi: 10.1523/JNEUROSCI.0019-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long F, Schipani E, Asahara H, Kronenberg H, Montminy M. The CREB family of activators is required for endochondral bone development. Development. 2001;128:541–550. doi: 10.1242/dev.128.4.541. [DOI] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35:605–623. doi: 10.1016/s0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- Love S. Oxidative stress in brain ischemia. Brain Pathol. 1999;9:119–131. doi: 10.1111/j.1750-3639.1999.tb00214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabuchi T, Kitagawa K, Kuwabara K, Takasawa K, Ohtsuki T, Xia Z, Storm D, Yanagihara T, Hori M, Matsumoto M. Phosphorylation of cAMP response element-binding protein in hippocampal neurons as a protective response after exposure to glutamate in vitro and ischemia in vivo. J. Neurosci. 2001;21:9204–9213. doi: 10.1523/JNEUROSCI.21-23-09204.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantamadiotis T, Lemberger T, Bleckmann SC, Kern H, Kretz H, Martin Villalba A, Tronche F, Kellendonk C, Gau D, Kapfhammer J. Disruption of CREB function in brain leads to neurodegeneration. Nat. Genet. 2002;31:47–54. doi: 10.1038/ng882. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Cheng A. Neurohormetic phytochemicals: Low-dose toxins that induce adaptive neuronal stress responses. Trends Neurosci. 2006;29:632–639. doi: 10.1016/j.tins.2006.09.001. [DOI] [PubMed] [Google Scholar]
- Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996;274:1678–1683. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- McLin JP, Steward O. Comparison of seizure phenotype and neurodegeneration induced by systemic kainic acid in inbred, outbred, and hybrid mouse strains. Eur. J. Neurosci. 2006;24:2191–2202. doi: 10.1111/j.1460-9568.2006.05111.x. [DOI] [PubMed] [Google Scholar]
- Obrietan K, Impey S, Storm DR. cAMP response element-mediated gene expression in transgenic reporter gene mouse strain. Methods Enzymol. 2002;345:570–584. doi: 10.1016/s0076-6879(02)45048-3. [DOI] [PubMed] [Google Scholar]
- Patel M. Mitochondrial dysfunction and oxidative stress: cause and consequence of epileptic seizures. Free Radic. Biol. Med. 2004;37:1951–1962. doi: 10.1016/j.freeradbiomed.2004.08.021. [DOI] [PubMed] [Google Scholar]
- Perluigi M, Joshi G, Sultana R, Calabrese V, De Marco C, Coccia R, Cini C, Butterfield DA. In vivo protective effects of ferulic acid ethyl ester against amyloid-beta peptide 1–42-induced oxidative stress. J. Neurosci. Res. 2006;84:418–426. doi: 10.1002/jnr.20879. [DOI] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation: II. Motor seizure, Electroenceph. Clin. Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Riccio A, Ahn S, Davenport CM, Blendy JA, Ginty DD. Mediation by a CREB family transcription factor of NGF-dependent survival of sympathetic neurons. Science. 1999;286:2358–2361. doi: 10.1126/science.286.5448.2358. [DOI] [PubMed] [Google Scholar]
- Rudolph D, Tafuri A, Gass P, Hämmerling GJ, Arnold B, Schütz G. Impaired fetal T cell development and perinatal lethality in mice lacking the cAMP response element binding protein. Proc. Natl.Acad. Sci. USA. 1998;95:4481–4486. doi: 10.1073/pnas.95.8.4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayre LM, Perry G, Smith MA. Oxidative stress and neurotoxicity. Chem. Res. Toxicol. 2008;2:172–188. doi: 10.1021/tx700210j. [DOI] [PubMed] [Google Scholar]
- Sherer TB, Betarbet R, Testa CM, Seo BB, Richardson JR, Kim JH, Miller GW, Yagi T, Matsuno-Yagi A, Greenamyre JT. Mechanism of toxicity in rotenone models of Parkinson's disease. J. Neurosci. 2003;23:10756–10764. doi: 10.1523/JNEUROSCI.23-34-10756.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St. Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jäger S, Handschin C, Zheng K, Lin J, Yang W, Simon DK, Bachoo R, Spiegelman BM. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- Tejada S, Sureda A, Roca C, Gamundí A, Esteban S. Antioxidant response and oxidative damage in brain cortex after high dose of pilocarpine. Brain Res. Bull. 2007;71:372–375. doi: 10.1016/j.brainresbull.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Tretter L, Sipos I, Adam-Vizi V. Initiation of neuronal damage by complex I deficiency and oxidative stress in Parkinson's disease. Neurochem. Res. 2004;29:569–577. doi: 10.1023/b:nere.0000014827.94562.4b. [DOI] [PubMed] [Google Scholar]
- Uldry M, Yang W, St-Pierre J, Lin J, Seale P, Spiegelman BM. Complementary action of the PGC-1 coactivators in mitochondrial biogenesis and brown fat differentiation. Cell Metab. 2006;3:333–341. doi: 10.1016/j.cmet.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Votyakova TV, Reynolds IJ. Ca2+induced permeabilization promotes free radical release from rat brain mitochondria with partially inhibited complex I. J. Neurochem. 2005;93:526–537. doi: 10.1111/j.1471-4159.2005.03042.x. [DOI] [PubMed] [Google Scholar]
- Walton M, Woodgate AM, Muravlev A, Xu R, During MJ, Dragunow M. CREB phosphorylation promotes nerve cell survival. J. Neurochem. 1999;73:1836–1842. [PubMed] [Google Scholar]
- Walton MR, Dragunow I. Is CREB a key to neuronal survival? Trends Neurosci. 2000;23:48–53. doi: 10.1016/s0166-2236(99)01500-3. [DOI] [PubMed] [Google Scholar]
- Warner DS, Sheng H, Batinić-Haberle I. Oxidants, antioxidants and the ischemic brain. J. Exp. Biol. 2004;207:3221–3231. doi: 10.1242/jeb.01022. [DOI] [PubMed] [Google Scholar]
- Wild AC, Moinova HR, Mulcahy RT. Regulation of gamma-glutamylcysteine synthetase subunit gene expression by the transcription factor Nrf2. J. Biol. Chem. 1999;274:33627–33636. doi: 10.1074/jbc.274.47.33627. [DOI] [PubMed] [Google Scholar]
- Wild AC, Gipp JJ, Mulcahy T. Overlapping antioxidant response element and PMA response element sequences mediate basal and beta-naphthoflavone-induced expression of the human gamma-glutamylcysteine synthetase catalytic subunit gene. Biochem. J. 1998;332:373–381. doi: 10.1042/bj3320373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z, Dudek H, Miranti CK, Greenberg ME. Calcium influx via the NMDA receptor induces immediate early gene transcription by a MAP kinase/ERK-dependent mechanism. J. Neurosci. 1996;16:5425–5436. doi: 10.1523/JNEUROSCI.16-17-05425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Shao XG, Sun H, Li YN, Yang J, Deng YC, Huang YG. Activation of cerebral peroxisome proliferator-activated receptors gamma exerts neuroprotection by inhibiting oxidative stress following pilocarpine-induced status epilepticus. Brain Res. 2008;1200C:146–158. doi: 10.1016/j.brainres.2008.01.047. [DOI] [PubMed] [Google Scholar]
- Zou J, Crews F. CREB and NF-kappaB transcription factors regulate sensitivity to excitotoxic and oxidative stress induced neuronal cell death. Cell. Mol. Neurobiol. 2006;26:385–405. doi: 10.1007/s10571-006-9045-9. [DOI] [PMC free article] [PubMed] [Google Scholar]