

Abstract
Impairments in cognitive control, such as those involved in working memory, are associated with dysfunction of the dorsolateral prefrontal cortex (DLPFC) in individuals with schizophrenia. This dysfunction appears to result, at least in part, from abnormalities in GABA-mediated neurotransmission. In this paper, we review recent findings indicating that the altered DLPFC circuitry in subjects with schizophrenia reflects changes in the expression of genes that encode selective presynaptic and postsynaptic components of GABA neurotransmission. Specifically, using a combination of methods, we found that subjects with schizophrenia exhibited expression deficits in GABA-related transcripts encoding presynaptic regulators of GABA neurotransmission, neuropeptide markers of specific subpopulations of GABA neurons, and certain subunits of the GABAA receptor. In particular, alterations in the expression of the neuropeptide somatostatin suggested that GABA neurotransmission is impaired in the Martinotti subset of GABA neurons that target the dendrites of pyramidal cells. In contrast, none of the GABA-related transcripts assessed to date were altered in the DLPFC of monkeys chronically exposed to antipsychotic medications, suggesting that the effects observed in the human studies reflect the disease process and not its treatment. In concert with previous findings, these data suggest that working memory dysfunction in schizophrenia may be attributable to altered GABA neurotransmission in specific DLPFC microcircuits.
Keywords: Postmortem, Neuropeptides, Somatostatin, GABAA receptor
Introduction
Although psychosis is usually the presenting and most striking clinical feature of schizophrenia, disturbances in cognition have been regarded as central to the illness since its initial description as dementia praecox. These impairments are thought to be the core features of the illness (Elvevåg and Goldberg, 2000) for the following reasons. First, a characteristic pattern of cognitive deficits occurs with high frequency, is relatively stable over time, and is independent of the psychotic symptoms of the illness (Gold, 2004). Indeed, if cognitive deficits are defined as the failure to achieve the expected level of cognitive functioning, then almost all patients with schizophrenia are cognitively impaired (Keefe et al., 2005). Second, cognitive abnormalities have been found throughout the life span of affected individuals, including during childhood and adolescence and at the initial onset of psychosis (Saykin et al., 1994; Green, 1998; Davidson et al., 1999; Cosway et al., 2000). Third, the unaffected relatives of individuals with schizophrenia also exhibit similar, although milder, cognitive deficits (Egan et al., 2001; Sitskoorn et al., 2004). Finally, the degree of cognitive dysfunction is the best predictor of long-term functional outcome for affected individuals (Green, 1996; Harvey et al., 1998). Thus, the development and implementation of effective treatments for cognitive deficits remains a major goal in schizophrenia research (Hyman and Fenton, 2003).
Of the demonstrated types of cognitive impairments in schizophrenia, substantial research has focused on working memory, typically defined as the ability to transiently maintain and manipulate a limited amount of information in order to guide thought or behavior (Miller and Cohen, 2001). Working memory involves several component processes including storage buffers for different domains of information [e.g., visuo-spatial scratch pad (short-term storage of visual information) and phonological loop (articulatory rehearsal and phonological coding of verbal information)] and a central executive component that controls the manipulation of information within the storage buffers (Baddeley, 1992). Although more work is needed to understand the functional integrity of all working memory components in schizophrenia (e.g., visuo-spatial scratchpad and episodic buffer), research in this area suggests two robust conclusions. First, individuals with schizophrenia exhibit relatively little impairment on tasks that depend primarily on the phonological loop and have intact activation of the brain regions (e.g., ventral lateral prefrontal cortex and posterior parietal cortex) that mediate the components of the phonological loop (Barch, 2006). Second, in contrast, measures of central executive function or cognitive control, especially the manipulation of transiently stored information, are clearly disturbed in subjects with schizophrenia (Cannon et al., 2005; Tan et al., 2005). Thus, the widely-reported alterations in working memory function in schizophrenia may be considered representative of deficits in a larger class of cognitive control processes.
Working memory impairments in schizophrenia are accompanied by altered activation of the dorsolateral prefrontal cortex (DLPFC), a brain region known to be associated with cognitive control (Goldman-Rakic, 1999; Weinberger et al., 2001; Callicott et al., 2003). For example, under appropriate conditions of cognitive activation, decreased blood flow or glucose utilization in the DLPFC appears to be a robust finding in schizophrenia (Weinberger et al., 1986; 1988; Taylor, 1996), although these disturbances are less reliably found in the resting state (Buchsbaum, 1990; Andreasen et al., 1992; Gur and Gur, 1995). Most interpretations of these results converge on the idea that DLPFC dysfunction in schizophrenia is task-specific and related to working memory impairment or cognitive control (Goldman-Rakic, 1987; Carter et al., 1998; Smith and Jonides, 1999). Studies examining neural activity using fMRI indicate that during working memory tasks, subjects with schizophrenia exhibit an altered relationship between working memory load, behavioral performance and DLPFC activation (Van Snellenberg et al., 2006). Although a direct experimental demonstration is still needed, the available data are consistent with the hypothesis that subjects with schizophrenia exhibit a left-shifted, inverted-U function between task difficulty and DLPFC activation (Callicott et al., 2003; Manoach, 2003). That is, schizophrenia is not associated with a simple increase or decrease in the degree of DLPFC activation while performing a working memory task; instead, subjects with schizophrenia exhibit greater DLPFC activation at relatively low working memory loads and relatively intact performance, but reduced activation at relatively high loads and impaired performance, perhaps reflecting differences in how subjects manage the demands of the task.
Several findings support the relevance of these alterations in DLPFC function to the disease process of schizophrenia. First, subjects with other psychotic disorders (MacDonald et al., 2005) or major depression (Barch et al., 2003) show normal activation of the DLPFC when performing working memory tasks, which indicates that the abnormalities observed in schizophrenia are, at least in part, specific to the clinical syndrome of schizophrenia. Second, deficits in activation of the DLPFC, but not of other cortical regions, during working memory tasks predict the severity of cognitive disorganization symptoms in subjects with schizophrenia (Perlstein et al., 2001). Third, reduced working memory capacity has been suggested to be rate limiting in the performance of other cognitive tasks in schizophrenia (Silver et al., 2003). Thus, working memory deficits seem to be a central feature of schizophrenia, and identifying the neural alterations in the DLPFC that produce these functional alterations is essential for understanding the underlying disease process.
GABA Neurotransmission and DLPFC Dysfunction in Schizophrenia
The dysfunction of the DLPFC in schizophrenia could reflect, at least in part, disturbances in inhibitory circuitry mediated by GABA-containing interneurons (Lewis et al., 2005) since studies in monkeys indicate that normal working memory function depends upon GABA-mediated circuitry in the DLPFC (Sawaguchi et al., 1989; Rao et al., 2000). The idea that alterations in GABA neurotransmission might contribute to cortical dysfunction in schizophrenia was initially suggested by findings of decreased glutamic acid decarboxylase (GAD) activity (Bird et al., 1979) and decreased GABA reuptake (Simpson et al., 1989) in postmortem studies. Over the past decade, studies across multiple labs, using DNA microarray, real-time quantitative PCR or in situ hybridization, have consistently found reduced levels of the transcript for the 67 kilodalton isoform of glutamic acid decarboxylase (GAD67), the principal synthesizing enzyme for GABA, in the DLPFC of subjects with schizophrenia (Akbarian et al., 1995; Guidotti et al., 2000; Mirnics et al., 2000; Volk et al., 2000; Vawter et al., 2002; Hashimoto et al., 2005; 2008; Straub et al., 2007). Indeed, reduced GAD67 mRNA expression in the DLPFC is perhaps the most widely and consistently replicated pathological disturbance in schizophrenia (Torrey et al., 2005; Akbarian and Huang, 2006). The deficit in GAD67 mRNA seems to be accompanied by a corresponding decrease in the cognate protein, although this has been less extensively studied (Guidotti et al., 2000). In contrast, cortical mRNA and protein levels of the other synthesizing enzyme for GABA, GAD65, are not changed in schizophrenia (Guidotti et al., 2000), nor is the density of GAD65-immunoreactive axon terminals (Benes et al., 2000). GAD67 accounts for the majority of GABA synthesis in the cortex (Mason et al., 2001; Battaglioli et al., 2003), whereas GAD65 contributes to GABA synthesis only under conditions of high synaptic demand (Battaglioli et al., 2003; Patel et al., 2006). Consistent with these findings, elimination of the GAD65 gene in mice does not alter cortical levels of GABA (Asada et al., 1996), whereas reductions in GAD67 mRNA are associated with marked decreases in cortical GAD activity and GABA content (Asada et al., 1997). In addition, levels of the mRNA for the GABA membrane transporter (GAT1), a protein responsible for reuptake of released GABA into nerve terminals, is also decreased in the DLPFC of subjects with schizophrenia (Ohnuma et al., 1999; Volk et al., 2001; Hashimoto et al., 2008). In concert, these findings suggest that schizophrenia is accompanied by reductions in both the synthesis and re-uptake of cortical GABA.
