

Abstract
The progesterone receptor (PR) is estrogen regulated, and PR levels in breast tumors can be used to predict the success of endocrine therapies targeting the estrogen receptor (ER). Tanaproget is a non-steroidal progestin agonist with very high PR binding affinity and excellent in vivo potency. When appropriately radiolabeled, it might be used to image PR-positive breast tumors non-invasively, by positron emission tomography (PET). We describe the synthesis and PR binding affinities of a series of fluoroalkyl-substituted 6-aryl-1,4-dihydrobenzo[d][1,3]oxazine-2-thiones, analogs of Tanaproget. Some of these compounds have subnanomolar binding affinities, higher than that of either Tanaproget itself or the high affinity PR ligand R5020. Structure-binding affinity relationships can be rationalized by molecular modeling of ligand complexes with PR, and the enantioselectivity of binding has been predicted. These compounds are being further evaluated as potential diagnostic PET imaging agents for breast cancer, and enantiomerically pure materials of defined stereochemistry are being prepared.
Introduction
The progesterone receptor (PR) is an estrogen-regulated protein found in female reproductive tissues and in many breast tumors, and it can serve as a target for various endocrine therapies, including the treatment of the breast cancer.1-6 PR could also be used as a target for diagnostic imaging and radiotherapy of breast cancer by the administration of a suitably radiolabeled PR ligand that accumulates in receptor-positive tumors, where it can be detected and quantified by imaging. Such agents could be used to delineate the PR positivity of tumors in vivo and in situ, in a non-invasive, comprehensive fashion, even in metastatic tumors and lymph nodes that are inaccessible to surgical or needle biopsy. This information can sometimes be used to evaluate tumor aggressiveness and predict the likelihood of responsiveness to endocrine therapies.7-10 In a related manner, a hormone receptor ligand, labeled with a different radionuclide (e.g., an Auger electron emitting isotope) that accumulates in a tumor through a receptor-mediated uptake process (especially one like PR that localizes activity in the nuclear chromatin fraction) could deliver a cytotoxic dose of high linear energy transfer radiation selectively to the tumor cells, ablating the tumor while limiting widespread radiation toxicity. Therefore, the development of PR ligands with potential for both diagnostic imaging and radiotherapy is an area of great current interest.
Diagnostic imaging of steroid receptors in breast tumors by positron emission tomography (PET) is well established and has been achieved using steroids labeled with fluorine-18. The most extensive studies have been done using 16α-[18F]fluoroestradiol (FES) for imaging the estrogen receptor (ER).7-10 A PR-based radioligand, however, would have some advantages over an ER-based one: There is a better correlation between PR status and responsiveness to endocrine therapies than there is with ER status;11-13 a PR-based ligand could be used after the initiation of anti-estrogen hormonal therapy, whereas an ER-based one would not be useful when tumor ER is saturated by the hormonal agent.14 Moreover, PR-based ligands may benefit from the increased PR levels induced by the transient agonistic effect of tamoxifen during the initial course of tamoxifen treatment of breast tumor.15-17 In fact, monitoring an increase in PR levels in a tumor by PET imaging after a brief challenge with an estrogen could form the basis of a new “estrogen challenge test” for assessing the functionality of tumor ER. Such an estrogen challenge test has been done by monitoring changes in tumor uptake of 2-[18F]fluoro-2-deoxyglucose (FDG) either a week after the initiation of tamoxifen therapy7, 8 or a day after a dose of estradiol.10 In both cases, an increase in FDG uptake after the challenge was very highly predictive of ultimate patient benefit from endocrine therapies.7, 8, 10
Studies are currently underway with [18F]fluoro furanyl norprogesterone (FFNP),18 a high affinity steroidal progestin that appears to have promise as an agent for PET imaging of PR. A number of related steroidal progestins labeled with fluorine, bromine, and iodine have also been prepared.19-23 To broaden the types of progestins to be evaluated for PET imaging of breast tumors, we embarked on a program to identify structurally novel, non-steroidal PR imaging agents, because such ligands can also have high receptor binding affinity and good pharmacokinetics, and they often have much less cross reactivity with other steroid receptors.
