

Abstract
Evidence of dysregulation of the CREB/CRE transcriptional pathway in animal models of Huntington’s disease (HD) suggests that strategies designed to augment CRE-mediated transcription may be of therapeutic value. Here, we investigated the consequences of CREB activation and repression in chemical and transgenic mouse models of HD. In the 3-nitropropionic acid (3-NP) model, CREB phospho-activation in the striatum was potently repressed within the neurotoxic “core” region prior to cell death. Conversely, marked expression of phospho-CREB, as well the CREB-regulated cytoprotective gene Bcl-2, was detected in the “penumbral” region. To examine potential contributory roles for the CREB/CRE transcriptional pathway in striatal degeneration, we used both CREB loss- (A-CREB) and gain- (VP16-CREB) of-function transgenic mouse strains. 3-NP-induced striatal lesion size and motor dysfunction were significantly increased in A-CREB mice compared to controls. Conversely, striatal damage and motor deficits were diminished VP16-CREB mice. Furthermore, transgenic A-CREB significantly accelerated motor impairment in the YAC128 mouse model of HD. Together, these results indicate that CREB functionality is lost during the early stages of striatal cell stress and that the repression of CREB-mediated transcription contributes to the pathogenic process.
INTRODUCTION
Huntington’s disease (HD) is a neurodegenerative disorder characterized by striatal and cortical atrophy, and a typical onset of motor and cognitive symptoms in midlife (reviewed in Gil and Rego, 2008). HD is an autosomal dominant genetic disease caused by a poly-glutamine repeat size greater than 35 in the huntingtin protein (reviewed in Semaka et al., 2006). Both the normal function of huntingtin and the mechanism of toxicity of poly-glutamine expanded huntingtin are yet to be definitively established. However, transcriptional dysregulation, excitotoxicity, oxidative stress, mitochondrial dysfunction, disruption of axonal transport and synaptic dysfunction have all been implicated in the HD-induced neuronal dysfunction and death (Cha, 2007; reviewed in Roze et al, 2008b). Given that many of these pathophysiological processes likely result from HD-evoked alterations in transcriptional networks, an understanding of the underlying cellular signaling state of the distressed striatum is merited.
In this regard, a particularly interesting set of observations has shown that HD pathogenesis leads to dysregulation of the CREB/cAMP response element (CRE) transcriptional pathway (Cui et al., 2006; DeMarch et al., 2007, 2008; Lee et al., 2005; Nucifora et al., 2001; Obrietan and Hoyt, 2004). CREB is a member of the basic leucine zipper family of transcription factors and plays diverse roles in an array of physiological processes (Huang et al., 2008; Krönke et al., 2003; Marie et al., 2005; Redmond et al., 2002; Shieh et al., 1998; Suzuki et al., 2007; Tao et al., 1998; Wilson et al., 1996). Within the nervous system, CREB regulates an array of neuroprotective processes, including the expression of trophic factors, antiapoptotic genes, detoxifying enzymes, and mitochondrial biogenesis (Shieh et al., 1998; Tao et al., 1998; St. Pierre et al., 2006; Wilson et al., 1996). Therefore, manipulation of the CREB/CRE transcriptional pathway represents a promising target for amelioration of HD pathology.
To begin our analysis of CREB and striatal cell death, we employed the YAC128 mouse strain and the 3-nitropropionic acid (3-NP) model of striatal neuron toxicity. YAC128 mice express a 128 CAG repeat and show a progressive motor deficit from 6 months of age (Slow et al., 2003). 3-NP is a potent, irreversible, mitochondrial toxin that triggers a marked reduction in ATP production and the rapid appearance of striatal lesions (Beal et al., 1993). The lesions are similar to those seen in HD and thus, 3-NP has been used to model HD pathology (Saulle et al., 2004) and test potential therapeutics. To investigate the role of CREB in HD pathology and to assess the potential neuroprotective effects of CREB activation, we generated mice transgenic for repressive form of CREB (A-CREB) and constitutively active form of CREB (VP16-CREB). These transgenes are highly expressed in medium spiny neurons in the striatum, a primary locus of the HD pathology. Here we provide evidence that CREB is a key regulator of neuroprotection in both the chemical and genetic model of HD.
MATERIALS AND METHODS
Animals
The generation of the TRE-A-CREB/GFP line was previously described by Lee et al., (2007). To generate mice transgenic for TRE-regulated VP16-CREB and enhanced green fluorescence protein (GFP), we generated a pTRE VP16-CREB/eGFP expression vector. To this end VP16-CREB was isolated from a pcDNA3 expression vector (provided by Dr. Soren Impey, Oregon Health Sciences University) via HindIII and Xba I digestion. The fragment was then blunted with Klenow and subcloned into SmaI-digested pIRES2-eGFP (Invitrogen). Primers (Forward: 5′ CGGCGCGGCCGCGCCACCATGGGAGCCCGGGAGATCTGGATCTGG 3′, Reverse: 5′ CGGCGCGGCCGCTTTACTTGTACAGCTCGTCCATGCC 3′ were used to amplify VP16-CREB/IRES/eGFP. A Kozak sequence (GCCACCATGG) was added upstream of start codon (bold) of VP16 CREB to improve protein expression, and a NotI digestion site (GCGGCCGC) was added to the 5′ end to allow for facile subcloning of the construct into the pTRE-Tight vector (Clontech, Mountain View, CA, USA). Next, to release the TRE promoter, VP16-CREB/IRES/eGFP and SV 40 polyA sequence, the vector was digested with XhoI. Transgenic C57BL/6 blastocytes were generated at the Ohio State University Transgenic Facility.
A bitransgenic system was used to generate tetracycline-inducible bicistronic A-CREB/eGFP and VP16-CREB/eGFP mice. To drive transgene expression, the A-CREB/eGFP and VP16-CREB/eGFP mice were crossed with CaMKIIα promoter-tTA transgenic mice (Mayford et al., 1996). To inhibit transgene expression, doxycycline (100 mg/L) was added to the drinking water of VP16-CREB mice and WT littermates from 2 weeks of age until 1 week before 3-NP injection. YAC128 mice were acquired from Jackson Lab. To generate tritransgenic mice for YAC128:: tTA::A-CREB, male mice transgenic for YAC128 were crossed with females transgenic for tTA::A-CREB. The genotypes were determined using PCR. All experiments involving animals were approved by the Ohio State University Animal Care and Use Committee.
3-NP intoxication
3-nitropropionic acid (3-NP, Sigma, St. Louis, MO) was dissolved in saline (25 mg/mL) and passed through a 0.2-μm filter. We followed the 3-NP injection protocol described by Huang et al. (2006). Briefly, 3-NP was injected intraperitoneally twice daily for 2 days at 12 h intervals (8:00 A.M. and 8:00 P.M.) at a dose of 60 mg/kg for the first two injections and 80 mg/kg for the second two injections. Animals were maintained on a standard 12 hr light/dark cycle, with lights on at 6:00 A.M.