As in other cortical regions and species (McBain and Fisahn, 2001; Markram et al., 2004), GABA neurons in the primate DLPFC comprise subclasses that can be distinguished on the basis of molecular, electrophysiological and anatomical properties. For example, the calcium-binding proteins, parvalbumin (PV) and calretinin (CR), and the neuropeptide somatostatin (SST) are, with a few exceptions, expressed in separate populations of primate cortical GABA neurons (Condé et al., 1994; Gabbott and Bacon, 1996; DeFelipe, 1997). These subtypes tend to exhibit different membrane firing properties (Kawaguchi and Kubota, 1993; Krimer et al., 2005; Zaitsev et al., 2005) and to have axons with different arborization patterns and synaptic targets (DeFelipe, 1997). For example, the axon terminals of fast-spiking, PV-containing chandelier and basket neurons principally target the axon initial segments and cell body/proximal dendrites, respectively, of pyramidal neurons (Williams et al., 1992; Melchitzky et al., 1999), the low-threshold spiking, SST-containing Martinotti cells innervate the distal dendrites of pyramidal neurons (DeLima and Morrison, 1989; Kawaguchi and Kubota, 1996; Ma et al., 2006), and GABA neurons that express CR (Condé et al., 1994; Gabbott and Bacon, 1996) have a regular-spiking, adaptive firing pattern (Kawaguchi, 1995; Zaitsev et al., 2005) and give rise to axon terminals that target other GABA neurons or the dendritic spines and shafts of pyramidal cells (Melchitzky et al., 2005). Thus, the functional consequences of a deficit in GAD67 in schizophrenia depend on the specific subpopulations of GABA neurons that are affected.
At the cellular level, the density of neurons with detectable levels of GAD67 mRNA was significantly decreased in schizophrenia subjects (Akbarian et al., 1995; Volk et al., 2000), whereas in neurons with detectable levels of GAD67 mRNA, the expression level per neuron did not differ from control values (Volk et al., 2000). These observations suggest that the majority of DLPFC GABA neurons express normal levels of GAD67 mRNA in subjects with schizophrenia, but approximately 25-35% of GABA neurons lack detectable levels of this transcript. Levels of PV mRNA, which is expressed in ∼25% of primate DLPFC GABA neurons (Condé et al., 1994), were significantly decreased in layers 3 and 4, but not in layers 2 or 5-6, of the DLPFC in subjects with schizophrenia (Hashimoto et al., 2003). The expression of PV mRNA per neuron was significantly decreased, but neither the density of neurons with detectable levels of PV mRNA nor the density of PV-immunoreactive neurons (Woo et al., 1997; Beasley et al., 2002) was changed in subjects with schizophrenia. These findings indicate that the neurons are still present and suggest that GAD67 mRNA expression is markedly reduced in PV-expressing neurons that also have reduced, but still detectable, levels of PV mRNA. This interpretation was confirmed by dual label in situ hybridization studies which found that ∼50% of PV mRNA+ neurons lacked detectable levels of GAD67 mRNA in the DLPFC of subjects with schizophrenia (Hashimoto et al., 2003). In contrast, neither the expression of calretinin (CR) mRNA (which is found in ∼50% of primate DLPFC GABA neurons (Condé et al., 1994)), the density of CR-immunoreactive neurons, nor the density of CR-immunoreactive axon terminals was changed in subjects with schizophrenia (Woo et al., 1997; Cotter et al., 2002; Hashimoto et al., 2003). These cell type-specific differences have been suggested to be a consequence of hypofunction of the NMDA receptor on PV neurons in schizophrenia (Lisman et al., 2008), but other findings suggest that the effects of NMDA receptor antagonists on the phenotype of cortical PV neurons are more likely to be mediated by NMDA receptors on other neurons (Gonzalez-Burgos and Lewis, 2008). In either case, it is important to note that abnormalities in PV neurons alone might not completely account for the deficits in expression of GAD67 and GAT1 mRNAs, as such changes were also observed in cortical layers 1, 2 and 5, where relatively few PV-expressing GABA neurons are located (Condé et al., 1994), and where no changes in the expression of PV mRNA were found (Hashimoto et al., 2003).
In addition, the levels of GABAA receptors in the DLPFC also appear to be abnormal in subjects with schizophrenia. For example, increased musci-mol-binding in pyramidal neuron cell bodies (Hanada et al., 1987; Benes et al., 1996) and increased GABAA receptor α2 subunits in the axon initial segments of pyramidal neurons (Volk et al., 2002) might represent compensatory receptor upregulation in response to decreased GABA release from GABA neurons, especially those that express PV (Lewis et al., 2005). However, the reports of decreased mRNA levels for the GABAA receptor γ2 and δ subunits (Huntsman et al., 1998; Vawter et al., 2002) suggest that the downregulation of GABAA receptors containing these subunits might also contribute to disturbances in DLPFC inhibitory circuitry in schizophrenia.
Alterations in Markers of GABA Neurotransmission are Cell and Receptor Subtype Specific
Together, the findings reviewed above suggested that the working memory impairments and DLPFC dysfunction in schizophrenia might reflect shifts in the expression of genes that encode selective presynaptic and postsynaptic components of GABA neurotransmission, but that the extent of the abnormalities might be broader than those described above. Consistent with this hypothesis, we found selective transcript alterations in the DLPFC of subjects with schizophrenia in a study of 80 GABA-related transcripts using a customized DNA microarray platform with enhanced sensitivity (Hashimoto et al., 2008). Specifically, we found significant expression deficits of GABA-related mRNAs whose protein products can be classified into three groups: 1) presynaptic regulators of GABA neurotransmission (GAD67 and GAT1), 2) neuropeptides [somatostatin (SST), neuropeptide Y (NPY) and cholecystokinin (CCK)], and 3) GABAA receptor subunits (α1, α4, β3, γ2 and δ). These differences by microarray were confirmed by parallel studies using real-time qPCR and/or in situ hybridization in the same subjects.
These gene expression deficits appear to reflect the disease process of schizophrenia, or at least not to be the consequence of other factors commonly associated with the illness, such as treatment with antipsychotic medications. For example, the expression of GAD67, GAT1 and SST mRNAs was unaltered in the DLPFC of monkeys chronically exposed to haloperidol and benztropine (Volk et al., 2000; 2001; Morris et al., 2008); long-term exposure of monkeys to typical (haloperidol) or atypical (olanzapine) antipsychotics did not alter mRNA levels for GAD67, SST, or GABAA α1 subunit in the DLPFC (Hashimoto et al., 2008); and subjects with schizophrenia who were not receiving antipsychotics at the time of death all showed decreased expression for the 10 GABA-related transcripts mentioned above (Hashimoto et al., 2008). Furthermore, the deficits in expression of these GABA-related transcripts is unlikely to be due solely to substances that influence GABA neurotransmission, such as alcohol, benzodiazepines, and valproic acid, because we did not observe a significant difference in expression changes for any transcripts between the subject pairs with or without a comorbid alcohol disorder or those with or without the use of benzodiazepines and/or valproic acid in the schizophrenia subjects at the time of death (Hashimoto et al., 2008).