Tanaproget (1), recently described by Wyeth Pharmaceuticals,24, 25 is a non-steroidal progestin agonist, one of a number of progestins of novel structure that have recently been described.26 1 (Tanaproget) is constituted of two moieties, a 5-cyanopyrrole and a benzoxazin-2-thione, and it is a PR agonist having an in vitro potency equivalent to that of the best steroidal progestins, but superior in selectivity with respect to the other members of the steroid receptor family.24-27 We anticipated that these characteristics might also make 1 a highly effective and selective in vivo probe, which, when appropriately radiolabeled with a positron-emitting radionuclide, might be useful for PET imaging of PR-positive breast tumors.
The synthesis of 1 is relatively simple, and based on structure-affinity relationships developed at Wyeth24 and a PR X-ray crystal structure they obtained,25 it was known that the pyrrole nitrogen can either be unsubstituted or substituted with small groups, such as a methyl group (e.g., 1 itself). The geminal dimethyl groups on the benzoxazin-2-thione ring can also be replaced by larger groups; even substitution with a 2-thienyl group gives a high affinity compound.24 By contrast, the benzoxazin-2-thione N-H cannot be substituted because it forms an important hydrogen bond interaction with an amino acid residue in the binding pocket.25 The structure of 1 and potential fluorine-substituted derivatives are given in Scheme 1. The most promising sites for the attachment of groups bearing a positron-emitting radionuclide are the pyrrole nitrogen (Region 1) and the geminal dimethyl groups (Region 2) on the benzoxazinthione ring. In this work, we describe the synthesis of various fluoroalkyl analogs of 1 and assess their binding affinity for PR.
Scheme 1. Tanaproget (1) and potential fluorine-substituted derivatives.

Results and Discussion
Synthesis
The N-fluoroalkyl substituted pyrrole nitrogen derivatives 5a and 5b were synthesized as shown in Scheme 2. The key intermediate 2 was prepared according to the published procedure.24 Alkylation of 2 with the corresponding fluoroalkyl reagents, followed by thionation with Lawesson's reagent in toluene, gave target molecules 5a and 5b in high yield.
Scheme 2. Synthesis of fluorine-substituted derivatives 5a and 5b.

Reaction conditions and reagents: (a) 2-FCH2CH2OTf (3a) or 3-FCH2CH2CH2OTf (3b), K2CO3, DMF, rt, 16 h, 74%, 84%; (b) Lawesson's reagent, toluene, reflux, 2 h, 78%, 82%.
Compounds 16 and 17 were prepared as shown in Scheme 3. The unsymmetrical homoallylic alcohol 7 precursor was prepared as a racemate by reaction of methyl ketone 6 with allylmagnesium chloride, followed by cyclization with carbonyl diimidazole (CDI) in THF. A coupling reaction of arylbromide 8 with N-BOC-pyrrole boronic acid in the presence of Pd(0) gave pyrrole 9. The 5-cyano functional group on pyrrole 10 was installed by treatment with chlorosulfonyl isocynate (CSI), followed by addition of DMF as a quench. Oxidation of 10 with a catalytic amount of OsO4, followed by treatment with NaIO4, afforded the aldehyde 11,28 which was reduced with sodium borohydride (NaBH4) to give the alcohol 12. Deprotection of the BOC group on the pyrrole nitrogen by thermolysis at 160 °C under neat conditions then afforded the pyrrole 13. Fluoride substitution of 13 was accomplished by treatment with DAST (diethylaminosulfur trifluoride). Methylation of 14 with methyl iodide in the presence of potassium carbonate in DMF, followed by thionation with Lawesson's reagent in toluene, gave target molecule 16 in good yield. Compound 17 was synthesized by direct thionation of 14 with Lawesson's reagent.
Scheme 3. Synthesis of fluorine-substituted derivatives 16 and 17.

