

Abstract
Background
Current treatments for chronic hepatitis C virus (HCV) infection employing pegylated interferon plus ribavirin are successful in approximately 50% of patients, demonstrating a need for more effective therapy. Consensus interferon (CIFN) is a recombinant type I interferon (IFN) that has demonstrated efficacy in patients who fail to respond to conventional IFN based therapy, suggesting it has distinct biological properties from native IFN species. Here we evaluated the host cell antiviral response and anti-HCV actions induced by IFN-alpha2a (IFN-α2a), pegylated-IFN-alpha2b (PEG-IFN) or CIFN treatment of cultured immortalized human hepatocytes, Huh7 human hepatoma cells, and Huh7 cells harboring genetically distinct HCV RNA replicons or infected with HCV 2A.
Methods
Cultured cells were treated with each IFN at relevant dosing based upon the pharmacologic attainable in vivo serum maximum (Cmax) IFN concentrations. Gene expression and antiviral properties were measured using protein, RNA and virus quantification assays.
Results
Treatment with CIFN maximally triggered JaK-STAT signaling in association with enhancement of ISG expression. The increased antiviral potency of CIFN over IFN-α2a and PEG-IFN associated with enhancement of the IFN-induced blockade upon viral protein synthesis, protection of the cellular IPS-1 protein from proteolysis by HCV, and reduced replication of an IFN-resistant HCV replicon variant. Microarray analyses revealed that treatment with CIFN induced a pattern of ISG expression in cultured hepatocytes that was distinct from either IFN-α2a or PEG-IFN.
Conclusion
CIFN exhibits increased anti-HCV potency over IFN-α2a and PEG-IFN through maximal and distinct induction of ISG expression and enhancement of the intracellular innate antiviral response while providing therapeutic protection of IPS-1 from HCV proteolysis. CIFN may therefore offer a treatment regimen whose actions impart translational control programs and restoration of the RIG-I/IPS-1 pathway of innate immune amplification that could be considered for treating previous treatment failures.
Keywords: Hepatitis C Virus, interferon, RIG-I, IPS-1, MAVS, HCV, therapy
Introduction
Hepatitis C virus (HCV) infection represents a serious global health problem wherein the World Health Organization estimates that approximately 2.2% of the world population is infected with HCV[1]. IFN-α2a or IFN-α2b, pegylated versions of each (PEG-IFN-α2a or Peg-IFN-α2b), and consensus IFN (CIFN) are each approved for HCV treatment in the United States in which the current standard of care consists of PEG-IFN-α2a or PEG-IFN-α2b with ribavirin [2]. Among these IFN preparations, CIFN a synthetic type I IFN produced from the consensus sequence of several naturally occurring IFN-α subtypes [3]. Previous in vivo studies have demonstrated the efficacy of CIFN in the treatment of chronic HCV infection [4-8]. When compared against IFN-α2b alone, administration of overall lower doses of CIFN resulted in greater reduction of serum HCV RNA levels in treated patients [4]. However, the distinctions by which each IFN type mediates antiviral actions against HCV have not been evaluated. Problematically, only about 50% of treated individuals respond to IFN therapy overall [7-9]. This low response rate necessitates a continual push to improve IFN therapy application and treatment regimen. A number of studies have linked the poor response rate of HCV to IFN therapy with the ability of the virus to evade and antagonize the intracellular antiviral defenses that are triggered by infection and/or induced by IFN [10]. However, the molecular mechanisms of IFN action against HCV are not well understood, thus impeding the development of improved therapeutic strategies to combat the HCV pandemic.
IFNs are potent cytokines and key players in stimulating the innate antiviral immune response. IFNs mediate antiviral effects through the transcriptional activation of IFN-stimulated genes (ISGs) [11]. ISGs are primarily induced by intracellular signaling triggered by IFN through the α/β IFN receptor, which activates the canonical Janus kinase-signal transducer and activator of transcription (Jak-STAT) pathway [12]. IFN triggers the phosphorylation of STAT1 and STAT2 to mediate STAT activation and formation of the ISGF3 complex consisting of STAT1, STAT2, and IRF-9 [13, 14]. ISGF3 binds to the interferon stimulated response element (ISRE) within the promoter region of ISGs to induce gene expression. ISG products impart cellular actions that limit viral replication and cell to cell virus spread, and that indirectly modulate the maturation of the adaptive immune response [15-19]. IFNs are naturally produced during infection by HCV or other viruses. In the case of HCV, viral RNA recognition by the retinoic acid-inducible gene-I (RIG-I) protein triggers a signaling cascade through the essential interferon promoter simulator-1 (IPS-1) protein, leading to activation of the interferon regulatory factor-3 (IRF-3) transcription factor and its induction of α/β IFN expression [16-20]. HCV can antagonize RIG-I signaling of IRF-3 through the actions of the viral NS3/4A protease, which targets and cleaves IPS-1 disrupting IRF-3 activation [20-24]. Thus, therapeutic application of IFN to mediate high, sustained ISG expression and concomitantly promote or restore RIG-I signaling in infected cells might provide increased antiviral potency against HCV.
Clinical studies indicate that IFNα-2a and IFNα-2b or PEG-IFNα-2a and PEG-IFNα-2b have similar efficacies in the treatment of HCV, with the pegylated-interferons achieving better antiviral responses than their non-pegylated counterparts when given in combination with ribavirin (reviewed in [27]). In vitro studies found that CIFN had increased antiviral and immune modulation activity as compared to IFNα-2a and IFNα-2b, which had very similar activities [28]. The primary objective of this study was to define the signaling distinctions and antiviral properties of IFN-α2a, PEG-IFN-α2b and CIFN toward regulating ISG expression and controlling HCV infection in vitro. Our rationale for studying these IFNs is that each has been used for the treatment of HCV infection, and they comprise a representative set of the standard and pegylated IFNs that are currently approved for anti-HCV therapy applications [2]. Our studies reveal unique properties of CIFN that confer potent and differential ISG expression that associates with enhanced suppression of HCV replication. Our results suggest that strategies aimed at modifying HCV treatment protocols for maximal expression of specific ISGs should be considered for improving IFN therapy.
Materials and Methods
Cell culture and virus
Huh7 and Huh7.5 cells are human hepatoma cells [29, 30]. PH5CH8 cells are immortalized primary human hepatocytes, a kind gift from Dr. N. Kato (Okayama University Graduate School of Medicine, Dentistry, and Pharmaceutical Sciences, Okayama, Japan) [31]. Cells were propagated in Dulbecco's Modified Eagle's Medium (Fisher) supplemented with 10% fetal bovine serum, minimal essential amino acids and Sigma antibiotic-antimycotic solution. Huh7-L2198S, Huh7-K2040, and Huh7-HP cells are clonal Huh7 cell lines harboring genetic variants of an HCV genotype 1b subgenomic RNA replicon described previously [32-34]. The HCV replicon cell lines were maintained in culture medium supplemented with 200 μg G418/mL. For IFN treatment of HCV-replicon cells, G418 containing media was removed, cells washed once with PBS and replaced with culture medium containing the indicated concentrations of each IFN. IFN-α2a was obtained from PBL Interferon Source (Piscataway, NJ), PEG-Intron™ (PEG-IFN) from Schering-Plough Corp. (Kenilworth, NJ), and CIFN (INFERGEN®; Interferon alfacon-1) from InterMune Inc. (Brisbane, CA). The pharmacologic Cmax concentration of IFN-α2a and Peg-IFN-α2b was determined from the product sheet provided by the manufacturer. The pharmacologic Cmax concentration of CIFN was obtained following FDA-approved clinical dosing schedules, which corresponded to 80 pg/mL [35].