The within subject pair differences in transcript levels were significantly correlated between GAD67 and SST (r=0.71, P <0.005), GAD67 and NPY (r=0.72, P <0.004), and SST and NPY (r=0.81, P <0.001), suggesting that GAD67 mRNA expression is also decreased in the subset of GABA neurons that express both SST and NPY (Hendry et al., 1984; Kubota et al., 1994). Thus, the presence of SST- and NPY-containing neurons predominantly in layers 2 and 6 (Hendry et al., 1984; Kubota et al., 1994) may account for the deficits in GAD67 mRNA expression in these layers that cannot be explained by the expression deficits in PV-containing neurons.
The idea that the deficit in SST mRNA expression is restricted to a subset of those neurons was supported by in situ hybridization studies which revealed that SST mRNA levels were significantly lower in DLPFC layers 2 - superficial 6 of subjects with schizophrenia, but not in layer 1, deep layer 6 or the underlying white matter (Morris et al., 2008). Interestingly, this pattern corresponds to differences in developmental birthdates of subsets of SST neurons. Specifically, the proliferation of early germinal zones over successive rounds of cell division during cortical development gives rise to post-mitotic migratory neurons. The earliest generated cells comprise the preplate which, later in development, is split into the marginal zone (adult layer 1) and the subplate (adult deep layer 6 and superficial white matter) by the later born neurons of the cortical plate (adult layers 2 - superficial 6) (Kostovic and Rakic, 1980; Luskin and Shatz, 1985; Chun and Shatz, 1989; Bayer and Altman, 1990). Thus, it appears that the subpopulation of the early generated preplate neurons that expresses SST (Chun and Shatz, 1989) and resides in layer 1, deep layer 6 and the superficial white matter are not affected in schizophrenia, whereas the later developing cortical plate neurons that express SST mRNA are affected.
Given that the cortical plate forms during the second trimester of gestation, these findings raise the possibility that the alterations in SST neurons reflect the effect of adverse environmental events during that time frame [e.g., maternal influenza (Brown, 2006)] that have been associated with an increased risk for schizophrenia. However, because early developmental insults appear to be neither necessary nor sufficient in the etiology of schizophrenia (Lewis and Levitt, 2002), other factors must also be contributory. For example, both SST- and PV-containing cortical GABA neurons are affected in schizophrenia, whereas those that contain CR are not. Thus, factors shared by SST and PV neurons but that differ from CR neurons [e.g., place and timing of neuron birth, transcription factors regulating cell fate, etc; see Wonders and Anderson (2006) for review] might also contribute to cell type-specific vulnerability. Of course, other features intrinsic to, or associated with, the connectivity of adult SST and PV cortical neurons might contribute to their greater vulnerability relative to other SST neurons. For example, approximately 50% of SST neurons, and many PV neurons, express trkB (Gorba and Wahle, 1999), the principal receptor of BDNF, whereas CR neurons do not. Interestingly, mice with genetically engineered reductions in the expression of trkB mRNA have significantly lower levels of cortical SST, PV and GAD67 mRNAs, but no change in CR mRNA expression. Given that the expression level of trkB mRNA (Hashimoto et al., 2005; Weickert et al., 2005) is reduced in the DLPFC of subjects with schizophrenia, reduced neurotrophin signaling through the trkB receptor might be an “upstream” event that contributes to reduced expression of SST, PV and GAD67 mRNAs in the illness.
Interneurons that contain both SST and NPY include the Martinotti cells that give rise to axons which project to layer 1 where they synapse on the apical dendrites of pyramidal neurons (Kawaguchi and Kubota, 1997; Reyes et al., 1998; Gibson et al., 1999; Ma et al., 2006). Thus, disturbances in the SST/NPY-containing Martinotti class of GABA neurons could contribute to the dysfunction of DLPFC circuitry associated with working memory impairments in schizophrenia. For example, disturbances in sensory-gating in schizophrenia have been correlated with impaired working memory performance (Silver and Feldman, 2005), consistent with the idea suggesting that the inability to filter distracting stimuli disrupts working memory (Miller and Cohen, 2001). In computational models, GABA neurons that target pyramidal neuron dendrites protect against distracting stimuli by boosting the inhibition on dendrites of nearby pyramidal neurons that are selective for other stimuli (Wang et al., 2004). Similarly, experimental studies have shown that high frequency trains from pyramidal neurons produce facilitating excitatory inputs to Martinotti cells that, by virtue of their synapses onto the dendrites of neighboring pyramidal neurons, cause disynaptic inhibition (Silberberg and Markram, 2007). Together, these findings suggest that Martinotti cells, by mediating the disynaptic inhibition of neighboring pyramidal neurons selective for other stimuli, can filter distracting stimuli during working memory tasks. Consequently, dysfunction of SST/NPY-containing Martinotti cells in schizophrenia could contribute to working memory impairments.
CCK is heavily expressed in GABA neurons that do not contain PV or SST (Lund and Lewis, 1993; Kawaguchi and Kondo, 2002), but that do express the cannabinoid 1 (CB1) receptor. The highly correlated within subject pair expression differences between CCK and GAD67 (r=0.84, P <0.001) suggest a deficit in GABA synthesis in CCK-containing GABA neurons. Interestingly, levels of CB1 receptor mRNA and protein were also reduced in the same subjects with schizophrenia, suggesting that the down regulation of this receptor, whose activation powerfully suppresses GABA release, could partially compensate for a deficit in GABA synthesis (Eggan et al., 2008). The axon terminals of CCK-positive basket neurons converge with those from PV-containing neurons on the perisomatic domain of pyramidal neurons (Kawaguchi and Kondo, 2002). Interestingly, recent studies indicate that CCK enhances the output of PV-containing basket neurons, while concurrently suppressing GABA release from CCK-containing basket neurons (Foldy et al., 2007; Karson et al., 2008). Thus, alterations in CCK may alter the balance between these two sources of perisomatic inputs to pyramidal neurons (Freund and Katona, 2007) and contribute to alterations in the neural synchrony in the DLPFC that are associated with normal working memory function (Lewis et al., 2005).
The deficits in expression of specific subunits of GABAA receptors suggest that both phasic and tonic inhibition may be altered in the DLPFC of subjects with schizophrenia. On the other hand, no changes in transcripts for GABAB receptors were observed, although a reduction in cortical GABAB receptor 1 protein levels in schizophrenia have been reported (Ishikawa et al., 2005). The α1 and γ2 GABAA subunits co-assemble into receptors that are enriched in postsynaptic sites where they mediate phasic inhibition (Nusser et al., 1998; Mangan et al., 2005). On the other hand, GABAA receptors containing the δ subunit are selectively localized to extrasynaptic sites (Nusser et al., 1998; Wei et al., 2003; Mangan et al., 2005) where, by virtue of their high affinity for GABA, they mediate the tonic inhibition provided by ambient levels of GABA in extracellular space (Wei et al., 2003; Farrant and Nusser, 2005). Interestingly, the within subject pair expression differences were significantly correlated between α1 and γ2 (r=0.84, P <0.001), α1 and δ (r=0.81, P <0.001), and γ2 and δ (r=0.85, P <0.001) mRNAs (Hashimoto et al., 2008). Given the predominant localization of the α1, γ2, and δ subunits to dendrites (Hendry et al., 1994; Fritschy and Mohler, 1995), the highly correlated expression deficits for these transcripts suggest coordinated downregulation of GABAA receptors mediating phasic and tonic inhibition in the dendritic domain of DLPFC pyramidal neurons in schizophrenia. In contrast, the differences in tissue levels of these subunit mRNAs were not correlated with any of the presynaptic transcripts that were altered in schizophrenia (Hashimoto et al., 2008), suggesting that distinct processes contribute to these abnormalities. Consistent with this hypothesis, variants in the GABAA receptor α1 subunit gene have been associated with both schizophrenia and altered expression levels of the α1 subunit mRNA (Petryshen et al., 2005). Furthermore, a targeted deletion of the GABAA receptor α1 subunit gene in mice resulted in altered cortical expression of other GABAA receptor subunits, such as increased α2 and decreased γ2 protein levels, as well as decreased levels of SST mRNA (Kralic et al., 2002; Ponomarev et al., 2006). Together, these findings suggest that the deficit in α1 subunit expression could be a causal process for at least some of the GABA-related gene expression changes in the DLPFC of subjects with schizophrenia.