We also verified that 3-NP-induced succinate dehydrogenase (SDH) inhibition was not altered by expression of the A-CREB or VP-16 CREB transgenes. SDH activity was measured by enzyme histochemistry of frozen coronal brain sections (12 μm-thick) from A-CREB::tTA, VP-16::tTA bitransgenic and tTA monotransgenic mice sacrificed 1 hr after injection of 80 mg/kg 3-NP or saline vehicle. Brain sections were incubated at 37°C for 2 hours in 0.55 mM nitroblue tetrazolium, 0.5 mM succinate as described (Higgins et al., 1999; Tabrizi et al., 2000). Importantly, we found that there was no staining in the absence of succinate substrate or in the presence of 1 mM 3NP added directly to the incubation solution. The optical density of the stained sections was quantified using Metamorph software (Universal Imaging, Downingtown, PA). Striatal optical density in sections from 3-NP injected mice was reduced to 26% of saline-injected mice, indicating that the 3-NP injection paradigm potently inhibited brain SDH activity. The ratio of cortical and striatal SDH activity (optical density) was compared among 3-NP treated tTA monotransgenic, VP-16 CREB::tTA, and A-CREB::tTA bitransgenic mice. Since CREB transgene expression is concentrated in the striatum (Fig. 4A and 7A), an effect of the transgenes on the 3-NP-evoked inhibition of SDH activity should be reflected in the SDH cortex/striatum activity ratio, relative to the ratio in tTA monotransgenic mice. The ratio (cortex/striatum) of SDH activity for tTA monotransgenic mice was 1.65 ± 0.10, VP-16 CREB::tTA mice was 1.63 ± 0.10, and A-CREB::tTA mice was 1.74 ± 0.17 (mean ± sem, n = 3 mice per genotype; n.s. by ANOVA). Since there were no differences in the cortex/striatum ratio among the mouse lines, we conclude that neither transgenic repression nor activation modulates the capacity of 3-NP to affect SDH activity.
Figure 4. CREB repression increases 3-NP-evoked cell death.

A–B. Striatal expression of the A-CREB/GFP transgenic construct. Low magnification image reveals marked GFP expression within the striatum (A), and double labeling for GFP and NeuN (B) was used to confirm that the transgene is specifically expressed in neurons. Scale bars: 400 μm for A and 20 μm for B. C. Cresyl violet image shows increased striatal damage in tTA::A-CREB mice compared to A-CREB mice lacking the tTA driver. Data were collected 1 day after last 3-NP injection. Scale bar: 400 μm. D. Quantitative analysis of striatal damage in tTA::A-CREB and A-CREB monotransgenic mice. Numbers in the bars refers to the number of animals analyzed. *, p < 0.05.
Figure 7. VP16-CREB confers neuroprotection and improves motor performance.

A. Striatal expression of the VP16-CREB/GFP transgenic construct. Immunohistochemical labeling for GFP show marked transgene expression in the striatum. Scale bar: 400 μm. B. Top: Time course depicting the 3-NP injection and sacrifice schedule. Bottom: Representative cresyl violet images reveal a decrease in the degree of 3-NP-induced striatal damage in tTA::VP16-CREB mice compared to WT mice. Scale bars: 400 μm and 100 μm for the low and high magnification images, respectively. C. Quantitative analysis shows that 3-NP evoked striatal damage was decreased in tTA::VP16-CREB mice compared to VP16-CREB lacking the tTA driver. D. Double-transgenic mice exhibited significantly increased rotarod fall latency compared to monotrasgenic VP16-CREB littermates. *, p < 0.05; ***, p < 0.001.
Tissue processing
Mice were anesthetized with a ketamine (150 mg/kg)/xylazine (30 mg/kg) cocktail and transcardially perfused with saline, followed by 4% paraformaldehyde in 0.01 M phosphate buffered saline (PBS), pH 7.4. Brains were postfixed for 4 hrs and then cryoprotected in 30% sucrose in 0.01 M PBS. Sequential coronal sections (40 μm) through the striatum were prepared using a cryotome. For the measurement of striatal damage, sections were processed for cresyl violet using the labeling method described in Choi et al. (2007).
Immunohistochemistry
For immunohistochemical labeling, sections (40 μm-thick) were blocked with 10% normal goat serum or 5% normal horse serum in PBS, followed by overnight incubation (4°C) with rabbit polyclonal anti-phospho-CREB antibody (pCREB, 1:1000; Cell Signaling Technology, Beverly, MA), rabbit polyclonal anti-GFP antibody (1:4000; Acquired from Dr. Luc G. Berthiaume, University of Alberta, Canada), goat anti-Bcl-2 antibody (1:500; Santa Cruz Biotechnology, Santa Cruz, CA) or a rabbit polyclonal antibody against the N-terminal fragment of α-spectrin (1:4,000, Higuchi et al., 2005). Sections were then processed using the ABC staining method (Vector Laboratories, Burlingame, CA) and nickel-intensified diaminobenzidine (DAB; Vector Laboratories) was used to visualize the signal. Photomicrographs were captured as described above.
For immunofluorescence labeling, sections were blocked with 10% normal goat serum or 5% normal horse serum in PBS, followed by overnight incubation with mouse anti-NeuN antibody (1:2000; Millipore, Billerica, MA), rabbit anti-GFP antibody (1:2000), goat anti-Bcl-2 antibody (1:500; Santa Cruz Biotechnology) or rabbit anti-pCREB antibody (1:1000; Cell Signaling Technology). Next, sections were incubated (2 h at room temperature) with secondary antibodies conjugated with Alexa 488 and/or Alexa 594 (1:1000; Invitrogen, San Diego, CA) and then mounted with Cytoseal (Richard-Allan Scientific, Kalamazoo, MI). Fluorescence images were captured using a Zeiss (Oberkochen, Germany) 510 Meta confocal microscope (2-μm-thick optical section).
Fluoro-Jade B staining
Sections (40 μm) were initially mounted on gelatin-coated slides, rehydrated and incubated in 0.06% potassium permanganate solution for 10 min. Next, the sections were incubated in 0.004% Fluoro-Jade B (Millipore, Bedford, MA) solution containing 0.1% glacial acetic acid for 20 min at room temperature, washed, and coverslipped with distrene plasticizer xylene (DPX; Electron Microscopy Sciences, Fort Washington, PA).