HCV was produced from Huh7.5 cells transduced with RNA from the JFH-1 infectious clone of HCV 2a exactly as described [23]. Virus stocks were prepared from supernatants of infected cell cultures and virus concentrated using Centricon Plus-70 filters (Millipore, Lowell, MA). HCV 2a titers were determined by focus forming assay. Cell supernatants were serially diluted 10-fold in complete DMEM and used to infect 104 naïve Huh7.5 cells per well in 48-well plates. The inoculum was incubated with cells for 1-3 h at 37°C and then supplemented with fresh culture medium. The level of HCV infection was determined 2 days post-infection by immunohistochemical staining for HCV proteins using the Vector® VIP substrate kit for peroxidase detection (Vector Laboratories, Burlingame, CA). The viral titer was determined by the average number of HCV-positive foci (ffu) detected at the highest dilutions.
Protein Analysis
For evaluation of protein expression, cell lysates were prepared exactly as described previously [18], except for experiments evaluating IPS-1 expression in which cells were lysed in RIPA buffer (10mM Tris, 150mM NaCl, 0.02% NaN3, 1% Na-deoxycholate, 1% Triton X-100, 0.1% SDS, 10μL/mL Sigma protease inhibitor cocktail, and 10μL/mL phosphatase inhibitor cocktail set II (Calbiochem, Gibbstown, NJ). Immunoblot analysis was conducted on 20-30 μg of total cell lysates with antiserum specific to PKR, ISG56, ISG15, RIG-I or IPS-1 and have been described previously [23, 32, 33]. Antibodies against actin, IRF-1, IRF-9, IRF-7, and total STAT2 were purchased from Santa Cruz (Santa Cruz, CA). Antibodies against total STAT1 or phosphorylated STAT1 were purchased from Cell Signaling (Danvers, MA). Two well-characterized anti-HCV patient serum were obtained with informed consent and Institutional Review Board approval through Dr. W. Lee (UT Southwestern Medical Center, Dallas, TX) and were used for detection of HCV proteins as described [32]. Secondary antibodies coupled to horseradish peroxidase were used for visualization of proteins by chemiluminescence. Each figure panel represents one membrane that was probed sequentially with multiple antibodies by stripping the antibodies from the membrane with 0.2M NaOH for 5 minutes, washing and blocking in 5% nonfat milk/PBS/Tween for 30 minutes prior to addition of the next antibody probe. Figure 1b represents one membrane that was rearranged after immunoblot analysis in order to be consistent with figure 1a, where Huh7 cells are on the left and PH5CH8 cells are on the right side of the panel. The data for each immunoblot analysis are representative of at least two independent experiments.
Figure 1.
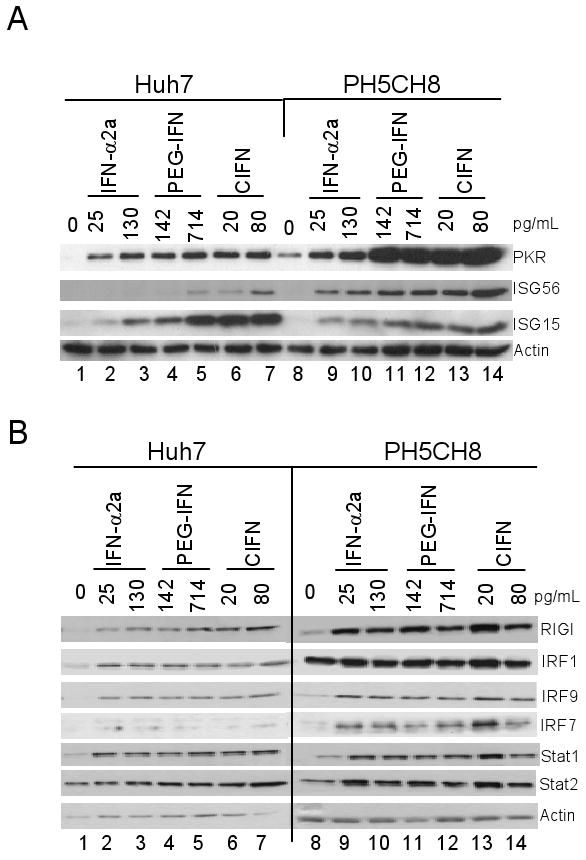
CIFN induces enhanced ISG expression. Huh7 or PH5CH8 cells were cultured in media alone (0) or with the indicated concentrations of each IFN (shown above each lane in pg/ml). 24 hours later immunoblot analysis was conducted to evaluate the expression of ISGs, including select antiviral ISGs (A) or ISGs and signaling proteins involved in activation and amplification of the IFN response (B). Protein expression levels were determined by densitometry analysis in which the protein levels in each lane were normalized to their respective actin control band. The fold change in protein abundance compared to the untreated control is shown under each respective lane.
For electrophoretic mobility-shift assays (EMSA), nuclear proteins were extracted and subjected to EMSA as previously described [36]. Extracts were incubated for 30 minutes at room temperature with a 32P-labeled double-stranded oligonucleotide probe corresponding to the ISRE from the ISG15 promoter. Bound complexes were analyzed via native 6% Tris-glycine-polyacrylamide gel electrophoresis. Gels were dried and bands visualized by autoradiography and quantitated using densitometry. For control samples, demonstrating that the observed shifted band represents the ISGF-3/ISRE DNA complex, we performed antibody blocking experiments by incubating IFNα-2a treated PH5CH8 nuclear extracts with 2μg anti-STAT1 or anti-IRF-9 antibody for 15 minutes at room temperature prior to addition of labeled probe.
Enzyme-Linked Immunosorbent Assay (ELISA)
In order to quantify neomycin phosphotransferase II (NPT II) protein levels in HCV subgenomic replicon cell lines a NPT II ELISA was performed as described by the kit manufacturer (Agdia, Elkhart, Indiana). Huh7-K2040 and Huh7-HP cells were respectively plated at 4.5×103 or 6×103 cells per well of a 96 well dish. Cells were cultured for 48 hours in media alone or with media containing serial dilutions of each IFN ranging from 2000-4 pg/mL. Cell extracts were prepared using the provided PEB1 extraction buffer and 20μL of total cell extract added to the provided NPT II antibody-coated 96-well microtiter plate in triplicate and ELISA performed exactly as instructed by the kit manufacturer. Bound NPT II protein in each well was measured using a plate reader with a 450nm absorbance filter. Standard curves were generated and used to quantify the NPT II concentration.