Conclusion
Although the functional significance of these alterations in GABA-related transcript levels depends on the extent to which they are translated into changes at the level of the cognate proteins, these findings inform our understanding of the nature and extent of altered GABA neurotransmission in the DLPFC of subjects with schizophrenia (FIG. 1). The deficits in expression of GAD67, GAT1 and PV mRNAs in PV-containing GABA neurons in the DLPFC of subjects with schizophrenia (Volk et al., 2000; 2001; Hashimoto et al., 2003) are associated with lower levels of GAT1 protein in the presynaptic terminals of PV-containing chandelier neurons (Pierri et al., 1999) and the upregulation of GABAA receptor α2 subunit in the postsynaptic axon initial segments of pyramidal neurons (Volk et al., 2002). Although the CR-containing GABA neurons appear to be unaffected, alterations in GABA neurotransmission do appear to be present in SST/NPY-containing Martinotti cells and CCK-containing basket neurons, which predominately target the distal dendrites and cell bodies of pyramidal neurons, respectively. Furthermore, gene expression deficits for α1 and γ2 GABAA receptor subunits and for δ and α4 subunits suggest decreased synaptic (phasic) and extrasynaptic (tonic) inhibition, respectively, in pyramidal neuron dendrites. GABA-mediated regulation at the dendritic domain of pyramidal neurons is important for the selection and integration of excitatory inputs from different cortical and subcortical areas, whereas GABA inputs at the perisomatic domain, including the axon initial segment and cell body, are critical for control of the timing and synchronization of pyramidal neuron firing (Markram et al., 2004; Somogyi and Klausberger, 2005; Freund and Katona, 2007). Therefore, the findings summarized in FIG. 1 suggest altered GABA-mediated regulation of both inputs to and outputs from DLPFC pyramidal neurons in subjects with schizophrenia. These alterations are certain to affect information processing in DLPFC circuitry and thus are likely to be major contributors to the working memory impairments in this illness. Understanding the interactions between, and the pathophysiological consequences of, these alterations in markers of GABA neurotransmission may lead to the identification of new drug targets for improving the cognitive deficits of schizophrenia.
FIGURE 1.
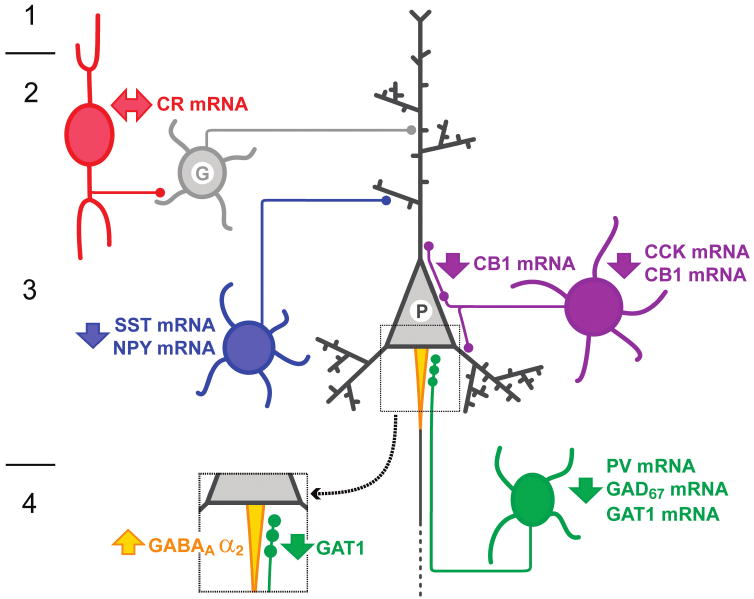
Schematic summary of some alterations in GABA-mediated circuitry in the DLPFC of subjects with schizophrenia. Altered GABA neurotransmission by PV-containing neurons (green) is indicated by gene expression deficits in these neurons and associated changes in their synapses, a decrease in GAT1 expression in their terminals and an upregulation of GABAA receptor α2 subunit at the axon initial segments of pyramidal neurons (enlarged square). Decreased expression of both SST and NPY mRNAs indicates alterations in SST and/or NPY-containing neurons (blue) that target the distal dendrites of pyramidal neurons. Decreased CCK mRNA levels, and CB1 receptor mRNA and protein levels, indicate an alteration of CCK-containing large basket neurons (purple) that represent a separate source of perisomatic inhibition from PV-containing neurons. Gene expression in CR-containing GABA neurons (red) does not seem to be altered. These changes appear to be accompanied by a downregulation of GABAA receptor subunits, including the α1 and α2 subunits present in receptors that mediate synaptic (phasic) inhibition and the α4 and δ subunits present in receptors that mediate extrasynaptic (tonic) inhibition. G, generic GABA neuron; P, pyramidal neuron; I-IV, layers of the DLPFC.
Acknowledgments
Cited studies conducted by the authors were supported by NIH grants MH43784 and MH45156, by an investigator-initiated grant from Eli Lilly, and by a NARSAD Young Investigator Award.
References
- Akbarian S, Huang HS. Molecular and cellular mechanisms of altered GAD1/GAD67 expression in schizophrenia and related disorders. Brain Res Rev. 2006;52:293–304. doi: 10.1016/j.brainresrev.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Akbarian S, Kim JJ, Potkin SG, Hagman JO, Tafazzoli A, Bunney WE, Jr, Jones EG. Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Arch Gen Psychiatry. 1995;52:258–266. doi: 10.1001/archpsyc.1995.03950160008002. [DOI] [PubMed] [Google Scholar]
- Andreasen NC, Rezai R, Alliger R, Swayze VW, II, Flaum M, Kirchner P, Cohen G, O'Leary DS. Hypofrontality in neuroleptic-naive patients and in patients with chronic schizophrenia: assessment with xenon 133 single-photon emission computed tomography and the Tower of London. Arch Gen Psychiatry. 1992;49:943–958. doi: 10.1001/archpsyc.1992.01820120031006. [DOI] [PubMed] [Google Scholar]
- Asada H, Kawamura Y, Maruyama K, Kume H, Ding R, Ji FY, Kanbara N, Kuzume H, Sanbo M, Yagi T, Obata K. Mice lacking the 65 kDa isoform of glutamic acid decarboxylase (GAD65) maintain normal levels of GAD67 and GABA in their brains but are susceptible to seizures. Biochem Biophys Res Comm. 1996;229:891–895. doi: 10.1006/bbrc.1996.1898. [DOI] [PubMed] [Google Scholar]
- Asada H, Kawamura Y, Maruyama K, Kume H, Ding R, Kanbara N, Kuzume H, Sanbo M, Yagi T, Obata K. Cleft palate and decreased brain γ-aminobutyric acid in mice lacking the 67-kDa isoform of glutamic acid decarboxylase. Proc Natl Acad Sci USA. 1997;94:6496–6499. doi: 10.1073/pnas.94.12.6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baddeley A. Working memory. Science. 1992;255:556–559. doi: 10.1126/science.1736359. [DOI] [PubMed] [Google Scholar]
- Barch DM. What can research on schizophrenia tell us about the cognitive neuroscience of working memory? Neuroscience. 2006;139:73–84. doi: 10.1016/j.neuroscience.2005.09.013. [DOI] [PubMed] [Google Scholar]
- Barch DM, Sheline YI, Csernansky JG, Snyder AZ. Working memory and prefrontal cortex dysfunction: specificity to schizophrenia compared with major depression. Biol Psychiatry. 2003;53:376–384. doi: 10.1016/s0006-3223(02)01674-8. [DOI] [PubMed] [Google Scholar]
- Battaglioli G, Liu H, Martin DL. Kinetic differences between the isoforms of glutamate decarboxylase: implications for the regulation of GABA synthesis. J Neurochem. 2003;86:879–887. doi: 10.1046/j.1471-4159.2003.01910.x. [DOI] [PubMed] [Google Scholar]
- Bayer SA, Altman J. Development of layer I and the subplate in the rat neocortex. Exp Neurol. 1990;107:48–62. doi: 10.1016/0014-4886(90)90062-w. [DOI] [PubMed] [Google Scholar]
- Beasley CL, Zhang ZJ, Patten I, Reynolds GP. Selective deficits in prefrontal cortical GABAergic neurons in schizophrenia defined by the presence of calcium-binding proteins. Biol Psychiatry. 2002;52:708–715. doi: 10.1016/s0006-3223(02)01360-4. [DOI] [PubMed] [Google Scholar]