Measurement of striatal damage and pCREB expression
The striatal boarders were defined laterally and dorsally by a corpus callosum, medially by the lateral ventricle and midline, and ventrally, by a horizontal plane through the anterior commisure. These and other neuroanaotomical landmarks (as described by Fernagut et al., 2002a and Fernagut et al., 2002b) were used to ensure that consistency was maintained across all sets of animals for our analysis of 3-NP-induced striatal lesions. Analysis was performed through the rostral (bregma: +1.50 mm) caudal (bregma: −0.50 mm) extent of the striatum. The volume of 3-NP-induced striatal damage was measured in every 5th striatal section (200 μm intervals). Photomicrographs (4X) of cresyl violet-labeled sections were captured using a 16-bit digital camera (Micromax YHS 1300; Princeton Instruments, Trenton, NJ) mounted on a Leica DM IRB microscope. For each section, the total striatal area and the damaged area (which was characterized by the extensive loss of cresyl violet signaling) were digitally outlined and area measurements were calculated using MetaMorph software. Volumes were calculated by summing the cross-sectional areas in each section and the percent of striatal damage in each animal was calculated by dividing damaged volume by total striatal volume. Data are presented as the mean ± SEM, and significance was assessed using a two-tailed Student’s t-test.
To examine activated CREB expression following 3-NP injection, mid-striatal (bregma +0.9 to −0.2 mm) coronal sections were immunolabeled and the pattern of pCREB expression was examined. Labeling revealed a loss of pCREB within the ostensible lesion ‘core’. Both the neuroanatomical location and the size of the region demarcated by the loss of pCREB paralleled the 3-NP-evoked lesion pattern revealed using Cresyl violet and Fluoro Jade-B labeling. Surrounding this was an ~ 300 μm zone where relatively high pCREB levels were observed: this region is referred to as the ‘penubra’. Lastly, we denoted a ‘medial’ striatal region adjacent to the penumbra, which did not exhibit marked signs of cell stress. To quantitate pCREB levels, densitometric analysis was performed within these three regions; for the core, intensity was measured within a 200 μm digital circle placed in the center of the core; penumbra pCREB levels were determined by measuring the intensity within concentric rings that defined the inner and outer boarder (300 μm) of the penumbra. Measurements within the medial striatal region were collected within a 200 μm digital circle, which was positioned next to the concentric rings. Values were corrected against background immunolabeling within the corpus callosum, and normalized to a value of 1 for the core region. Analysis was performed using MetaMorph software.
Rotarod assessments
Mice were trained on an accelerating rotarod (Columbus Instruments) with 3 trials per day for 3 consecutive days. Only mice which attained steady baseline of performance (300 seconds for the fall latency on the 3rd day) were used. The testing was executed from 1 day before to 3 or 4 days after the first 3-NP injection. Mice were tested three times a day with a 1 h interval between each test. YAC128::tTA::A-CREB animals were trained with 3 trials per day for 3 consecutive days (2–20 rpm) and rotarod performance was tested 3 times with 1 hr interval (4–40 rpm). The longest latency to fall off the rotarod for the three daily trials was used for the analysis. Data are presented as the mean ± SEM, and significance was assessed using a two-tailed Student’s t-test.
Primary neuronal culture/LDH release assay
Striatal tissue was dissected from Sprague–Dawley rat pups (E18–E19) and enzymatically digested into a single-cell suspension using the methods and reagents described by Lee et al. (2005). Cell were seeded at a density of 2.2 × 105/cm2 onto poly-d-lysine-(> 540 kDa; Sigma) coated 24-well plates and maintained as described in Lee et al. (2005). The LDH release assay was performed as described by Koh and Choi (1987). The value for LDH release from the vehicle-treatment condition was normalized to a value of 1. Data are presented as the mean ± SEM, and significance was assessed using a two-tailed Student’s t-test.
Western blotting
Cultured neurons were rinsed with ice-cold PBS and then lysed in ice-cold RIPA buffer (150 mM NaCl, 50 mM Tris pH 7.4, 1 mM EDTA, 1% Triton X-100, 0.1% SDS, 5 mM NaF, 25 μM sodium vanadate supplemented with protease inhibitor cocktail: complete mini tablet, Roche Diagnostics). Protein content was determined via a Bradford assay (Bio-Rad, Hercules, CA). Twenty μg of protein were loaded onto a 10% SDS-PAGE gel, electrophoresed using standard procedures, and the protein was transblotted onto polyvinylidene fluoride (Immobilon P; Millipore). Following blocking with 10% (wt/vol) powdered milk, membranes were incubated (4°C overnight) with rabbit anti-pCREB antibody (1:2000, Cell Signaling Technology). Samples were then incubated with an HRP-conjugated anti-rabbit IgG secondary antibody (1:2500, Perkin-Elmer, Wellesley, MA) and the signal was detected using the Renaissance Chemiluminescent Detection System (New England Nuclear). As a protein loading control, membranes were also probed with a goat polyclonal antibody against total erk-1 and erk-2 (1:2000, Santa Cruz Biotechnology). Membranes were then incubated with an alkaline phosphatase-conjugated secondary antibody and the signal was detected using the Western-CDP Star Chemiluminescent Detection System (Perkin-Elmer).
RESULTS
To induce striatal lesions, mice received a 2-day 3-NP injection paradigm: on the first day, mice were injected twice with 60 mg/kg of 3-NP over a 12 hr interval, and with 80 mg/kg at a 12 h interval on the second day (Huang et al., 2006). With this regimen, cresyl violet labeling revealed a distinct lesion ‘core’ within the lateral striatum 1 day following the final 3-NP injection (Fig. 1A). Maximal cell loss was observed within the mid-striatum (bregma +0.9 to 0.2 mm), with moderate cell loss detected within rostral and caudal regions of the striatum. The pattern of striatal cell loss is consistent with other work examining the effects of 3-NP using the C57/Bl6 line of mice (Fernagut et al., 2002a).
Figure 1. 3-NP intoxication regulates striatal CREB phosphorylation.