RNA methods and viral RNA quantitation
Total RNA was isolated from cells using the Trizol LS reagent (Invitrogen, Carlsbad, California). HCV and human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) RNA levels were determined by one-step quantitative real-time RT-PCR (qRT-PCR) using the ABI Prism 7500 sequence detection system in the presence of SYBR green with 100-250ng total RNA per reaction and each reaction done in triplicate. The primer pairs used for the detection of HCV were 5′-TGC GGA ACC GGT GAG TACA-3′(sense) and 5′ CGG GTT GAT CCA AGA AAG GA-3′(antisense). Relative mRNA expression levels were calculated using the comparative cycle threshold (CT) method with human GAPDH as the internal standard [37].
35S-Methionine labeling and immunoprecipitation analysis
Huh7.5 cells were infected with the HCV 2a at an MOI of 0.5 and passaged. At this point 100% of the cells were infected, as determined by parallel anti-HCV immunostaining. The cells were then plated at 105 cells per well of a 6-well dish and 24 hours later cells were washed. Culture media were then replaced with media containing the respective pharmacologic Cmax concentration of IFNs. After a further 24 hours, media was removed and cells were starved of methionine for 2 hours. Cells were labeled by adding 100 μCi of [35S] methionine to the culture media for 1 hour. Cells were then incubated for 10 minutes on ice in IP-lysis buffer (25mM Tris-HCL (pH7.5), 150mM NaCl, 1%NP40, okadaic acid, protease inhibitor (Sigma) and phosphatase inhibitor cocktail set II (Calbiochem). Total cell lysates were precleared with 40μL of protein A agarose for 3 hours at 4°C and recovered lysates subject to immunoprecipitation using 1μL of anti-HCV serum and 40 μL of Protein-A agarose beads (Roche). The reactions were incubated for 3 hours at 4°C, immunecomplexes washed 5 times with lysis buffer and 40 μl of SDS-sample buffer were added. Samples were boiled for 5 minutes and proteins separated on a 10% SDS-polyacrylamide gel (SDS-PAGE). The gel was fixed, soaked in amplify buffer (Amersham, Piscataway, NJ) for 30 minutes, dried and exposed to phosphor screen for 3-6 days. The radiosignal was visualized and quantified by phosphorimager analysis.
Microarray expression analysis
PH5CH8 cells were plated at a density of 106 cells per 100 mm dish, allowed to recover for 48 hours, and medium then removed and replaced with media containing the respective pharmacologic Cmax of the indicated IFN. Cells were harvested in a reverse time course following 20, 8, and 4 hr treatment with the various IFNs, or mock-treatment with media alone (control). Four independent samples were generated for each IFN and mock treatment at every time point. Expression analysis was performed using Affymetrix (Santa Clara, CA) Genechip® U133 plus 2.0 arrays in accordance with manufacturer-recommended protocols. Data were exported from Affymetrix MAS5 software and only those probe sets called “present” in all replicates of at least one condition were used for further analysis in order to purge the data set of unreliable information. Statistical analysis was performed in Partek Pro 5.1 (Partek, St. Paul, MN) or GeneSpring 6.1 (Silicon Genetics, Redwood City, CA). Statistical significance of expression differences was determined by 2-way ANOVA (time, IFN treatment, interaction of time and IFN treatment) with untreated samples excluded, and a Bonferonni p < 0.05 used as a multiple test correction. Principle Component Analysis (PCA) was used to visualize inter-sample variability associated with the expression profiles of the genes deemed to have statistically significant differences in expression with the various IFN treatments. Probe sets identified in the 2-way ANOVA as significantly different in IFN treatment were used in a 1-way ANOVA with a Tukey post hoc test. In this second statistical test, p < 0.05 was considered significant and ANOVA were conducted for each time point.
Results
Differential ISG expression induced by therapeutic IFNs
To define the response of human hepatocytic cells to clinically-relevant IFNs, we examined the induction of ISGs and IRFs after IFN treatment in Huh7 and PH5CH8 cells. Immunoblot analysis of cell lysates after 24 hours of treatment with increasing doses of IFN-α2a, PEG-IFN and CIFN revealed that CIFN treatment comparatively induced the highest expression of ISG56, ISG15 and PKR, known ISGs that impart antiviral actions against HCV or other RNA viruses (Figure 1A) [18, 32, 38]. Pharmacological studies have determined that the maximum IFN serum levels obtained in patients occur 2 hours following clinical dosing schedules and correspond to 130 pg/mL IFN-α2a, 320 pg/mL PEG-IFN and 80 pg/mL CIFN (Cmax) [35]. We found that maximal induction of PKR and ISG15 expression occurred at 130 pg/mL IFN-α2a, 714 pg/mL PEG-IFN and 20 pg/mL CIFN in both cell lines. In Huh7 cells induction of ISG56 was only appreciably induced at 24 hours post treatment with 80 pg/mL of CIFN (Figure 1A). The Huh7 and PH5CH8 cell lines were also evaluated for expression of proteins which are involved in driving IFN signaling and ISG expression [39]. As seen in Figure 1B, after 24 hours of treatment with increasing IFN doses, we observed the induced expression and similar high abundance of IRF-1, IRF-9, total STAT1 and total STAT2 within the treated cells.
Effects of CIFN Treatment on the Jak-STAT Signaling Pathway
In order to determine the mechanism by which CIFN directed enhanced ISG expression in the Huh7 and PH5CH8 cell lines, we evaluated the Jak-STAT pathway in IFN-treated cells. STAT1 is activated by IFN signaling upon phosphorylation of tyrosine 701, wherein activated STAT-1 leads to increased STAT and IRF-9 abundance [16]. As shown in Figure 2A, treatment of cells with the Cmax of each IFN resulted in the accumulation of active STAT1 phosphorylated on tyr-701. Treatment with CIFN induced the highest abundance of phosphorylated STAT1, with maximum levels reached at approximately 60 minutes in Huh7 cells and 30 minutes after treatment in PH5CH8 cells. There were no appreciable differences in total STAT1 levels induced among the different IFNs. We also evaluated kinetics of induction of IRF-9, observing increased expression at 240 minutes post IFN treatment with the highest levels induced by CIFN (Figure 2A).
Figure 2.