- Benes FM, Vincent SL, Marie A, Khan Y. Up-regulation of GABA-A receptor binding on neurons of the prefrontal cortex in schizophrenic subjects. Neuroscience. 1996;75:1021–1031. doi: 10.1016/0306-4522(96)00328-4. [DOI] [PubMed] [Google Scholar]
- Benes FM, Todtenkopf MS, Logiotatos P, Williams M. Glutamate decarboxylase(65)-immunoreactive terminals in cingulate and prefrontal cortices of schizophrenic and bipolar brain. J Chem Neuroanat. 2000;20:259–269. doi: 10.1016/s0891-0618(00)00105-8. [DOI] [PubMed] [Google Scholar]
- Bird ED, Spokes EGS, Iversen LL. Increased dopamine concentration in limbic areas of brain from patients dying with schizophrenia. Brain. 1979;102:347–360. doi: 10.1093/brain/102.2.347. [DOI] [PubMed] [Google Scholar]
- Brown AS. Prenatal infection as a risk factor for schizophrenia. Schizophr Bull. 2006;32:200–202. doi: 10.1093/schbul/sbj052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchsbaum M. The frontal lobes, basal ganglia and temporal lobes as sites for schizophrenia. Schizophr Bull. 1990;16:379–389. doi: 10.1093/schbul/16.3.379. [DOI] [PubMed] [Google Scholar]
- Callicott JH, Mattay VS, Verchinski BA, Marenco S, Egan MF, Weinberger DR. Complexity of prefrontal cortical dysfunction in schizophrenia: more than up or down. Am J Psychiatry. 2003;160:2209–2215. doi: 10.1176/appi.ajp.160.12.2209. [DOI] [PubMed] [Google Scholar]
- Cannon TD, Glahn DC, Kim J, Van Erp TG, Karlsgodt K, Cohen MS, Nuechterlein KH, Bava S, Shirinyan D. Dorsolateral prefrontal cortex activity during maintenance and manipulation of information in working memory in patients with schizophrenia. Arch Gen Psychiatry. 2005;62:1071–1080. doi: 10.1001/archpsyc.62.10.1071. [DOI] [PubMed] [Google Scholar]
- Carter CS, Perlstein W, Ganguli R, Brar J, Mintun M, Cohen JD. Functional hypofrontality and working memory dysfunction in schizophrenia. Am J Psychiatry. 1998;155:1285–1287. doi: 10.1176/ajp.155.9.1285. [DOI] [PubMed] [Google Scholar]
- Chun JJM, Shatz CJ. Interstitial cells of the adult neocortical white matter are the remnant of the early generated subplate neuron population. J Comp Neurol. 1989;282:555–569. doi: 10.1002/cne.902820407. [DOI] [PubMed] [Google Scholar]
- Condé F, Lund JS, Jacobowitz DM, Baimbridge KG, Lewis DA. Local circuit neurons immunoreactive for calretinin, calbindin D-28k, or parvalbumin in monkey prefrontal cortex: distribution and morphology. J Comp Neurol. 1994;341:95–116. doi: 10.1002/cne.903410109. [DOI] [PubMed] [Google Scholar]
- Cosway R, Byrne M, Clafferty R, Hodges A, Grant E, Abukmeil SS, Lawrie SM, Miller P, Johnstone EC. Neuropsychological change in young people at high risk for schizophrenia: results from the first two neuropsychological assessments of the Edinburgh High Risk Study. Psychol Med. 2000;30:1111–1121. doi: 10.1017/s0033291799002585. [DOI] [PubMed] [Google Scholar]
- Cotter D, Landau S, Beasley C, Stevenson R, Chana G, MacMillan L, Everall I. The density and spatial distribution of GABAergic neurons, labelled using calcium binding proteins, in the anterior cingulate cortex in major depressive disorder, bipolar disorder, and schizophrenia. Biol Psychiatry. 2002;51:377–386. doi: 10.1016/s0006-3223(01)01243-4. [DOI] [PubMed] [Google Scholar]
- Davidson M, Reichenberg A, Rabinowitz J, Weiser M, Kaplan Z, Mark M. Behavioral and intellectual markers for schizophrenia in apparently healthy male adolescents. Am J Psychiatry. 1999;156:1328–1335. doi: 10.1176/ajp.156.9.1328. [DOI] [PubMed] [Google Scholar]
- DeFelipe J. Types of neurons, synaptic connections and chemical characteristics of cells immunoreactive for calbindin-D28K, parvalbumin and calretinin in the neocortex. J Chem Neuroanat. 1997;14:1–19. doi: 10.1016/s0891-0618(97)10013-8. [DOI] [PubMed] [Google Scholar]
- DeLima AD, Morrison JH. Ultrastructural analysis of somatostatin-immunoreactive neurons and synapses in the temporal and occipital cortex of the macaque monkey. J Comp Neurol. 1989;283:212–227. doi: 10.1002/cne.902830205. [DOI] [PubMed] [Google Scholar]
- Egan MF, Goldberg TE, Gscheidle T, Weirich M, Rawlings R, Hyde TM, Bigelow L, Weinberger DR. Relative risk for cognitive impairments in siblings of patients with schizophrenia. Biol Psychiatry. 2001;50:98–107. doi: 10.1016/s0006-3223(01)01133-7. [DOI] [PubMed] [Google Scholar]
- Eggan SM, Hashimoto T, Lewis DA. Reduced cortical cannabinoid 1 receptor mRNA and protein expression in schizophrenia. Arch Gen Psychiatry. 2008;65:772–784. doi: 10.1001/archpsyc.65.7.772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elvevåg B, Goldberg TE. Cognitive impairment in schizophrenia is the core of the disorder. Crit Rev Neurobiol. 2000;14:1–21. [PubMed] [Google Scholar]
- Farrant M, Nusser Z. Variations on an inhibitory theme: phasic and tonic activation of GABAA receptors. Nat Rev Neurosci. 2005;6:215–229. doi: 10.1038/nrn1625. [DOI] [PubMed] [Google Scholar]
- Foldy C, Lee SY, Szabadics J, Neu A, Soltesz I. Cell type-specific gating of perisomatic inhibition by cholecystokinin. Nat Neurosci. 2007;10:1128–1130. doi: 10.1038/nn1952. [DOI] [PubMed] [Google Scholar]
- Freund TF, Katona I. Perisomatic inhibition. Neuron. 2007;56:33–42. doi: 10.1016/j.neuron.2007.09.012. [DOI] [PubMed] [Google Scholar]
- Fritschy JM, Mohler H. GABAA-receptor heterogeneity in the adult rat brain: differential regional and cellular distribution of seven major subunits. J Comp Neurol. 1995;359:154–194. doi: 10.1002/cne.903590111. [DOI] [PubMed] [Google Scholar]
- Gabbott PLA, Bacon SJ. Local circuit neurons in the medial prefrontal cortex (areas 24a,b,c, 25 and 32) in the monkey: II. Quantitative areal and laminar distributions. J Comp Neurol. 1996;364:609–636. doi: 10.1002/(SICI)1096-9861(19960122)364:4<609::AID-CNE2>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Gibson JR, Beierlein M, Connors BW. Two networks of electrically coupled inhibitory neurons in neocortex. Nature. 1999;402:75–79. doi: 10.1038/47035. [DOI] [PubMed] [Google Scholar]
- Gold JM. Cognitive deficits as treatment targets in schizophrenia. Schizophr Res. 2004;72:21–28. doi: 10.1016/j.schres.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS. Circuitry of primate prefrontal cortex and regulation of behavior by representational memory. In: Plum F, Mountcastle V, editors. Handbook of Physiology. American Physiological Society; Bethesda, MD: 1987. pp. 373–417. [Google Scholar]
- Goldman-Rakic PS. The physiology approach: functional architecture of working memory and disordered cognition in schizophrenia. Biol Psychiatry. 1999;46:650–661. doi: 10.1016/s0006-3223(99)00130-4. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Burgos G, Lewis DA. GABA neurons and the mechanisms of network oscillations: implications for understanding cortical dysfunction in schizophrenia. Schizophr Bull. 2008;34:944–961. doi: 10.1093/schbul/sbn070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorba T, Wahle P. Expression of trkB and trkC but not BDNF mRNA in neurochemically identified interneurons in rat visual cortex in vivo and in organotypic cultures. Eur J Neurosci. 1999;11:1179–1190. doi: 10.1046/j.1460-9568.1999.00551.x. [DOI] [PubMed] [Google Scholar]