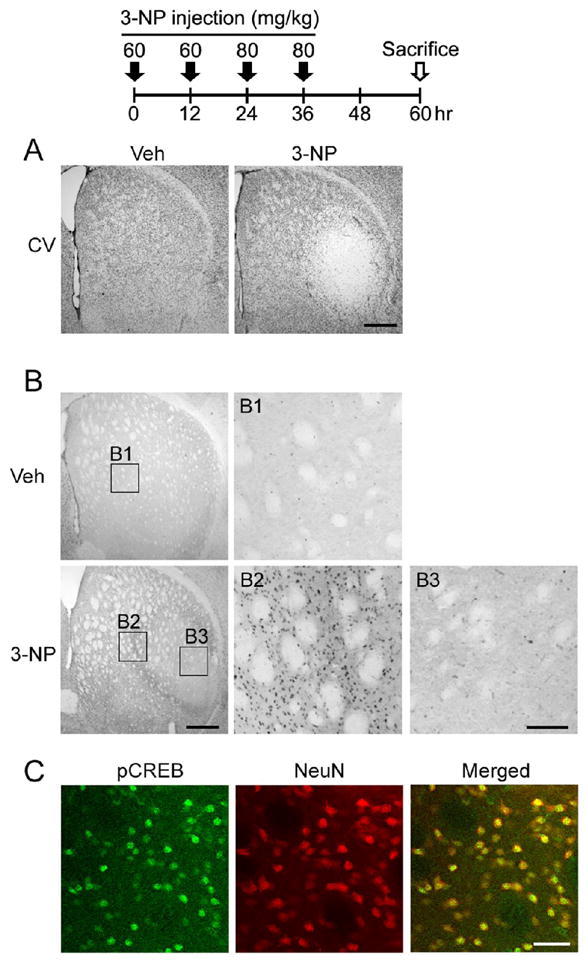
Top: time course depicting the 3-NP injection and sacrifice schedule for experiments presented in A–C. A. Cresyl violet (CV) histology reveals marked 3-NP-induced cell damage within the lateral striatum. Tissue was collected from mice sacrificed 1 day after the last 3-NP injection. Uniform CV labeling was detected throughout the striatum of vehicle-infused tissue. Scale bar = 400 μm. B. Immunoreactivity for the Ser-133 phosphorylated form of CREB (pCREB). Top) Representative section from a vehicle-injected animal: note the modest level of pCREB expression. Boxed region (B1) is magnified to the right. Bottom). Marked pCREB expression was detected within the penumbral region surrounding the foci of 3-NP-evoked striatal damaged (magnified in B2). Limited pCREB expression was observed within the 3-NP ‘core’ (magnified in B3). Scale bars: 400 μm in the low magnification image and 100 μm in the enlarged image. C. Representative immunofluorescence image within the penumbral region shows that pCREB co-labels with NeuN, indicating CREB is activated in striatal neurons. Scale bar = 50 μm.
To investigate the role of the CREB/CRE transcriptional pathway in striatal vulnerability to 3-NP, we first examined the expression pattern of the Ser-133 phosphorylated form of CREB (pCREB). Using the 3-NP paradigm described above, and killing mice 1 day after the last injection, we found that pCREB levels were markedly elevated in a ‘penumbral’ region surrounding the ‘core’ area of 3-NP-induced cell damage (Fig. 1B). Other groups have used similar terminology to describe a 3-NP evoked lesion ‘core’ where profound cell death occurs, and a ‘penumbral’ region, where gliosis and limited neuronal survival is observed (Ryu et al., 2003). Double-immunofluorescence labeling for pCREB and NeuN showed that the activated form of CREB was specific to neurons in the penumbral region (Fig. 1C). Expression was significantly elevated relative to the damaged ‘core’ region as well as more medial striatal regions, which did not exhibit marked signs of cell stress (Fig. 2).
Figure 2. 3-NP-induced pCREB in the striatum.

Top: time course depicting the 3-NP injection and sacrifice schedule A. Concentric rings are used to depict the 3-NP-induced pattern of CREB phosphorylation within the core (C), penumbral (P) and medial (M) striatal regions. Scale bar: 400 μm. B. Quantitative densitometric analysis of pCREB expression within the 3 regions. pCREB levels were significantly higher in the penumbra than in the either the core or medial regions. Animals were sacrificed 24 hrs after the last 3-NP injection. Data were acquired from 7 animals. p values were generated using the two-tailed Student’s t-test.
While there is extensive striatal damage 24h following the last 3-NP injection (Fig. 1A and Fig. 3A1), when we examined the striatum at 12 h after the final 3-NP injection, neuronal death measured by Fluoro-jade B (FJB) staining was not detected (Fig. 3A). Given that FJB is a highly sensitive marker of dead and dying cells (Fig. 3A1), the absence of a FJB signal at the 12 hr time point indicates that striatal tissue was still viable. This analysis was complemented by immunolabeling for calpain enzymatic activity. Calpain activity was of interest because it is a marker of cell stress and has been shown to participate in both apoptotic (Mattson, 2000; Rami, 2003) and nonapoptotic (Mattson 2000; Pang et al., 2003) neuronal death. To this end, tissue collected from both the 12- and 24-hr post 3-NP injection time points were immunolabled with an antibody that specifically detects a 136-kDa fragment of α-spectrin (fodrin), which is cleaved by activated calpain (Higuchi et al., 2005). Interestingly, at the 12 hr time point modest calpain activity was detected within the core region, whereas at 24 hrs a marked pattern of calpain activity was detected, and individual immunoreactive cells could be clearly identified (Fig. 3C). These data support the FJB labeling data, indicating that 3-NP evokes a progressive deterioration of striatal cells which culminates in cell death by 24 hrs following the last 3-NP injection.
Figure 3. CREB activity is repressed prior to 3-NP-induced neuronal damage.

Top: time course depicting the 3-NP injection and sacrifice schedule for experiments presented in A and B. A. Fluoro-Jade B (FJB) histology did not detect striatal neuronal damage 12 hrs after the last 3-NP injection. Scale bar: 400 μm. A1 Inset: Representative FJB labeling from a mouse sacrificed 24 hrs after the last 3-NP injection. Note the marked cell death (denoted by bright green fluorescence) within the dorso-lateral striatum. B. pCREB immunolabeling of the adjacent section to the FJB-labeled section in A revealed that CREB activity was elevated within the penumbral and repressed within the core region. Scale bars: 400 μm in the low magnification image and 100 μm in the enlarged image. C. Representative calpain activity anaysis under control conditions (C1) and 12-hrs (C2) and 24-hrs (C3) post-NP injection. Boxed regions are shown to the right. Scale bar: 400 μm. D–E. In cultured striatal neurons, LDH release was used to assay 3-NP-evoked cell death. Following 2 days of incubation, 10 mM 3-NP led to a significant increase in cell death relative to the control treatment condition (0 3-NP); after 3 days of incubation, both 5 and 10 mM 3-NP elicited significant cell death relative to the control treatment condition. *, p < 0.05; **, p < 0.01. F. After 2 days of incubation with 3-NP, cultured striatal cells were analyzed for pCREB via Western blotting. Administration of 1 mM 3-NP led to a marked increase in pCREB, however, treatment with 5 and 10 mM 3-NP did not increase pCREB levels. Of note, 5 mM 3-NP did not evoke significant cell death following 2 days of treatment, thus indicating that the relative reduction in pCREB preceded cell death. To probe for total protein levels, the membrane was stripped and labeled for total erk 1 and erk 2 (Total ERK). Data are representative of triplicate determinations.