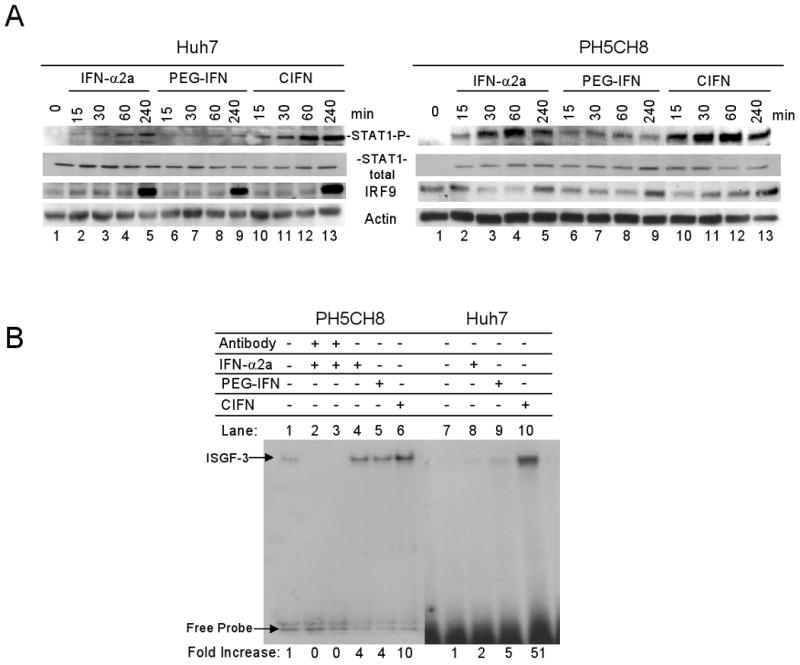
CIFN maximally activates the Jak-STAT pathway. (A) The abundance of phosphotyrosine 701 STAT-1 (activated STAT-1), total STAT-1 and IRF-9 was measured by immunoblot analysis after treatment with the different IFNs at the Cmax dose for the time in minutes above each lane. Treatment of Huh7 (left panel set) and PH5CH8 (right panel set) is shown. (B) EMSA of the ISGF-3 transcription factor complex in PH5CH8 (lanes1-6) and Huh7 (lanes 7-10) cells treated with the Cmax of each IFN for 30 minutes. Arrows indicate the positions of the unbound probe and ISGF-3 complex, as shown by IRF-9 and Stat1 antibody blocking analysis. Densitometry analysis was used to quantitate bands corresponding to the ISGF-3 complex, displayed as fold-increase relative to the untreated controls which were set to 1 (lanes 1 and 7). Results shown are representative of three independent experiments.
We also assessed the affect of the different therapeutic IFNs on the formation of ISGF3 and its DNA binding activity to the ISRE. Cells were treated with the Cmax of each IFN and nuclear proteins evaluated for their ability to bind to the ISRE from the ISG15 promoter in an EMSA. As shown in Figure 2B, IFN treatment induced the formation of a gel-shift complex. Complex formation was abrogated by prior incubation of the reaction with antibodies specific to STAT1 or IRF9, thus defining the complex as ISGF3 [16]. CIFN treatment of PH5CH8 or Huh7 cells respectively resulted in an approximate 10 and 50-fold increase in ISGF3/DNA complex formation compared to nontreated cells, wherein lower levels of ISGF3/DNA complex formation were observed in cells treated with IFN-α2a or PEG-IFN. Together these results indicate that CIFN has increased signaling potency over IFN-α2a or PEG-IFN.
Therapeutic IFNs have Differential Antiviral Effects Against HCV RNA Replication
The antiviral potency of IFN has been directly linked to the strength of Jak-STAT pathway signaling [16], suggesting that differential signaling actions by the IFNs could impart distinct properties to limit HCV replication. We therefore assessed the antiviral actions of each IFN for their ability to suppress the replication of genetically distinct HCV 1b subgenomic RNA replicons in Huh7 cells. For these analyses we compared IFN actions within cells harboring IFN-sensitive (Huh7-K2040 cells) or IFN-resistant (Huh7-HP cells) replicon variants [33, 34]. We treated each replicon cell line with the Cmax of the respective IFN for 24, 48 or 96 hours and evaluated host and viral protein expression in the treated cells. HCV protein levels within the Huh7-K2040 cells decreased within 24 hours of treatment with each of the IFNs (Figure 3A, left panel), consistent with the induction of ISGs and the IFN-sensitivity of this HCV replicon variant [34]. By contrast, our analyses demonstrated that the HCV-HP replicon was relatively resistant to IFN but that CIFN treatment suppressed the abundance of the HCV NS5A protein within 96 hours of treatment of the Huh7-HP cells (Figure 3A). Compared to the rapid but transient activation of the Jak-STAT pathway that is mediated by IFN treatment [10], ISG expression by CIFN was maintained throughout the 96 hr treatment in association with the suppression of HCV protein abundance.
Figure 3.

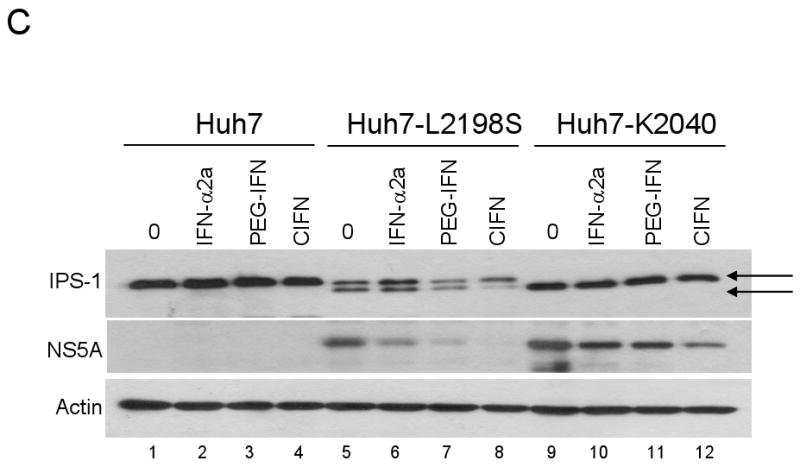
Comparison of the antiviral effects rendered by the IFNs on HCV RNA replicon containing cells. (A) Immunoblot analysis of Huh7 cells (lanes 1-4 on both panels) and Huh7 cells harboring the HCV RNA replicons after incubation with the Cmax of each IFN for the indicated times. HCV NS5A protein levels were quantified by densitometry analysis and the values are displayed below the corresponding lane as a percentage of the protein within matched control cells cultured with media alone. Control values from nontreated cells were set at 100%. (B) ELISA of NPT II protein levels from HCV-K2040 (green) and HCV-HP (red) IFN treated cells. The calculated EC50 of each IFN is displayed under the corresponding graph. Data are representative of three independent experiments. (C) Immunoblot analysis of endogenous IPS-1, NS5A and actin expression in control Huh7 cells (lanes1-4), Huh7-L2198S cells (lanes 5-8) and Huh7-K2040 cells (lanes 9-12) cultured with media alone (0) or Cmax of each IFN for 72 hours. Arrows denote the full length (FL) and cleaved forms of IPS-1.
The reduction of viral protein expression by CIFN was linked to enhanced levels of ISG56 and PKR (Figure 3A), known antiviral proteins that suppress translation from the HCV internal ribosome entry site to suppress protein synthesis and viral RNA replication [18, 32, 40]. We therefore evaluated the relative potency of the different IFNs toward suppressing HCV RNA replication of the different replicon variants. In the replicon system the HCV IRES directs the synthesis of the NPT II gene conferring neomycin resistance [32, 41]. We therefore measured the HCV IRES-driven NPT II protein levels in cells treated with increasing concentrations (4-2000 pg/ml) of each IFN. The effective concentration of each IFN required to reduce NPT II levels by 50% (EC50) after 48 hour treatments was calculated. We found that CIFN had an EC50 of 18.7 pg/mL in Huh7-K2040 cells compared to 37.1 and 50.9 pg/ml for IFN-α2a and PEG-IFN, respectively. The EC50 of CIFN treatment of the IFN-resistant Huh7-HP cells was 54.6 pg/mL compared to 169.2 pg/mL for IFN-α2a and 360.2 pg/mL for PEG-IFN treatment (Figure 3B). These results demonstrate that CIFN has potent antiviral activity against distinct HCV RNA replicon variants and induces antiviral activity at much lower doses than treatment with IFN-α2a or PEG-IFN.