- Green MF. What are the functional consequences of neurocognitive deficits in schizophrenia? Am J Psychiatry. 1996;153:321–330. doi: 10.1176/ajp.153.3.321. [DOI] [PubMed] [Google Scholar]
- Green MF. Schizophrenia From a Neurocognitive Perspective: Probing the Impenetrable Darkness. Allyn and Bacon; Boston, MA: 1998. [Google Scholar]
- Guidotti A, Auta J, Davis JM, Gerevini VD, Dwivedi Y, Grayson DR, Impagnatiello F, Pandey G, Pesold C, Sharma R, Uzunov D, Costa E. Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder. Arch Gen Psychiatry. 2000;57:1061–1069. doi: 10.1001/archpsyc.57.11.1061. [DOI] [PubMed] [Google Scholar]
- Gur RC, Gur RE. Hypofrontality in schizophrenia: RIP. Lancet. 1995;345:1383–1384. doi: 10.1016/s0140-6736(95)92591-0. [DOI] [PubMed] [Google Scholar]
- Hanada S, Mita T, Nishino N, Tanaka C. [3H] Muscimol binding sites increased in autopsied brains of chronic schizophrenics. Life Sci. 1987;40:239–266. doi: 10.1016/0024-3205(87)90341-9. [DOI] [PubMed] [Google Scholar]
- Harvey PD, Howanitz E, Parrella M, White L, Davidson M, Mohs RC, Hoblyn J, Davis KL. Symptoms, cognitive functioning, and adaptive skills in geriatric patients with lifelong schizophrenia: a comparison across treatment sites. Am J Psychiatry. 1998;155:1080–1086. doi: 10.1176/ajp.155.8.1080. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Volk DW, Eggan SM, Mirnics K, Pierri JN, Sun Z, Sampson AR, Lewis DA. Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. J Neurosci. 2003;23:6315–6326. doi: 10.1523/JNEUROSCI.23-15-06315.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T, Bergen SE, Nguyen QL, Xu B, Monteggia LM, Pierri JN, Sun Z, Sampson AR, Lewis DA. Relationship of brain-derived neurotrophic factor and its receptor TrkB to altered inhibitory prefrontal circuitry in schizophrenia. J Neurosci. 2005;25:372–383. doi: 10.1523/JNEUROSCI.4035-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T, Arion D, Unger T, Maldonado-Aviles JG, Morris HM, Volk DW, Mirnics K, Lewis DA. Alterations in GABA-related transcriptome in the dorsolateral prefrontal cortex of subjects with schizophrenia. Mol Psychiatry. 2008;13:147–161. doi: 10.1038/sj.mp.4002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendry SHC, Jones EG, Emson PC. Morphology, distribution, and synaptic relations of somatostatin- and neuropeptide Y-immunoreactive neurons in rat and monkey neocortex. J Neurosci. 1984;4:2497–2517. doi: 10.1523/JNEUROSCI.04-10-02497.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendry SHC, Huntsman MM, Viñuela A, Mohler H, de Blas AL, Jones EG. GABAA receptor subunit immunoreactivity in primate visual cortex: distribution in macaques and humans and regulation by visual input in adulthood. J Neurosci. 1994;14:2383–2401. doi: 10.1523/JNEUROSCI.14-04-02383.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntsman MM, Tran BV, Potkin SG, Bunney WE, Jones EG. Altered ratios of alternatively spliced long and short γ2 subunit mRNAs of the γ-amino butyrate type A receptor in prefrontal cortex of schizophrenics. Proc Natl Acad Sci USA. 1998;95:15066–15071. doi: 10.1073/pnas.95.25.15066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman SE, Fenton WS. Medicine. What are the right targets for psychopharmacology? Science. 2003;299:350–351. doi: 10.1126/science.1077141. [DOI] [PubMed] [Google Scholar]
- Ishikawa M, Mizukami K, Iwakiri M, Asada T. Immunohistochemical and immunoblot analysis of γ-aminobutyric acid B receptor in the prefrontal cortex of subjects with schizophrenia and bipolar disorder. Neurosci Lett. 2005;383:272–277. doi: 10.1016/j.neulet.2005.04.025. [DOI] [PubMed] [Google Scholar]
- Karson MA, Whittington KC, Alger BE. Cholecystokinin inhibits endocannabinoid-sensitive hippocampal IPSPs and stimulates others. Neuropharmacology. 2008;54:117–128. doi: 10.1016/j.neuropharm.2007.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y. Physiological subgroups of nonpyramidal cells with specific morphological characteristics in layer II/III of rat frontal cortex. J Neurosci. 1995;15:2638–2655. doi: 10.1523/JNEUROSCI.15-04-02638.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, Kondo S. Parvalbumin, somatostatin and cholecystokinin as chemical markers for specific GABAergic interneuron types in the rat frontal cortex. J Neurocytol. 2002;31:277–287. doi: 10.1023/a:1024126110356. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Kubota Y. Correlation of physiological subgroupings of nonpyramidal cells with parvalbumin- and calbindinD28k-immunoreactive neurons in layer V of rat frontal cortex. J Neurophysiol. 1993;70:387–396. doi: 10.1152/jn.1993.70.1.387. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Kubota Y. Physiological and morphological identification of somatostatin- or vasoactive intestinal polypeptide-containing cells among GABAergic cell subtypes in rat frontal cortex. J Neurosci. 1996;16:2701–2715. doi: 10.1523/JNEUROSCI.16-08-02701.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, Kubota Y. GABAergic cell subtypes and their synaptic connections in rat frontal cortex. Cereb Cortex. 1997;7:476–486. doi: 10.1093/cercor/7.6.476. [DOI] [PubMed] [Google Scholar]
- Keefe RS, Eesley CE, Poe MP. Defining a cognitive function decrement in schizophrenia. Biol Psychiatry. 2005;57:688–691. doi: 10.1016/j.biopsych.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Kostovic I, Rakic P. Cytology and the time of origin of interstitial neurons in the white matter in infant and adult human and monkey telencephalon. J Neurocytol. 1980;9:219–242. doi: 10.1007/BF01205159. [DOI] [PubMed] [Google Scholar]
- Kralic JE, Korpi ER, O'Buckley TK, Homanics GE, Morrow AL. Molecular and pharmacological characterization of GABAA receptor alpha1 subunit knockout mice. J Pharmacol Exp Ther. 2002;302:1037–1045. doi: 10.1124/jpet.102.036665. [DOI] [PubMed] [Google Scholar]
- Krimer LS, Zaitsev AV, Czanner G, Kroner S, Gonzalez-Burgos G, Povysheva NV, Iyengar S, Barrionuevo G, Lewis DA. Cluster analysis-based physiological classification and morphological properties of inhibitory neurons in layers 2-3 of monkey dorsolateral prefrontal cortex. J Neurophysiol. 2005;94:3009–3022. doi: 10.1152/jn.00156.2005. [DOI] [PubMed] [Google Scholar]
- Kubota Y, Hattori R, Yui Y. Three distinct subpopulations of GABAergic neurons in rat frontal agranular cortex. Brain Res. 1994;649:159–173. doi: 10.1016/0006-8993(94)91060-x. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Levitt P. Schizophrenia as a disorder of neurodevelopment. Annu Rev Neurosci. 2002;25:409–432. doi: 10.1146/annurev.neuro.25.112701.142754. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci. 2005;6:312–324. doi: 10.1038/nrn1648. [DOI] [PubMed] [Google Scholar]