Reaction condition and reagents: (a) allylmagnesium chloride, ether, -78 °C to rt, 12 h, 83%; (b) CDI, THF, rt, overnight, 89%; (c) N-BOC-pyrrole boronic acid, Pd(PPh3)4, aq. Na2CO3, toluene, reflux, 16 h, 73%; (d) CSI, DMF, THF, -78 °C, 1 h 30 min, 76%; (e) i) OsO4, NMO, THF:water (10:1), rt, 5 h, ii) NaIO4, THF:water (4:1), rt, 1 h, 80%; (f) NaBH4, EtOH, rt, 2 h, 82%; (g) neat, 160 °C, 20 min, 78%; (h) DAST, CH2Cl2, -78 °C, 1 h, 39%; (i) Lawesson's reagent, toluene, 120 °C, 2 h, 75-79%; (j) MeI,K2CO3, DMF, rt, 16 h, 75%.
Fluoropropyl compounds 24 and 25, which have one more carbon compared to fluoroethyl compounds 16 and 17, were prepared as shown in Scheme 4. Treatment of the allyl derivative 8 with 9-borabicyclo[3.3.1]nonane (9-BBN) afforded the corresponding alcohol 18,29 which was then treated with the DAST to give the fluorine-substituted compound 19. Fluoropropyl compound 19 was reacted with N-BOC-pyrrole boronic acid in the presence of Pd(0) to provide pyrrole 20, which was treated with CSI and DMF in sequence to furnish nitrile compound 21. Removal of the BOC group from pyrrole by heating neat at 160 °C gave compound 22, and addition of methyl iodide to substitute on the pyrrole nitrogen yielded methylated compound 23. The desired compounds 24 and 25 were prepared by thionation with Lawesson's reagent in toluene, as shown in Scheme 4.
Scheme 4. Synthesis of fluorine-substituted derivatives 24 and 25.

Reaction condition and reagents: a) 9-BBN, THF, rt, 20 h, 89%; b) DAST, CH2Cl2, -78 °C, 1 h, 70%; c) N-BOC-pyrrol boronic acid, Pd(PPh3)4, aq. K2CO3, toluene, 80 °C, 16 h, 72%; d) CSI, DMF, THF, -78 °C, 1 h 30 min, 70%; e) neat, 160 °C, 20 min, 68%; f) MeI, K2CO3, DMF, 90 °C, 16 h, 78%, g) Lawesson's reagent, toluene, reflux, 2 h, 71-78%.
Progesterone Receptor Binding Affinities and Binding Selectivity
The relative binding affinities of the tanaproget derivatives as PR ligands were determined by a competitive radiometric binding assay using [3H]R5020 as tracer and R5020 as a standard, as previously described.19, 30 The binding affinities are expressed as relative binding affinity (RBA) values, with the RBA of the R5020 standard set to 100 (R5020 binds to PR with a Kd of 0.4 nM). The values given are the average ± SD of two or more independent determinations. The results are shown in Table 1. We also prepared a sample of 1, and it was found to have an RBA value of 151, which corresponds to the binding affinity reported by Wyeth.24
Table 1.
Relative binding affinities (RBAs) of Tanaproget Derivatives.a
 | |||
|---|---|---|---|
| compoundb | R1 | R2 | RBA (R5020 = 100) |
| Tanaproget (1) | CH3 | CH3 | 151 ± 39 |
| 5a | CH2CH2F | CH3 | 18.5 ± 5.2 |
| 5b | CH2CH2CH2F | CH3 | 0.99 ± 0.28 |
| 16b | CH3 | CH2CH2F | 151 ± 13 |
| 17b | H | CH2CH2F | 198 ± 30 |
| 24b | CH3 | CH2CH2CH2F | 90.9 ± 27 |
| 25b | H | CH2CH2CH2F | 189 ± 40 |
Relative binding affinity (RBA) values were determined by a competitive radiometric binding assay, using [3H]R5020 as a tracer. For details, see methods and our prior publication.19, 30 Values are expressed as percentages relative to the affinity of the standard, R5020 = 100%. From the binding affinity of R5020 (KD = 0.4 nM), one can calculate the KI of these compounds (KI = 0.4 nM/[RBA] × 100).