Interestingly, at 12 hr post 3-NP injection time point, pCREB expression was repressed in the presumptive core region, relative to the rest of the striatum (Fig. 3B and 3B1). Furthermore, an increase in pCREB was observed within a penumbra-like region at this time point. This pCREB expression pattern is similar to what is observed at the 24 h time point, indicating that pCREB repression preceded cell death. To directly test the idea that 3-NP can trigger CREB repression prior to neuronal death, we cultured striatal neurons from embryonic day 18 rats and monitored time- and dose-dependent 3-NP-induced cell death using a lactate dehydrogenase (LDH) release assay. Incubation with 10 mM 3-NP for 2 and 3 days triggered significant cell death, while incubation with 5 mM 3-NP triggered significant cell death only following 3 days of incubation; 1 mM 3-NP did not evoke cell death at either the time point (Fig. 3C and 3D). Using these data as a starting point, we examined pCREB following 2 days of incubation with 3-NP. Interestingly, pCREB was increased by 1 mM 3-NP exposure, but repressed by incubation of 5 or 10 mM 3-NP (Fig. 3E). Since 5 mM 3-NP was not toxic at this 2-day time point, but ultimately led to cell death a day later, these data support the idea that active CREB repression precedes cell death. Thus, these in vitro and in vivo findings indicate that 3-NP intoxication disrupts CREB phosphorylation, and, in turn, raises the possibility that a subsequent decrease in CREB-mediated transcription contributes to the cell death process.
To test for a potential causative connection between CREB dysregulation and 3-NP-evoked toxicity, we recently generated a transgenic mouse line (Lee et al.,2007) that expresses a tetracycline-regulated dominant-negative form of CREB (A-CREB; Ahn et al., 1998), as well as the transgene marker protein GFP. Crossing the A-CREB mice with a tetracycline-regulated aCaMKII-tTA activator line (tTA::A-CREB: Mayford et al., 1996) revealed marked transgene expression in the striatum (Fig. 4A). Importantly, only NeuN-positive cells expressed the transgene (~ 64%: Lee et al., 2009), indicating that A-CREB expression was neuron-specific (Fig. 4B). Further, we recently reported that transgenic A-CREB represses CRE-mediated transcription and that tonic transgene expression does not alter striatal neuron viability (Lee et al., 2007; 2009). Using cresyl violet staining we observed a much higher degree of tissue damage in tTA::A-CREB mice (~ 3-fold) compared to A-CREB mice that lacked the driver strain (Fig. 4C and 4D). Tissue was processed 24 hrs after the last 3-NP injection. The damage extended in all directions from the lesion locus within the dorsolateral striatum. Next, as an assessment of the effects of A-CREB on neuroprotective gene expression, we analyzed expression of the putative CREB target gene Bcl-2 (Krönke et al., 2003; Wilson et al., 1996) 1 day after the last 3-NP injection. In wild type mice, we noted a marked increase in Bcl-2 expression within the penumbral region (Fig. 5A). Interestingly, in the penumbral region of tTA::A-CREB double transgenic mice, Bcl-2 expression was largely absent from GFP-positive cells (Fig. 5B), indicating that A-CREB represses Bcl-2 expression.
Figure 5. Neuroprotective gene expression within the penumbral region.

Top: time course depicting the 3-NP injection and sacrifice schedule for experiments presented in A and B. A. Relative to vehicle-injected mice, 3-NP intoxication led to a marked increase in Bcl-2 expression within the penumbral region. Dashed line approximates the core/penumbral boarder. Boxed region is shown to the right. Scale bar: 50 μm. B. Representative double immunofluorescence labeling within the penumbra for Bcl-2 and for the A-CREB transgene marker GFP. Of note, relative to Bcl-2 expression in the monotransgenic A-CREB mouse, modest Bcl-2 expression was observed in the tTA::A-CREB mouse. Further, merging the GFP and Bcl-2 images reveals a lack of overlapping signals, suggesting that A-CREB represses Bcl-2 expression. Scale bar = 20 μm.
Next, we assessed motor function in tTA::A-CREB bitransgenic mice and in A-CREB monotransgenic mice. For this study, latency to fall from an accelerating rotarod (2–20 rpm over 300 sec) was examined from 1 day before to 4 days after the first 3-NP injection. As shown in Figure 6A, rotarod performance of 3-NP-treated tTA::A-CREB mice was significantly compromised relative to 3-NP treated A-CREB mice. Of note, the latency of tTA::A-CREB to fall on day 2 of the 3NP injection was markedly shorter relative to A-CREB monotransgenic mice, and interestingly, over the subsequent 2-day period tTA::A-CREB did not show significant recovery in rotarod performance. This contrasts with A-CREB monotransgenic mice, where, on day 4, motor performance had returned to near maximal levels. Together, these data reveal a profound enhancement of 3-NP-induced toxicity and a concordant decrease in motor performance in mice with repressed CREB function.
Figure 6. A-CREB enhances motor deficits.

A. Rotarod analysis of 3-NP-evoked fall latency (in seconds) in tTA::A-CREB and A-CREB monotransgenic mice. tTA::A-CREB mice exhibited a significantly decreased fall latency compared to A-CREB monotransgenic mice. *, p < 0.05; **, p < 0.01. B. Mouse tail biopsy results for tTA (top), A-CREB/GFP (middle) and YAC128 (bottom) were used to determine the genotypes of three representative mice. Of note, mouse #3 expresses all three transgenes. PCR reactions were run in a 1% agarose gel and visualized using ethidium bromide. C. Rotarod analysis of WT, YAC128 and YAC128:: tTA::A-CREB transgenic mice. (Left) There was no significant difference in fall latency between wild type and YAC128 alone at 2 months of age. However, the YAC128::tTA::A-CREB line showed significantly impaired rotarod performance compared to WT mice. (Right) At 6 months of age, a statistically significant decrease in motor performance was observed in the YAC128 line, relative to WT mice. In addition, the YAC128 phenotype was enhanced by disruption of CREB. Eleven animals were analyzed for the WT line: 11 for the YAC128 line, and 12 for the YAC128:: tTA::A-CREB line. *: p < 0.05.
We next asked whether impaired CREB function affects disease progression in a genetic mouse model of HD. For this study, we used the YAC128 transgenic mouse line (Slow et al., 2003). To generate tri-transgenic mice for YAC128::tTA::A-CREB, we crossed the YAC128 line with tTA::A-CREB double-transgenic mice (representative PCR result are shown in Fig. 6B). Rotarod performance was monitored from 2 months of age in WT nontransgenic littermates, YAC128 transgenic mice and tri-transgenic mice (YAC128::tTA::A-CREB). At 2 months of age there was no significant difference in rotarod performance between control and YAC128 mice (Fig. 6C). However, there was a significant decrease in rotarod performance in tri-transgenic mice compared to control mice at this age. Moving to 6 months of age, the YAC128 mice exhibited an accelerated rate of falling relative to WT littermates. Importantly, the tritransgenic mice exhibited a significantly enhanced impairment relative to YAC128 mice. These results suggest that CREB function is also critical for the maintenance of motor performance in genetic model of HD animals.