Differential Restoration of IPS-1 by IFN treatment
The difference among the IFNs to suppress HCV protein abundance could be an important determinant modulating the status of the RIG-I pathway through viral protease cleavage of IPS-1. We therefore examined the ability of the IFNs to impart reduced viral protein expression concomitant with restoration of IPS-1 to its noncleaved, full-length form that is competent to mediate RIG-I signaling and IFN amplification [23]. For these analyses we compared IFN treatment among Huh7 cells harboring distinct HCV RNA replicons of low (Huh7-L2198S) or high replication fitness (Huh9-K2040 cells) due to the presence of different adaptive mutations within their viral protein-coding region [32, 33]. The replication fitness of the HCV replicon variants corresponded with the partial or complete proteolysis of endogenous IPS-1, respectively (Figure 3C). Following CIFN treatment IPS-1 was protected from cleavage in both HCV replicon cell lines correlating with a marked reduction in viral protein abundance. However, cleavage of IPS-1 was only minimally protected by either IFN-α2a or PEG-IFN treatment.
CIFN Regulation of HCV Infection
In order to determine how IFN impacts actual HCV infection, we examined HCV 2A JFH1 infectious virus production and viral RNA levels Huh7 cell cultures treated with the different IFNs [42]. Cells were infected with HCV 2A for 24 hours before media was replaced with Cmax concentrations of each IFN. Supernatants were harvested 24 hours after treatment and viral titer determined by focus forming assay. Total RNA from infected cells was also harvested 24 hours post-IFN treatment and HCV RNA levels quantitated using real-time RT-PCR. As shown in Figure 4A (upper panel), IFN treatment for 24 hours resulted in a significant decrease in infectious virus production that was greatest in cells treated with CIFN. In contrast, viral RNA levels remained relatively unchanged from mock-treated cells and among the IFN treated cells after 24 hrs of treatment (Figure 4A, lower panel). We determined that after 24 hours CIFN treatment resulted in an acute decrease of 94% reduction of infectious HCV 2A released into the culture supernatant as compared to parallel cultures of untreated infected cells, while viral RNA levels remained relatively unchanged at this time point.
Figure 4.

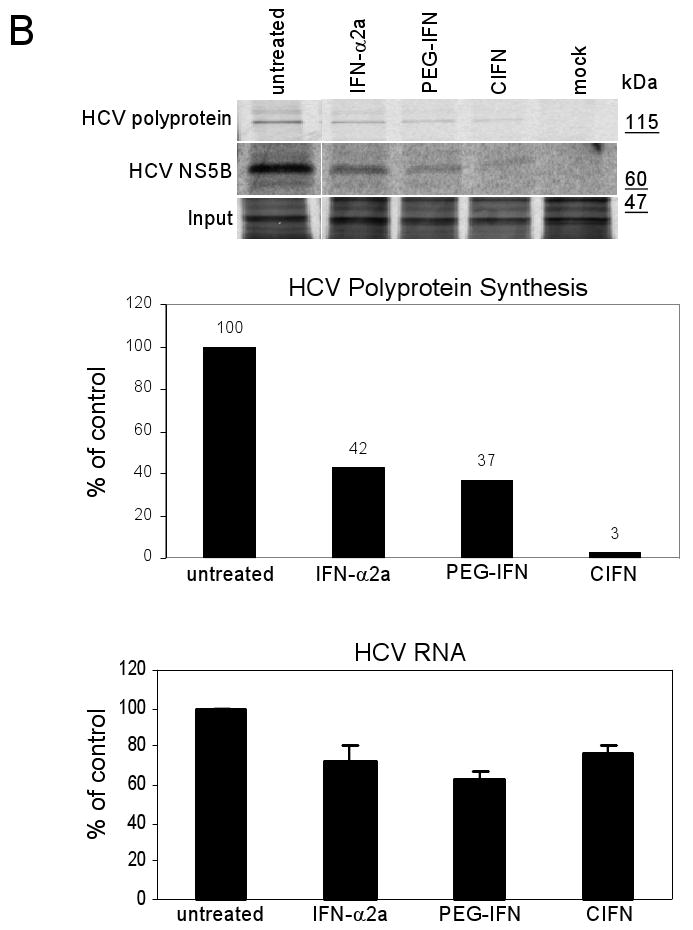
CIFN exhibits enhanced antiviral efficacy against the HCV 2a infectious clone. (A) Huh7 cells were infected with HCV 2a at an MOI of 1. 24 hours later media was replaced with fresh media alone (nontreated) or with fresh media containing the Cmax of each IFN. After a further 24 hours the viral titer was assessed by focus forming assay. Results are the mean number of infected foci (ffu) ±SD expressed as a percent of the untreated control (top panel). The bottom panel shows % decrease of HCV RNA, 24 hours post-treatment, as compared to untreated samples with GAPDH mRNA levels used as an internal standard. Results are representative of two independent experiments conducted in triplicate. (B) Cellular and viral protein synthesis. Noninfected Huh7 cells (mock) and HCV 2a-infected Huh7.5 cells were cultured for 24 hours in the presence of the Cmax of each IFN followed by [35S]methionine pulse-labeling for 1 hour. The bands shown were selected from the autoradiogram from SDS-PAGE analyses of the total cellular proteins (input) and HCV proteins (polyprotein and NS5B) recovered from cell extracts by immunoprecipitation. The incorporation of radiolabel into the HCV-polyprotein was quantified and values displayed as a percentage of the untreated sample, set to 100%. Molecular masses, in kDa, are indicated on the right side of the autoradiogram. HCV RNA levels were assessed after a continued IFN treatment of 96 hours by quantitative real-time RT-PCR and are presented as the percentage HCV RNA remaining relative to the untreated control (lower panel). The error bars represent the standard deviation of the means of HCV RNA levels from triplicate samples. P values denote significant reductions in CIFN treated samples compared to IFN-α2a or PEG-IFN treated samples (Student's t-test). Results are representative of two independent experiments.
Previous studies have shown that IFN treatment of cells induces cellular translational control programs which dominantly suppress translation from the HCV IRES [18, 34], consistent with our observation of an acute reduction in HCV production over viral RNA abundance in the IFN-treated cells. To determine the impact of the different IFNs on HCV protein synthesis, we conducted metabolic labeling studies and anti-HCV protein immunoprecipitation analyses of infected cells cultured alone or with the different IFNs. As shown in Figure 4B, 24 hours of IFN treatment rendered a specific reduction in HCV protein synthesis but had little overall effect on total cellular protein synthesis (input). The greatest suppression of viral protein synthesis was observed in cells treated with CIFN, which mediated greater than 90% decrease in the synthesis of the HCV-polyprotein and the fully processed NS5B protein as compared to their approximate reduction of 60% for IFN-α2a or PEG-IFN treatment. While HCV RNA levels decreased only modestly after 24 hours of treatment with either IFN (see Figure 4A, lower panel) prolonged IFN treatment through 96 hours resulted in a significant reduction of HCV RNA (Figure 4B, lower panel). These observations support previous studies indicating that IFN mediates a translational blockade of HCV replication, and suggest that prolonged suppression of viral protein abundance during IFN treatment results in an overall suppression viral RNA levels. These results demonstrate enhanced HCV translational-suppressive properties of CIFN compared to IFN-α2a or PEG-IFN.