- Lisman JE, Coyle JT, Green RW, Javitt DC, Benes FM, Heckers S, Grace AA. Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci. 2008;31:234–242. doi: 10.1016/j.tins.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund JS, Lewis DA. Local circuit neurons of developing and mature macaque prefrontal cortex: Golgi and immunocytochemical characteristics. J Comp Neurol. 1993;328:282–312. doi: 10.1002/cne.903280209. [DOI] [PubMed] [Google Scholar]
- Luskin MB, Shatz CJ. Studies of the earliest generated cells of the cat's visual cortex: cogeneration of the subplate and marginal zones. J Neurosci. 1985;5:1062–1075. doi: 10.1523/JNEUROSCI.05-04-01062.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Hu H, Berrebi AS, Mathers PH, Agmon A. Distinct subtypes of somatostatin-containing neocortical interneurons revealed in transgenic mice. J Neurosci. 2006;26:5069–5082. doi: 10.1523/JNEUROSCI.0661-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald AW, III, Carter CS, Kerns JG, Ursu S, Barch DM, Holmes AJ, Stenger VA, Cohen JD. Specificity of prefrontal dysfunction and context processing deficits to schizophrenia in never-medicated patients with first-episode psychosis. Am J Psychiatry. 2005;162:475–484. doi: 10.1176/appi.ajp.162.3.475. [DOI] [PubMed] [Google Scholar]
- Mangan PS, Sun C, Carpenter M, Goodkin HP, Sieghart W, Kapur J. Cultured hippocampal pyramidal neurons express two kinds of GABAA receptors. Mol Pharmacol. 2005;67:775–788. doi: 10.1124/mol.104.007385. [DOI] [PubMed] [Google Scholar]
- Manoach DS. Prefrontal cortex dysfunction during working memory performance in schizophrenia: reconciling discrepant findings. Schizophr Res. 2003;60:285–298. doi: 10.1016/s0920-9964(02)00294-3. [DOI] [PubMed] [Google Scholar]
- Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu C. Interneurons of the neocortical inhibitory system. Nat Rev Neurosci. 2004;5:793–807. doi: 10.1038/nrn1519. [DOI] [PubMed] [Google Scholar]
- Mason GF, Martin DL, Martin SB, Manor D, Sibson NR, Patel A, Rothman DL, Behar KL. Decrease in GABA synthesis rate in rat cortex following GABA-transaminase inhibition correlates with the decrease in GAD(67) protein. Brain Res. 2001;914:81–91. doi: 10.1016/s0006-8993(01)02778-0. [DOI] [PubMed] [Google Scholar]
- McBain CJ, Fisahn A. Interneurons unbound. Nat Rev Neurosci. 2001;2:11–23. doi: 10.1038/35049047. [DOI] [PubMed] [Google Scholar]
- Melchitzky DS, Sesack SR, Lewis DA. Parvalbumin-immunoreactive axon terminals in macaque monkey and human prefrontal cortex: laminar, regional and target specificity of Type I and Type II synapses. J Comp Neurol. 1999;408:11–22. [PubMed] [Google Scholar]
- Melchitzky DS, Eggan SM, Lewis DA. Synaptic targets of calretinin-containing axon terminals in macaque monkey prefrontal cortex. Neuroscience. 2005;130:185–195. doi: 10.1016/j.neuroscience.2004.08.046. [DOI] [PubMed] [Google Scholar]
- Miller EK, Cohen JD. An integrative theory of prefrontal cortex function. Annu Rev Neurosci. 2001;24:167–202. doi: 10.1146/annurev.neuro.24.1.167. [DOI] [PubMed] [Google Scholar]
- Mirnics K, Middleton FA, Marquez A, Lewis DA, Levitt P. Molecular characterization of schizophrenia viewed by microarray analysis of gene expression in prefrontal cortex. Neuron. 2000;28:53–67. doi: 10.1016/s0896-6273(00)00085-4. [DOI] [PubMed] [Google Scholar]
- Morris HM, Hashimoto T, Lewis DA. Alterations in somatostatin mRNA expression in the dorsolateral prefrontal cortex of subjects with schizophrenia or schizoaffective disorder. Cereb Cortex. 2008;18:1575–1587. doi: 10.1093/cercor/bhm186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusser Z, Sieghart W, Somogyi P. Segregation of different GABAA receptors to synaptic and extrasynaptic membranes of cerebellar granule cells. J Neurosci. 1998;18:1693–1703. doi: 10.1523/JNEUROSCI.18-05-01693.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnuma T, Augood SJ, Arai H, McKenna PJ, Emson PC. Measurement of GABAergic parameters in the prefrontal cortex in schizophrenia: focus on GABA content, GABAA receptor α-1 subunit messenger RNA and human GABA transporter-1 (HGAT-1) messenger RNA expression. Neuroscience. 1999;93:441–448. doi: 10.1016/s0306-4522(99)00189-x. [DOI] [PubMed] [Google Scholar]
- Patel AB, de Graaf RA, Martin DL, Battaglioli G, Behar KL. Evidence that GAD65 mediates increased GABA synthesis during intense neuronal activity in vivo. J Neurochem. 2006;97:385–396. doi: 10.1111/j.1471-4159.2006.03741.x. [DOI] [PubMed] [Google Scholar]
- Perlstein WM, Carter CS, Noll DC, Cohen JD. Relation of prefrontal cortex dysfunction to working memory and symptoms in schizophrenia. Am J Psychiatry. 2001;158:1105–1113. doi: 10.1176/appi.ajp.158.7.1105. [DOI] [PubMed] [Google Scholar]
- Petryshen TL, Middleton FA, Tahl AR, Rockwell GN, Purcell S, Aldinger KA, Kirby A, Morley CP, McGann L, Gentile KL, Waggoner SG, Medeiros HM, Carvalho C, Macedo A, Albus M, Maier W, Trixler M, Eichhammer P, Schwab SG, Wildenauer DB, Azevedo MH, Pato MT, Pato CN, Daly MJ, Sklar P. Genetic investigation of chromosome 5q GABAA receptor subunit genes in schizophrenia. Mol Psychiatry. 2005;10:1074–1088. doi: 10.1038/sj.mp.4001739. [DOI] [PubMed] [Google Scholar]
- Pierri JN, Chaudry AS, Woo TU, Lewis DA. Alterations in chandelier neuron axon terminals in the prefrontal cortex of schizophrenic subjects. Am J Psychiatry. 1999;156:1709–1719. doi: 10.1176/ajp.156.11.1709. [DOI] [PubMed] [Google Scholar]
- Ponomarev I, Maiya R, Harnett MT, Schafer GL, Ryabinin AE, Blednov YA, Morikawa H, Boehm SL, Homanics GE, Berman AE, Lodowski KH, Bergeson SE, Harris RA. Transcriptional signatures of cellular plasticity in mice lacking the α1 subunit of GABAA receptors. J Neurosci. 2006;26:5673–5683. doi: 10.1523/JNEUROSCI.0860-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SG, Williams GV, Goldman-Rakic PS. Destruction and creation of spatial tuning by disinhibition: GABAA blockade of prefrontal cortical neurons engaged by working memory. J Neurosci. 2000;20:485–494. doi: 10.1523/JNEUROSCI.20-01-00485.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A, Lujar R, Rozov BN, Somogyi P, Sakman B. Target-cell-specific facilitation and depression in neocortical circuits. Nat Neurosci. 1998;1:279–285. doi: 10.1038/1092. [DOI] [PubMed] [Google Scholar]
- Sawaguchi T, Matsumura M, Kubota K. Delayed response deficits produced by local injection of bicuculline into the dorsolateral prefrontal cortex in Japanese macaque monkeys. Exp Brain Res. 1989;75:457–469. doi: 10.1007/BF00249897. [DOI] [PubMed] [Google Scholar]