The 3-NP data presented above showing that CREB functional repression precedes cell death, and that transgenic disruption of CREB augments cellular toxicity raise the interesting possibility that therapeutics designed to upregulate CREB function will decrease striatal neurotoxicity. To explore this possibility, we generated mice transgenic for VP16-CREB, a constitutively active form of CREB that binds to the CRE promoter and drives tonic CRE-mediated gene expression (Barco et al., 2002). This construct is analogous to the A-CREB construct, in that it expresses a GFP marker, and, when crossed to the αCaMKII-tTA activator line, drives transgene expression in the striatum (Fig. 7A). With this CREB activator line, we measured the relative extent of 3-NP-induced striatal damage. For this experiment, transgene expression was repressed via the administration of doxycycline (100 μg/mL) from birth to 7 days prior to 3-NP injection. This measure was taken since previous work has shown that chronic expression of VP16-CREB leads to cell death and bouts of epileptiform activity (Lopez et al., 2007). VP16-CREB dramatically attenuated 3-NP striatal toxicity, as assessed via cresyl violet staining. Thus, at 3 days after the last 3-NP injection, significant cell damage was not detected in 67% of the tTA::VP16-CREB mice (n=9), whereas 100% of WT littermates exhibited striatal damage (n=5: Fig. 7B and 7C). To assess motor function, we examined rotarod performance from 1 day before to 3 days after the first 3-NP injection. For these experiments a more aggressive accelerating rotarod training paradigm (4–40 rpm over 300 sec) than the one used to test for A-CREB functionality (2–20 rpm) was used, since the motor deficit recovered in a few days after termination of 3-NP injection at the slower acceleration setting (See Fig. 6A). Motor performance in tTA::VP16-CREB mice was significantly better (i.e. longer latency to fall) than WT littermates (Fig. 7D). These data reveal that upregulation of CREB-mediated transcription provides both neuroprotection and improved motor outcome against striatal neurotoxicity.
DISCUSSION
Here we tested the potential role that dysregulation of the CREB/CRE transcriptional pathway plays in the progression of striatal neurodegeneration. To this end, we have focused on the temporal and spatial regulation of CREB activation following cytotoxic striatal stress, and utilized CREB loss- and gain-of-function transgenic mouse models. The findings reported here indicate that 1) a loss of CREB function preceded cell death, 2) abrogation of CREB-signaling enhances cytotoxicity, and 3) augmentation of CREB-mediated transcription confers protection against striatal neuronal death.
The rationale for focusing on the CREB/CRE pathway as a route by which to offset HD pathogenesis is grounded in an extensive literature showing that CREB plays a critical role in synapse formation, synaptic transmission, dendritic morphology and neuroprotective gene expression (Huang et al., 2008; Marie et al., 2005; Krönke et al., 2003; Redmond et al., 2002; Shieh et al., 1998; Suzuki et al., 2007;Tao et al.,1998; Wilson et al., 1996). Importantly, these physiological processes and dysregulation of CREB signaling are implicated in HD pathogenesis (DeMarch et al., 2008; Cui et al., 2006; Gines et al., 2003; Jiang et al., 2006; Nucifora et al., 2001; Obrietan and Hoyt 2004; Roze et al., 2008a; Sugars et al., 2004). Further, genetic deletion of CREB triggers an HD-like striatal pathology phenotype (Mantamadiotis et al., 2002), whereas the administration of rolipram, which elevates intracellular levels of CREB phosphorylation, slows the HD-like pathology in the R6/2 transgenic mouse strain (DeMarch et a., 2008).
Initially, we noted a distinctive pattern of CREB activation in response to 3-NP. pCREB was detected in the penumbral area both before and during active 3-NP-induced striatal neuronal loss, while CREB phosphorylation was repressed in the core region. Core CREB repression appeared to occur prior to cell death, thus suggesting an active mechanism of CREB repression. These findings are consistent with work using ischemia models where the activated form of CREB, as well as CREB-mediated gene transcription, is repressed in the infarct core, but increased in the peri-infarct area (Irving et al., 2000; Sugiura et al., 2004). Likewise, our work showing that Bcl-2 is upregulated in the penumbra, is paralleled by studies showing that ischemia elevates the BDNF (a putative CREB-regulated gene) expression outside of the ischemic core (Kokaia et al., 1995). Thus, an active program of CREB-dependent transcription may be a key component of a neuroprotective penumbral ‘barrier’ that limits the extent of neuronal cell damage. This supposition is supported here by work showing that the repression of CREB signaling dramatically extends the degree of 3-NP evoked striatal death. Further, apparently conflicting studies (Nucifora et al., 2001; Obrietan and Hoyt. 2004) regarding the regulation of CREB during HD pathogenesis may be resolved by the observations reported here. Hence, mild striatal pathology (i.e., within the 3-NP-induced penumbra) triggers neuroprotective CREB-dependent transcription, whereas more progressive striatal degeneration (i.e., within the 3-NP-induced core) leads to an active program of CREB repression, which, in turn facilitates the neuronal pathogenesis.
As a complement to the 3-NP studies, we reported impaired rotarod performance as early as 2 months of age in tri-transgenic mice (YAC128::tTA::A-CREB), compared to WT mice and at 6 months the tri-transgenic mice were significantly more impaired than YAC128 mice. These data are highly suggestive of an important role of CRE-dependent transcription in protection against the toxic effects of mutant huntingtin in vivo. Further, given the potent neuroprotective effect of VP16-CREB on 3-NP-induced striatal lesions, it is reasonable to postulate that transgenic upregulation of CREB-mediated gene expression would also confer a marked neuroprotective effect against mutant huntingtin. Future studies will examine gene expression patterns and striatal pathology in YAC128 mice that have either activated or repressed CREB.
In conclusion, the observation that the upregulation of CREB-dependent transcription reduces striatal cell death and facilitates behavioral recovery provides a strong rationale for the development of therapeutic approaches designed to affect CREB-dependent transcription.