Compounds 16, 17, 24 and 25 were tested as racemates.
The fluorine-substituted ligands show binding affinities to PR that range from poor to excellent, and we found that the nature of the substituent R1 on the pyrrole nitrogen, and the substituent R2 on the benzoxazin-2-thione moiety both had significant effects on PR binding affinity. While the methyl group on the pyrrole nitrogen is well tolerated, larger fluoroalkyl substituents at this position decreased the binding affinity considerably; e.g., the N-fluoroethyl compound 5a had an RBA of 18.5, and the N-fluoropropyl compound 5b had a much lower binding affinity (RBA = 0.99). The compounds with fluoroalkyl moieties on the benzoxazin-2-thione template, R2, bound very well, with the fluoroethyl moiety giving somewhat higher affinities for PR (e.g., 16, RBA = 151; 17, RBA= 198) than the fluoropropyl moiety (e.g., 24, RBA = 90.9; 25, RBA= 189).
It is well known that steroidal progestins often have substantial cross reactivity with other evolutionally related nuclear receptors, most notably with the androgen receptor (AR) and the glucocorticoid receptor (GR).31-37 Compound 1, however, has very low affinity for both AR and GR,24-27 as do some other non-steroidal progestins.31, 33, 36, 37 Notably, our lead compound, the fluoropropyl Tanaproget analog (25) binds to AR with an affinity less than 0.04% that of the potent androgen R1881 and to GR with less than 0.9% that of the potent glucocorticoid dexamethasone. Thus, its PR binding affinity as well as its binding selectivity are both excellent.
Molecular Modeling of Structure-Binding Affinity Relationships and Predicted Enantioselectivity
The binding affinities for the Tanaproget analogs shown in Table 1 show a strong dependence on the nature and position of the alkyl or fluoroalkyl substituent on the pyrrole-benzoxazin-2-thione ligand core. Thus, while the pyrrole N-methyl group of 1 is well tolerated (1, RBA = 151), the larger fluoroalkyl substituents are not (5a = 18.5, 5b = 0.99), indicating that the ligand-binding pocket of PR has very limited tolerance for more than a methyl group. By contrast, when fluoroethyl or propyl substituents replace one of the C4 methyl groups of the benzoxazin-2-thione unit, high affinity compounds are produced, with three compounds (16, 17, 25) having equivalent or higher binding affinity than that of compound 1 itself and indicating that this region of the PR ligand binding pocket has good bulk tolerance, as suggested by prior work.24
Intriguingly, there appears to be some “reciprocity” between the substituent on the pyrrole nitrogen and at the C4 position such that larger groups (fluoroethyl and fluoropropyl) are more readily accommodated at C4 when there is a smaller group (H) on the pyrrole nitrogen. Also, it is of note that while the pyrrole N-substituted compounds 5a and 5b are achiral, the high affinity Tanaproget analogs modified at C4 position of benzoxazin-2-thione are chiral, but thus far have only been studied as racemates. Thus, it is possible that one enantiomer will have higher binding affinity than the other.
To investigate further the structure-binding affinity correlations we have uncovered, including the apparent reciprocity between the pyrrole N-substituent and the C4-substituents, and to explore possible enantioselectivity, we modeled our new ligands into the ligand binding pocket of PR, using as a guide the X-ray crystal structure of PR complexed with 1 (PDB accession code 1zuc). All structures from Table 1 were docked into the ligand binding pocket using Autodock Vina,38 minimized, and further analyzed using commercial molecular modeling software.3, 30
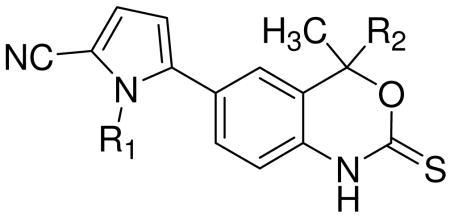
First focusing on N-substitution at the pyrrole, we investigated the effects of substituent size on ligand conformation by analyzing the torsional energetics of the ligand independent of the protein. For the unsubstituted (N-H) analog of 1, the torsional (tor), electronic (ele), and van der Waals (vdW) terms contribute significantly to the change in total energy (Fig. 1A); however, in all cases of N-substitution, the change in ligand energy is dominated by vdW forces (Fig. 1B). Increasing the size of the substituent magnifies this affect by increasing the ligand energy at small dihedral angles (Fig. 1C). By extension, the minimum energy conformation is shifted to a larger dihedral angle as the substituent size increases (Fig. 1C inset).