Gene Expression Differences between the various IFNs
To determine if CIFN conferred differential gene expression that might associate with its enhanced antiviral actions against HCV compared to IFN-α2a and PEG-IFN, genome-wide microarray analysis of the cellular transcriptome was performed following treatment of PH5CH8 cells with the different IFNs. We identified 8251 unique or redundant probe sets whose expression profile exhibited time dependent regulation by one or more of the IFNs. Using a 2-way ANOVA analysis, 86 probe sets were detected with significantly different expression levels at one or more time point following treatment with CIFN compared to IFN-α2a or PEG-IFN-α2b (Supplemental Table 1). Principle Component Analysis (PCA) was used to visualize inter-sample variability associated with the expression profiles of the ISGs detected by these 86 probe sets. PCA represents an orthogonal linear transformation of expression data to a new coordinate system, reducing variance dimensionality by retaining those characteristics of the data set that contribute most to its variance. Post hoc analysis of expression at each time point was used to associate expression differences identified by the 2-way ANOVA with differences between CIFN and IFN-α2a, CIFN and PEG-IFN-α2b, or IFN-α2a and PEG-IFN. Together PCA and post hoc analysis clearly demonstrated that, at all time points examined, the transcriptional responses of human hepatocytes to CIFN is radically different from the responses to IFN-α2a or PEG-IFN treatments, which clustered similarly to each other (Figure 5). In general, the genes observed to be differentially regulated by microarray analysis confirm and extend the IFN-dependent expression differences characterized by immunoblot analyses. Differentially regulated genes identified by microarray analysis include products known to participate in antiviral cell signaling (JAK2, MYD88, RIG-I, IRF-7), in elaborating direct antiviral responses (OAS proteins, Mx, ISG20, RIG-I, IFIT proteins, IFI proteins) and in the adaptive immune responses (IL15RA, WARS). Additionally, many proteins of unknown function are also observed (Supplemental Table 1). These observations represent the seminal analysis of IFN-induced gene expression over a time course in primary hepatocytes, thus providing insights into the hepatocyte response to different IFNs. Our results demonstrate that CIFN induces a distinct transcriptional response in hepatocytes compared to IFN-α2a and PEG-IFN.
Figure 5.
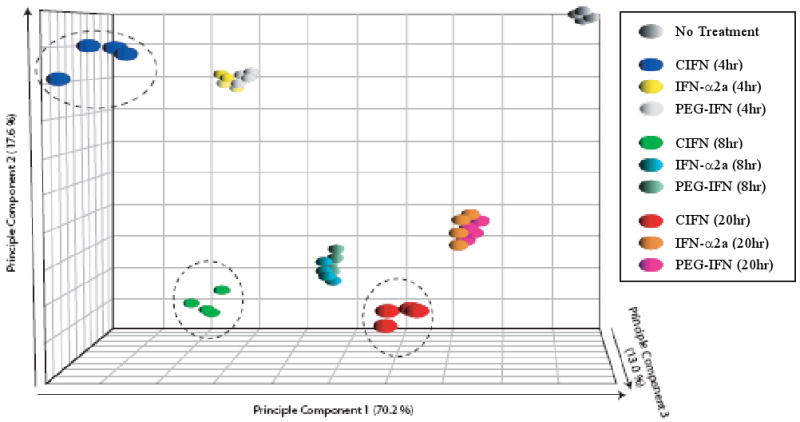
ISG expression induced by CIFN in human hepatocytes is distinct from IFN-α2a and PEG-IFN. The figure shows Principle Component Analysis (PCA) of the IFN-regulated transcriptome from PH5CH8 cells that were cultured in the presence of media alone (CTRL) or media containing the Cmax of each IFN for 4, 8 or 20 hours as indicated. The greatest determinant of inter-sample variance lies on the X coordinate (the first principal component), the second greatest variance on the Y coordinate (the second principal component), and the third greatest principle component on the Z coordinate. Higher-order principal components contributing less to intersample variability are ignored.
Discussion
The overall low response rate of HCV to IFN-based therapy suggests that HCV can evade or resist the antiviral actions of IFN, and that variable host responses to IFN may underlie treatment efficacy. The failure of IFN treatment has been associated with large quasi-species diversity and high viral load, suggesting that continual pressure from the host immune response may drive the outgrowth of highly fit HCV variants able to evade or resist IFN actions [43]. Thus, aggressive initial treatment may be a means of increasing success of therapy by limiting the emergence of HCV ‘evasion variants’. CIFN treatment of patients with relapsing HCV infection after previous standard of care treatment demonstrated that CIFN was faster than IFN-α2b plus ribavirin in the time taken to reach maximal response rate and had a lower prevalence of relapse of infection [8]. In accordance with this our study provides evidence that CIFN has enhanced ability over IFN-α2a or PEG-IFN to rapidly induce the expression of ISGs known to govern HCV infection, enhance immunity, and to direct the cellular antiviral response. CIFN treatment of hepatocytic cell lines led to robust activation of the Jak-STAT signaling pathway characterized by increased levels of STAT1 phosphorylation, ISGF-3 DNA binding and ISG expression. We also observed that despite inducing robust ISG expression (Figure 1), PEG-IFN treatment of cells rendered reduced levels of STAT1 tyr-701 and ISGF3 activation compared to CIFN (Figure 2). These data suggest that pegylation may alter the signaling properties of IFN, and/or that other, non-canonical pathways of IFN signaling can contribute to ISG expression. Indeed, IFN treatment can trigger signaling through STAT-independent cellular pathways, including MAP kinase and PKC pathways [reviewed in reference 11]. We speculate that signaling differences among the IFNs tested could include noncanonical IFN signaling pathways whose differential actions by CIFN might enhance the overall response to treatment.
Our study also demonstrates that CIFN has superior ability to suppress the replication of genotype 1b HCV RNA replicon variants, including the HCV-HP replicon that evolved under selective pressure from endogenous IFN to resist the antiviral actions of IFN-α2a [34]. We showed that when all three IFNs were used at Cmax concentrations CIFN effectively reduced viral protein levels of the highly fit HCV-K2040 and the HCV-HP replicons to an extent better than the other IFNs tested. Importantly, our data show that CIFN has increased potency compared to IFN-α2a and PEG-IFN against the HCV 2a JFH1 cell culture-infectious virus, and that overall, IFN imposes its antiviral actions against HCV by directing a dominant blockade to viral IRES function and RNA translation. This blockade of viral RNA translation imposes limitations to HCV protein production that impart subsequent reduction of intracellular viral RNA and infectious virus production. Moreover, we found that CIFN treatment resulted in protection of IPS-1 from proteolysis by HCV through reduced levels of the viral NS3/4A protease. Therefore CIFN may provide a therapeutic benefit by restoring the function of the RIG-I pathway to amplify endogenous IFN production and direct a diverse repertoire of ISG expression, further limiting virus replication and spread [44]. The anti-HCV actions of CIFN occurred at lower concentrations than of IFN-α2a or PEG-IFN, suggesting that CIFN might be amenable to lower dose administration in patients to obtain similar antiviral effects.