Acknowledgments
This work was supported by the National Institutes of Health (Grant number: NS47176) and a Post-Doctoral Fellowship for Y-S. C. from the Hereditary Disease Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn S, Olive M, Aggarwal S, Krylov D, Ginty DD, Vinson C. A dominant-negative inhibitor of CREB reveals that it is a general mediator of stimulus-dependent transcription of c-fos. Mol Cell Biol. 1998;18:967–977. doi: 10.1128/mcb.18.2.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barco A, Alarcon JM, Kandel ER. Expression of constitutively active CREB protein facilitates the late phase of long-term potentiation by enhancing synaptic capture. Cell. 2002;108:689–703. doi: 10.1016/s0092-8674(02)00657-8. [DOI] [PubMed] [Google Scholar]
- Beal MF, Brouillet E, Jenkins BG, Ferrante RJ, Kowall NW, Miller JM, Storey E, Srivastava R, Rosen BR, Hyman BT. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci. 1993;13:4181–4192. doi: 10.1523/JNEUROSCI.13-10-04181.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha JH. Transcriptional signatures in Huntington’s disease. Prog Neurobiol. 2007;83:228–248. doi: 10.1016/j.pneurobio.2007.03.004. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YS, Lin SL, Lee B, Kurup P, Cho HY, Naegele JR, Lombroso PJ, Obrietan K. Status epilepticus-induced somatostatinergic hilar interneuron degeneration is regulated by striatal enriched protein tyrosine phosphatase. J Neurosci. 2007;27:2999–3009. doi: 10.1523/JNEUROSCI.4913-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59–69. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- DeMarch Z, Giampà C, Patassini S, Bernardi G, Fusco FR. Beneficial effects of rolipram in the R6/2 mouse model of Huntington’s disease. Neurobiol Dis. 2008;30:375–387. doi: 10.1016/j.nbd.2008.02.010. [DOI] [PubMed] [Google Scholar]
- DeMarch Z, Giampà C, Patassini S, Martorana A, Bernardi G, Fusco FR. Beneficial effects of rolipram in a quinolinic acid model of striatal excitotoxicity. Neurobiol Dis. 2007;25:266–273. doi: 10.1016/j.nbd.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Fernagut PO, Diguet E, Stefanova N, Biran M, Wenning GK, Canioni P, Bioulac B, Tison F. Subacute systemic 3-nitropropionic acid intoxication induces a distinct motor disorder in adult C57Bl/6 mice: behavioural and histopathological characterisation. Neurosci. 2002a;114:1005–1017. doi: 10.1016/s0306-4522(02)00205-1. [DOI] [PubMed] [Google Scholar]
- Fernagut PO, Diguet E, Jaber M, Bioulac B, Tison F. Dopamine transporter knock-out mice are hypersensitive to 3-nitropropionic acid-induced striatal damage. Eur J Neurosci. 2002b;15:2053–2056. doi: 10.1046/j.1460-9568.2002.02047.x. [DOI] [PubMed] [Google Scholar]
- Gil JM, Rego AC. Mechanisms of neurodegeneration in Huntington’s disease. Eur J Neurosci. 2008;27:2803–2820. doi: 10.1111/j.1460-9568.2008.06310.x. [DOI] [PubMed] [Google Scholar]
- Gines S, Seong IS, Fossale E, Ivanova E, Trettel F, Gusella JF, Wheeler VC, Persichetti F, MacDonald ME. Specific progressive cAMP reduction implicates energy deficit in presymptomatic Huntington’s disease knock-in mice. Hum Mol Genet. 2003;12:497–508. doi: 10.1093/hmg/ddg046. [DOI] [PubMed] [Google Scholar]
- Higgins DS, Hoyt KR, Baic C, Vensel J, Sulka M. Metabolic and glutamatergic disturbances in the Huntington’s disease transgenic mouse. Ann NY Acad Sci. 1999;893:298–300. doi: 10.1111/j.1749-6632.1999.tb07841.x. [DOI] [PubMed] [Google Scholar]
- Higuchi M, Tomioka M, Takano J, Shirotani K, Iwata N, Masumoto H, Maki M, Itohara S, Saido TC. Distinct mechanistic roles of calpain and caspase activation in neurodegeneration as revealed in mice overexpressing their specific inhibitors. J Biol Chem. 2005;280:15229–15237. doi: 10.1074/jbc.M500939200. [DOI] [PubMed] [Google Scholar]
- Huang QY, Wei C, Yu L, Coelho JE, Shen HY, Kalda A, Linden J, Chen JF. Adenosine A2A receptors in bone marrow-derived cells but not in forebrain neurons are important contributors to 3-nitropropionic acid-induced striatal damage as revealed by cell-type-selective inactivation. J Neurosci. 2006;26:11371–11378. doi: 10.1523/JNEUROSCI.1907-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YH, Lin Y, Brown TE, Han MH, Saal DB, Neve RL, Zukin RS, Sorg BA, Nestler EJ, Malenka RC, Dong Y. CREB modulates the functional output of nucleus accumbens neurons: a critical role of N-methyl-D-aspartate glutamate receptor (NMDAR) receptors. J Biol Chem. 2008;283:2751–2760. doi: 10.1074/jbc.M706578200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irving EA, Barone FC, Reith AD, Hadingham SJ, Parsons AA. Differential activation of MAPK/ERK and p38/SAPK in neurones and glia following focal cerebral ischaemia in the rat. Brain Res Mol Brain Res. 2000;77:65–75. doi: 10.1016/s0169-328x(00)00043-7. [DOI] [PubMed] [Google Scholar]
- Jiang H, Poirier MA, Liang Y, Pei Z, Weiskittel CE, Smith WW, DeFranco DB, Ross CA. Depletion of CBP is directly linked with cellular toxicity caused by mutant huntingtin. Neurobiol Dis. 2006;23:543–51. doi: 10.1016/j.nbd.2006.04.011. [DOI] [PubMed] [Google Scholar]
- Koh JY, Choi DW. Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J Neurosci Methods. 1987;20:83–90. doi: 10.1016/0165-0270(87)90041-0. [DOI] [PubMed] [Google Scholar]
- Kokaia Z, Zhao Q, Kokaia M, Elmér E, Metsis M, Smith ML, Siesjö BK, Lindvall O. Regulation of brain-derived neurotrophic factor gene expression after transient middle cerebral artery occlusion with and without brain damage. Exp Neurol. 1995;136:73–88. doi: 10.1006/exnr.1995.1085. [DOI] [PubMed] [Google Scholar]
- Krönke G, Bochkov VN, Huber J, Gruber F, Blüml S, Fürnkranz A, Kadl A, Binder BR, Leitinger N. Oxidized phospholipids induce expression of human heme oxygenase-1 involving activation of cAMP-responsive element-binding protein. J Biol Chem. 2003;278:51006–51014. doi: 10.1074/jbc.M304103200. [DOI] [PubMed] [Google Scholar]