- Saykin AJ, Shtasel DL, Gur RE, Kester DB, Mozley LH, Stafiniak P, Gur RC. Neuropsychological deficits in neuroleptic naive patients with first-episode schizophrenia. Arch Gen Psychiatry. 1994;51:124–131. doi: 10.1001/archpsyc.1994.03950020048005. [DOI] [PubMed] [Google Scholar]
- Silberberg G, Markram H. Disynaptic inhibition between neocortical pyramidal cells mediated by Martinotti cells. Neuron. 2007;53:735–746. doi: 10.1016/j.neuron.2007.02.012. [DOI] [PubMed] [Google Scholar]
- Silver H, Feldman P. Evidence for sustained attention and working memory in schizophrenia sharing a common mechanism. J Neuropsychiatry Clin Neurosci. 2005;17:391–398. doi: 10.1176/jnp.17.3.391. [DOI] [PubMed] [Google Scholar]
- Silver H, Feldman P, Bilker W, Gur RC. Working memory deficit as a core neuropsychological dysfunction in schizophrenia. Am J Psychiatry. 2003;160:1809–1816. doi: 10.1176/appi.ajp.160.10.1809. [DOI] [PubMed] [Google Scholar]
- Simpson MDC, Slater P, Deakin JFW, Royston MC, Skan WJ. Reduced GABA uptake sites in the temporal lobe in schizophrenia. Neurosci Lett. 1989;107:211–215. doi: 10.1016/0304-3940(89)90819-7. [DOI] [PubMed] [Google Scholar]
- Sitskoorn MM, Aleman A, Ebisch SJ, Appels MC, Kahn RS. Cognitive deficits in relatives of patients with schizophrenia: a meta-analysis. Schizophr Res. 2004;71:285–295. doi: 10.1016/j.schres.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Smith EE, Jonides J. Storage and executive processes in the frontal lobes. Science. 1999;283:1657–1661. doi: 10.1126/science.283.5408.1657. [DOI] [PubMed] [Google Scholar]
- Somogyi P, Klausberger T. Defined types of cortical interneurone structure space and spike timing in the hippocampus. J Physiol. 2005;562:9–26. doi: 10.1113/jphysiol.2004.078915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub RE, Lipska BK, Egan MF, Goldberg TE, Callicott JH, Mayhew MB, Vakkalanka RK, Kolachana BS, Kleinman JE, Weinberger DR. Allelic variation in GAD1 (GAD67) is associated with schizophrenia and influences cortical function and gene expression. Mol Psychiatry. 2007;12:854–869. doi: 10.1038/sj.mp.4001988. [DOI] [PubMed] [Google Scholar]
- Tan HY, Choo WC, Fones CS, Chee MW. fMRI study of maintenance and manipulation processes within working memory in first-episode schizophrenia. Am J Psychiatry. 2005;162:1849–1858. doi: 10.1176/appi.ajp.162.10.1849. [DOI] [PubMed] [Google Scholar]
- Taylor SF. Cerebral blood flow activation and functional lesions in schizophrenia. Schizophr Res. 1996;19:129–140. doi: 10.1016/0920-9964(95)00000-3. [DOI] [PubMed] [Google Scholar]
- Torrey EF, Barci BM, Webster MJ, Bartko JJ, Meador-Woodruff JH, Knable MB. Neurochemical markers for schizophrenia, bipolar disorder, and major depression in postmortem brains. Biol Psychiatry. 2005;57:252–260. doi: 10.1016/j.biopsych.2004.10.019. [DOI] [PubMed] [Google Scholar]
- Van Snellenberg JX, Torres IJ, Thornton AE. Functional neuroimaging of working memory in schizophrenia: task performance as a moderating variable. Neuropsychology. 2006;20:497–510. doi: 10.1037/0894-4105.20.5.497. [DOI] [PubMed] [Google Scholar]
- Vawter MP, Crook JM, Hyde TM, Kleinman JE, Weinberger DR, Becker KG, Freed WJ. Microarray analysis of gene expression in the prefrontal cortex in schizophrenia: a preliminary study. Schizophr Res. 2002;58:11–20. doi: 10.1016/s0920-9964(01)00377-2. [DOI] [PubMed] [Google Scholar]
- Volk DW, Austin MC, Pierri JN, Sampson AR, Lewis DA. Decreased glutamic acid decarboxylase67 messenger RNA expression in a subset of prefrontal cortical γ-aminobutyric acid neurons in subjects with schizophrenia. Arch Gen Psychiatry. 2000;57:237–245. doi: 10.1001/archpsyc.57.3.237. [DOI] [PubMed] [Google Scholar]
- Volk DW, Austin MC, Pierri JN, Sampson AR, Lewis DA. GABA transporter-1 mRNA in the prefrontal cortex in schizophrenia: decreased expression in a subset of neurons. Am J Psychiatry. 2001;158:256–265. doi: 10.1176/appi.ajp.158.2.256. [DOI] [PubMed] [Google Scholar]
- Volk DW, Pierri JN, Fritschy JM, Auh S, Sampson AR, Lewis DA. Reciprocal alterations in pre- and postsynaptic inhibitory markers at chandelier cell inputs to pyramidal neurons in schizophrenia. Cereb Cortex. 2002;12:1063–1070. doi: 10.1093/cercor/12.10.1063. [DOI] [PubMed] [Google Scholar]
- Wang XJ, Tegner J, Constantinidis C, Goldman-Rakic PS. Division of labor among distinct subtypes of inhibitory neurons in a cortical microcircuit of working memory. Proc Natl Acad Sci USA. 2004;101:1368–1373. doi: 10.1073/pnas.0305337101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Zhang N, Peng Z, Houser CR, Mody I. Perisynaptic localization of delta subunit-containing GABAA receptors and their activation by GABA spillover in the mouse dentate gyrus. J Neurosci. 2003;23:10650–10661. doi: 10.1523/JNEUROSCI.23-33-10650.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weickert CS, Ligons DL, Romanczyk T, Ungaro G, Hyde TM, Herman MM, Weinberger DR, Kleinman JE. Reductions in neurotrophin receptor mRNAs in the prefrontal cortex of patients with schizophrenia. Mol Psychiatry. 2005;10:637–650. doi: 10.1038/sj.mp.4001678. [DOI] [PubMed] [Google Scholar]
- Weinberger DR, Berman KF, Zec RF. Physiologic dysfunction of dorsolateral prefrontal cortex in schizophrenia. I. Regional cerebral blood flow evidence. Arch Gen Psychiatry. 1986;43:114–124. doi: 10.1001/archpsyc.1986.01800020020004. [DOI] [PubMed] [Google Scholar]
- Weinberger DR, Berman KF, Illowsky BP. Physiological dysfunction of dorsolateral prefrontal cortex in schizophrenia: III. A new cohort and evidence for a mono-aminergic mechanism. Arch Gen Psychiatry. 1988;45:609–615. doi: 10.1001/archpsyc.1988.01800310013001. [DOI] [PubMed] [Google Scholar]
- Weinberger DR, Egan MF, Bertolino A, Callicott JH, Mattay VS, Lipska BK, Berman KF, Goldberg TE. Prefrontal neurons and the genetics of schizophrenia. Biol Psychiatry. 2001;50:825–844. doi: 10.1016/s0006-3223(01)01252-5. [DOI] [PubMed] [Google Scholar]
- Williams SM, Goldman-Rakic PS, Leranth C. The synaptology of parvalbumin-immunoreactive neurons in primate prefrontal cortex. J Comp Neurol. 1992;320:353–369. doi: 10.1002/cne.903200307. [DOI] [PubMed] [Google Scholar]
- Wonders CP, Anderson SA. The origin and specification of cortical interneurons. Nat Rev Neurosci. 2006;7:687–696. doi: 10.1038/nrn1954. [DOI] [PubMed] [Google Scholar]
- Woo TU, Miller JL, Lewis DA. Schizophrenia and the parvalbumin-containing class of cortical local circuit neurons. Am J Psychiatry. 1997;154:1013–1015. doi: 10.1176/ajp.154.7.1013. [DOI] [PubMed] [Google Scholar]
- Zaitsev AV, Gonzalez-Burgos G, Povysheva NV, Kroner S, Lewis DA, Krimer LS. Localization of calcium-binding proteins in physiologically and morphologically characterized interneurons of monkey dorsolateral prefrontal cortex. Cereb Cortex. 2005;15:1178–1186. doi: 10.1093/cercor/bhh218. [DOI] [PubMed] [Google Scholar]