Mathematical models derived from analyzing viral kinetics in HCV-infected patients during IFN-α therapy have shown that viral decline follows a bi-phasic pattern where the initial phase of rapid viral decline in the serum is attributed to acute blockade of de novo virus production [45]. The present study provides further support that IFN treatment mediates an acute suppression of viral RNA translation, possibly explaining the molecular mechanism of the first-phase of viral decline in treated patients. Our data show that CIFN imposed a more potent blockade of HCV protein synthesis, reducing viral protein levels to a greater extent than IFN-α2a or PEG-IFN, and ultimately suppressing viral RNA levels. Overall, our results support a model in which CIFN mediates its enhanced antiviral actions, in part, by acute induction of cellular translational control programs that disrupt viral protein synthesis, imposing limitations on viral RNA replication and infectious virus production.
CIFN binds the α/β IFN receptor with higher affinity and avidity that IFN-α2a, supporting the idea that it might trigger the intracellular signaling pathways, including the JAK-STAT pathway and other IFN-responsive pathways to mediate its distinct biological effects [3]. Our microarray analysis support these results demonstrating that while IFN-α2a or PEG-IFN induced a similar set of genes in immortalized primary human hepatocytes, the gene expression profile after CIFN treatment is quantitatively distinct. These results suggest that CIFN may impart multiple and distinct biological activities, against HCV, including the suppression of viral protein synthesis and general immune enhancement. Our findings correlate with published results from clinical studies evaluating the antiviral response of CIFN therapy in HCV infected patients. Collectively, these studies indicate that CIFN may be a useful therapeutic alternative for the effective treatment of HCV patients infected with high viral load, HCV genotype 1 or for non-responders and relapsers after standard PEG-IFN therapy [4-8].
In conclusion, we have shown that CIFN exerts anti-HCV efficacy in vitro with higher potency compared to IFN-α2a or PEG-IFN-α2b, and that the antiviral actions of IFN against HCV are attributed in part to a dominant suppression of HCV IRES function. Our study provides evidence of the complex nature of the IFN response, wherein the induction of specific ISGs may impart differential outcome of therapy and infection. Modification of IFN therapy regimens to impart the rapid and high level expression of ISGs known to suppress HCV replication could offer therapeutic benefits to improve treatment outcome.
Supplementary Material
Description and expression characteristics of the 86 ISGs identified as induced higher by CIFN in at least one time point compared to IFN-α2a or PEG-IFN in PH5CH8 cells. Cells were cultured in the presence of media alone (CTRL) or media containing the Cmax of each IFN for 4, 8 or 20 hours as indicated, and RNA expression assessed by microarray analysis. Gene expression differences were identified by 2-way ANOVA and a Bonferonni, p<0.05 used as a multiple test correction. The mean fold induction of each gene by each IFN treatment at every time point is listed.
Acknowledgments
We thank C. Wang and H. Tan for technical assistance and InterMune for CIFN. This work was supported by NIH grants AI060389, DA024563, AI40035 (Project 4), a grant from the Burroughs Wellcome Fund, and a gift from Mr. and Mrs. R. Batcheldor.
References
- 1.Global burden of disease (GBD) for hepatitis C. J Clin Pharmacol. 2004;44:20–29. doi: 10.1177/0091270003258669. [DOI] [PubMed] [Google Scholar]
- 2.Deutsch M, Hadziyannis SJ. Old and emerging therapies in chronic hepatitis C: an update. J Viral Hepat. 2008;15:2–11. doi: 10.1111/j.1365-2893.2007.00887.x. [DOI] [PubMed] [Google Scholar]
- 3.Blatt LM, Davis JM, Klein SB, Taylor MW. The biologic activity and molecular characterization of a novel synthetic interferon-alpha species, consensus interferon. J Interferon Cytokine Res. 1996;16:489–499. doi: 10.1089/jir.1996.16.489. [DOI] [PubMed] [Google Scholar]
- 4.Tong MJ, Reddy KR, Lee WM, et al. Treatment of chronic hepatitis C with consensus interferon: a multicenter, randomized, controlled trial. Consensus Interferon Study Group. Hepatology. 1997;26:747–754. doi: 10.1002/hep.510260330. [DOI] [PubMed] [Google Scholar]
- 5.Witthoeft T, Fuchs M, Ludwig D. Recent i.v.-drug users with chronic hepatitis C can be efficiently treated with daily high dose induction therapy using consensus interferon: an open-label pilot study. World J Gastroenterol. 2007;13:579–584. doi: 10.3748/wjg.v13.i4.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yasuda S, Miyata K. Interferon alfacon-1 (Advaferon): a novel synthetic interferon for the treatment of hepatitis C, its pharmacological and clinical profile. Nippon Yakurigaku Zasshi. 2002;120:421–426. doi: 10.1254/fpj.120.421. [DOI] [PubMed] [Google Scholar]
- 7.Sjogren MH, Sjogren R, Jr, Lyons MF, et al. Antiviral response of HCV genotype 1 to consensus interferon and ribavirin versus pegylated interferon and ribavirin. Dig Dis Sci. 2007;52:1540–1547. doi: 10.1007/s10620-007-9757-9. [DOI] [PubMed] [Google Scholar]
- 8.Miglioresi L, Bacosi M, Russo F, et al. Consensus interferon versus interferon-alpha 2b plus ribavirin in patients with relapsing HCV infection. Hepatol Res. 2003;27:253–259. doi: 10.1016/s1386-6346(03)00269-9. [DOI] [PubMed] [Google Scholar]
- 9.Fried MW, Shiffman ML, Reddy KR, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med. 2002;347:975–982. doi: 10.1056/NEJMoa020047. [DOI] [PubMed] [Google Scholar]
- 10.McHutchison JG, Gordon SC, Schiff ER, et al. Interferon alfa-2b alone or in combination with ribavirin as initial treatment for chronic hepatitis C. Hepatitis Interventional Therapy Group. N Engl J Med. 1998;339:1485–1492. doi: 10.1056/NEJM199811193392101. [DOI] [PubMed] [Google Scholar]
- 11.McHutchison JG. Understanding hepatitis C. Am J Manag Care. 2004;10:S21–29. [PubMed] [Google Scholar]
- 12.Feld JJ, Hoofnagle JH. Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature. 2005;436:967–972. doi: 10.1038/nature04082. [DOI] [PubMed] [Google Scholar]
- 13.Der SD, Zhou A, Williams BR, Silverman RH. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc Natl Acad Sci U S A. 1998;95:15623–15628. doi: 10.1073/pnas.95.26.15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heim MH. The Jak-STAT pathway: cytokine signalling from the receptor to the nucleus. J Recept Signal Transduct Res. 1999;19:75–120. doi: 10.3109/10799899909036638. [DOI] [PubMed] [Google Scholar]
- 15.Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu Rev Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 16.Sen GC. Viruses and interferons. Annu Rev Microbiol. 2001;55:255–281. doi: 10.1146/annurev.micro.55.1.255. [DOI] [PubMed] [Google Scholar]