- Lee B, Butcher GQ, Hoyt KR, Impey S, Obrietan K. Activity-dependent neuroprotection and cAMP response element-binding protein (CREB): kinase coupling, stimulus intensity, and temporal regulation of CREB phosphorylation at serine 133. J Neurosci. 2005;25:1137–1148. doi: 10.1523/JNEUROSCI.4288-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B, Cao R, Choi YS, Cho HY, Rhee AD, Hah CK, Hoyt KR, Obrietan K. The CREB/CRE transcriptional pathway: protection against oxidative stress-mediated neuronal cell death. J Neurochem. 2009;108:1251–1265. doi: 10.1111/j.1471-4159.2008.05864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B, Dziema H, Lee KH, Choi YS, Obrietan K. CRE-mediated transcription and COX-2 expression in the pilocarpine model of status epilepticus. Neurobiol Dis. 2007;25:80–91. doi: 10.1016/j.nbd.2006.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Kim CH, Simon DK, Aminova LR, Andreyev AY, Kushnareva YE, Murphy AN, Lonze BE, Kim KS, Ginty DD, Ferrante RJ, Ryu H, Ratan RR. Mitochondrial cyclic AMP response element-binding protein (CREB) mediates mitochondrial gene expression and neuronal survival. J Biol Chem. 2005;280:40398–40401. doi: 10.1074/jbc.C500140200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez de Armentia M, Jancic D, Olivares R, Alarcon JM, Kandel ER, Barco A. cAMP response element-binding protein-mediated gene expression increases the intrinsic excitability of CA1 pyramidal neurons. J Neurosci. 2007;27:13909–13918. doi: 10.1523/JNEUROSCI.3850-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantamadiotis T, Lemberger T, Bleckmann SC, Kern H, Kretz O, Martin Villalba A, Tronche F, Kellendonk C, Gau D, Kapfhammer J, Otto C, Schmid W, Schütz G. Disruption of CREB function in brain leads to neurodegeneration. Nat Genet. 2002;31:47–54. doi: 10.1038/ng882. [DOI] [PubMed] [Google Scholar]
- Marie H, Morishita W, Yu X, Calakos N, Malenka RC. Generation of silent synapses by acute in vivo expression of CaMKIV and CREB. Neuron. 2005;45:741–752. doi: 10.1016/j.neuron.2005.01.039. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol. 2000;1:120–129. doi: 10.1038/35040009. [DOI] [PubMed] [Google Scholar]
- Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996;274:1678–1683. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- Nucifora FC, Jr, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, Dawson TM, Ross CA. Interference by huntingtin and atrophin-1 with CBP-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–2428. doi: 10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- Obrietan K, Hoyt KR. CRE-mediated transcription is increased in Huntington’s disease transgenic mice. J Neurosci. 2004;24:791–796. doi: 10.1523/JNEUROSCI.3493-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang Z, Bondada V, Sengoku T, Siman R, Geddes JW. Calpain facilitates the neuron death induced by 3-nitropropionic acid and contributes to the necrotic morphology. J Neuropathol Exp Neurol. 2003;62:633–643. doi: 10.1093/jnen/62.6.633. [DOI] [PubMed] [Google Scholar]
- Rami A. Ischemic neuronal death in the rat hippocampus: the calpain-calpastatin-caspase hypothesis. Neurobiol Dis. 2003;13:75–88. doi: 10.1016/s0969-9961(03)00018-4. [DOI] [PubMed] [Google Scholar]
- Redmond L, Kashani A, Ghosh A. Calcium regulation of dendritic growth via Cam kinase IV and CREB-mediated transcription. Neuron. 2002;34:999–1010. doi: 10.1016/s0896-6273(02)00737-7. [DOI] [PubMed] [Google Scholar]
- Roze E, Betuing S, Deyts C, Marcon E, Brami-Cherrier K, Pagès C, Humbert S, Mérienne K, Caboche J. Mitogen- and stress-activated protein kinase-1 deficiency is involved in expanded-huntingtin-induced transcriptional dysregulation and striatal death. FASEB J. 2008a;22:1083–1093. doi: 10.1096/fj.07-9814. [DOI] [PubMed] [Google Scholar]
- Roze E, Saudou F, Caboche J. Pathophysiology of Huntington’s disease: from huntingtin functions to potential treatments. Curr Opin Neurol. 2008b;21:497–503. doi: 10.1097/WCO.0b013e328304b692. Review. [DOI] [PubMed] [Google Scholar]
- Ryu JK, Nagai A, Kim J, Lee MC, McLarnon JG, Kim SU. Microglial activation and cell death induced by the mitochondrial toxin 3-nitropropionic acid: in vitro and in vivo studies. Neurobiol Dis. 2003;12:121–132. doi: 10.1016/s0969-9961(03)00002-0. [DOI] [PubMed] [Google Scholar]
- Saulle E, Gubellini P, Picconi B, Centonze D, Tropepi D, Pisani A, Morari M, Marti M, Rossi L, Papa M, Bernardi G, Calabresi P. Neuronal vulnerability following inhibition of mitochondrial complex II: a possible ionic mechanism for Huntington’s disease. Mol Cell Neurosci. 2004;25:9–20. doi: 10.1016/j.mcn.2003.09.013. [DOI] [PubMed] [Google Scholar]
- Semaka A, Creighton S, Warby S, Hayden MR. Predictive testing for Huntington disease: interpretation and significance of intermediate alleles. Clin Genet. 2006;70:283–294. doi: 10.1111/j.1399-0004.2006.00668.x. Review. [DOI] [PubMed] [Google Scholar]
- Shieh PB, Hu SC, Bobb K, Timmusk T, Ghosh A. Identification of a signaling pathway involved in calcium regulation of BDNF expression. Neuron. 1998;20:727–740. doi: 10.1016/s0896-6273(00)81011-9. [DOI] [PubMed] [Google Scholar]
- Slow EJ, van Raamsdonk J, Rogers D, Coleman SH, Graham RK, Deng Y, Oh R, Bissada N, Hossain SM, Yang YZ, Li XJ, Simpson EM, Gutekunst CA, Leavitt BR, Hayden MR. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet. 2003;12:1555–1567. doi: 10.1093/hmg/ddg169. [DOI] [PubMed] [Google Scholar]
- St Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jäger S, Handschin C, Zheng K, Lin J, Yang W, Simon DK, Bachoo R, Spiegelman BM. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- Sugars KL, Brown R, Cook LJ, Swartz J, Rubinsztein DC. Decreased cAMP response element-mediated transcription: an early event in exon 1 and full-length cell models of Huntington’s disease that contributes to polyglutamine pathogenesis. J Biol Chem. 2004;279:4988–4899. doi: 10.1074/jbc.M310226200. [DOI] [PubMed] [Google Scholar]
- Sugiura S, Kitagawa K, Omura-Matsuoka E, Sasaki T, Tanaka S, Yagita Y, Matsushita K, Storm DR, Hori M. CRE-mediated gene transcription in the peri-infarct area after focal cerebral ischemia in mice. J Neurosci Res. 2004;75:401–407. doi: 10.1002/jnr.10881. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Zhou H, Neumaier JF, Pham TA. Opposing functions of CREB and MKK1 synergistically regulate the geometry of dendritic spines in visual cortex. J Comp Neurol. 2007;503:605–617. doi: 10.1002/cne.21424. [DOI] [PubMed] [Google Scholar]
- Tabrizi SJ, Workman J, Hart PE, Mangiarini L, Mahal A, Bates G, Cooper JM, Schapira AH. Mitochondrial dysfunction and free radical damage in the Huntington R6/2 transgenic mouse. Ann Neurol. 2000;47:80–86. doi: 10.1002/1531-8249(200001)47:1<80::aid-ana13>3.3.co;2-b. [DOI] [PubMed] [Google Scholar]
- Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron. 1998;20:709–726. doi: 10.1016/s0896-6273(00)81010-7. [DOI] [PubMed] [Google Scholar]
- Wilson BE, Mochon E, Boxer LM. Induction of bcl-2 expression by phosphorylated CREB proteins during B-cell activation and rescue from apoptosis. Mol Cell Biol. 1996;16:5546–5556. doi: 10.1128/mcb.16.10.5546. [DOI] [PMC free article] [PubMed] [Google Scholar]