- 17.Kolumam GA, Thomas S, Thompson LJ, Sprent J, Murali-Krishna K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J Exp Med. 2005;202:637–650. doi: 10.1084/jem.20050821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang C, Pflugheber J, Sumpter R, Jr, et al. Alpha interferon induces distinct translational control programs to suppress hepatitis C virus RNA replication. J Virol. 2003;77:3898–3912. doi: 10.1128/JVI.77.7.3898-3912.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shimazaki T, Honda M, Kaneko S, Kobayashi K. Inhibition of internal ribosomal entry site-directed translation of HCV by recombinant IFN-alpha correlates with a reduced La protein. Hepatology. 2002;35:199–208. doi: 10.1053/jhep.2002.30202. [DOI] [PubMed] [Google Scholar]
- 20.Guo JT, Bichko VV, Seeger C. Effect of alpha interferon on the hepatitis C virus replicon. J Virol. 2001;75:8516–8523. doi: 10.1128/JVI.75.18.8516-8523.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prabhu R, Joshi V, Garry RF, et al. Interferon alpha-2b inhibits negative-strand RNA and protein expression from full-length HCV1a infectious clone. Exp Mol Pathol. 2004;76:242–252. doi: 10.1016/j.yexmp.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 22.Meylan E, Curran J, Hofmann K, et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 23.Loo YM, Owen DM, Li K, et al. Viral and therapeutic control of IFN-beta promoter stimulator 1 during hepatitis C virus infection. Proc Natl Acad Sci U S A. 2006;103:6001–6006. doi: 10.1073/pnas.0601523103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Foy E, Li K, Sumpter R, Jr, et al. Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc Natl Acad Sci U S A. 2005;102:2986–2991. doi: 10.1073/pnas.0408707102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson CL, Owen DM, Gale M., Jr Functional and therapeutic analysis of hepatitis C virus NS3.4A protease control of antiviral immune defense. J Biol Chem. 2007;282:10792–10803. doi: 10.1074/jbc.M610361200. [DOI] [PubMed] [Google Scholar]
- 26.Lau DT, Fish PM, Sinha M, et al. Interferon regulatory factor-3 activation, hepatic interferon-stimulated gene expression, and immune cell infiltration in hepatitis C virus patients. Hepatology. 2008;47:799–809. doi: 10.1002/hep.22076. [DOI] [PubMed] [Google Scholar]
- 27.Hayashi N, Takehara T. Antiviral therapy for chronic hepatitis C: past, present, and future. J Gastroenterol. 2006;41:17–27. doi: 10.1007/s00535-005-1740-7. [DOI] [PubMed] [Google Scholar]
- 28.Ozes ON, Reiter Z, Klein S, Blatt LM, Taylor MW. A comparison of interferon-Con1 with natural recombinant interferons-alpha: antiviral, antiproliferative, and natural killer-inducing activities. J Interferon Res. 1992;12:55–59. doi: 10.1089/jir.1992.12.55. [DOI] [PubMed] [Google Scholar]
- 29.Sumpter R, Jr, Loo YM, Foy E, et al. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J Virol. 2005;79:2689–2699. doi: 10.1128/JVI.79.5.2689-2699.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blight KJ, McKeating JA, Rice CM. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol. 2002;76:13001–13014. doi: 10.1128/JVI.76.24.13001-13014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ikeda M, Sugiyama K, Mizutani T, et al. Human hepatocyte clonal cell lines that support persistent replication of hepatitis C virus. Virus Res. 1998;56:157–167. doi: 10.1016/s0168-1702(98)00063-x. [DOI] [PubMed] [Google Scholar]
- 32.Pflugheber J, Fredericksen B, Sumpter R, Jr, et al. Regulation of PKR and IRF-1 during hepatitis C virus RNA replication. Proc Natl Acad Sci U S A. 2002;99:4650–4655. doi: 10.1073/pnas.062055699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Foy E, Li K, Wang C, et al. Regulation of interferon regulatory factor-3 by the hepatitis C virus serine protease. Science. 2003;300:1145–1148. doi: 10.1126/science.1082604. [DOI] [PubMed] [Google Scholar]
- 34.Sumpter R, Jr, Wang C, Foy E, Loo YM, Gale M., Jr Viral evolution and interferon resistance of hepatitis C virus RNA replication in a cell culture model. J Virol. 2004;78:11591–11604. doi: 10.1128/JVI.78.21.11591-11604.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tan H, Derrick J, Hong J, et al. Global transcriptional profiling demonstrates the combination of type I and type II interferon enhances antiviral and immune responses at clinically relevant doses. J Interferon Cytokine Res. 2005;25:632–649. doi: 10.1089/jir.2005.25.632. [DOI] [PubMed] [Google Scholar]
- 36.Fredericksen B, Akkaraju GR, Foy E, et al. Activation of the interferon-beta promoter during hepatitis C virus RNA replication. Viral Immunol. 2002;15:29–40. doi: 10.1089/088282402317340215. [DOI] [PubMed] [Google Scholar]
- 37.Anonymous. ABI 7700 Sequence Detection System. Foster City, CA: Applied Biosystem: Applied Biosystem; 1997. User Bulletin No. 2, Relative quantification of gene expression. updated 2001. [Google Scholar]
- 38.Lenschow DJ, Lai C, Frias-Staheli N, et al. IFN-stimulated gene 15 functions as a critical antiviral molecule against influenza, herpes, and Sindbis viruses. Proc Natl Acad Sci U S A. 2007;104:1371–1376. doi: 10.1073/pnas.0607038104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gale M, Jr, Foy EM. Evasion of intracellular host defence by hepatitis C virus. Nature. 2005;436:939–945. doi: 10.1038/nature04078. [DOI] [PubMed] [Google Scholar]
- 40.Jiang D, Guo H, Xu C, et al. Identification of three interferon-inducible cellular enzymes that inhibit the replication of hepatitis C virus. J Virol. 2008;82:1665–1678. doi: 10.1128/JVI.02113-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lohmann V, Korner F, Koch J, et al. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999;285:110–113. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 42.Cai Z, Zhang C, Chang KS, et al. Robust production of infectious hepatitis C virus (HCV) from stably HCV cDNA-transfected human hepatoma cells. J Virol. 2005;79:13963–13973. doi: 10.1128/JVI.79.22.13963-13973.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Farci P, Strazzera R, Alter HJ, et al. Early changes in hepatitis C viral quasispecies during interferon therapy predict the therapeutic outcome. Proc Natl Acad Sci U S A. 2002;99:3081–3086. doi: 10.1073/pnas.052712599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yoneyama M, Kikuchi M, Natsukawa T, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 45.Neumann AU, Lam NP, Dahari H, et al. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science. 1998;282:103–107. doi: 10.1126/science.282.5386.103. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description and expression characteristics of the 86 ISGs identified as induced higher by CIFN in at least one time point compared to IFN-α2a or PEG-IFN in PH5CH8 cells. Cells were cultured in the presence of media alone (CTRL) or media containing the Cmax of each IFN for 4, 8 or 20 hours as indicated, and RNA expression assessed by microarray analysis. Gene expression differences were identified by 2-way ANOVA and a Bonferonni, p<0.05 used as a multiple test correction. The mean fold induction of each gene by each IFN treatment at every time point is listed.