Figure 1.
Ligand energy dependence on pyrrole- benzoxazin-2-thione core dihedral angle. At relatively small angles, several energy terms contribute to the change in total ligand energy when rotating around the bond connecting the N-H pyrrole and benzoxazin-2-thione core (Panel A). N-substitution at the pyrrole (Panel B), changes these torsional dynamics by drastically increasing the magnitude of the vdW energy term. This effect is magnified with increasing substituent size (Panel C), and results in a shift to larger dihedral angles for the minimum energy conformation (Panel C inset). (Note differences in the energy scales on the different panels.)
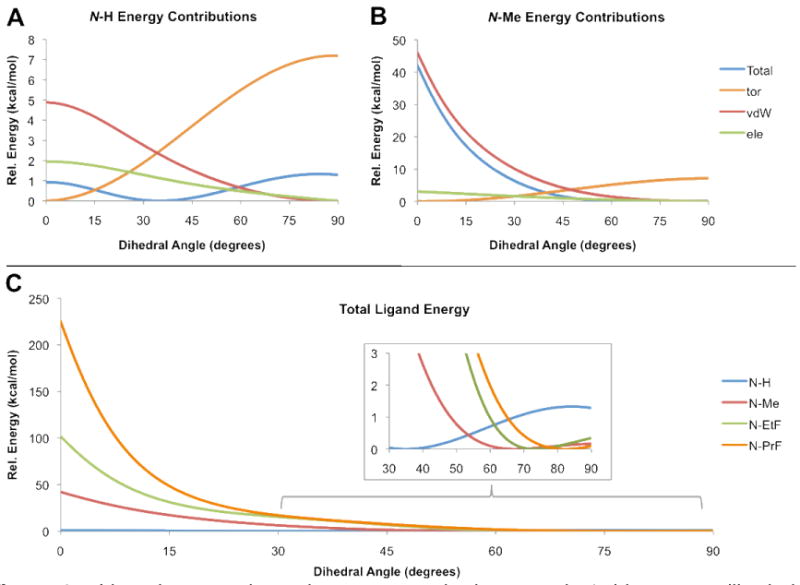
The effect of dihedral angle on binding affinity becomes obvious within the context of the protein, as shown for the docked poses of 1, 5a, and 5b in Figure 2A. The rotation of the pyrrole to accommodate larger substituents shifts the position of the nitrile away from Gln725, an important hydrogen bonding contact. Studying the energy associated with the interaction between the ligand and the protein when rotating the pyrrole in the binding pocket, however, suggested that small substituents potentially make other non-specific beneficial contacts, as shown in Figure 2B.
Figure 2.
Docking poses for N-substituted analogs of Tanaproget. (Panel A) The steric bulk of the pyrrole N-substituent for 1 (cyan), 5a (blue), and 5b (red) induces an increasingly large dihedral angle, rotating the nitrile away from Gln725, and resulting in reduced hydrogen bonding. (Panel B) Calculating the interaction energy, however, suggests that small substituents (e.g. N-Me and N-EtF) have less rotational freedom, but make beneficial contacts with the receptor compared to unsubstituted analogs.
To ensure that any change in the interaction energy was solely due to the substituent, we started with the docked conformation of 5a (N-EtF) and modified the N-substituent accordingly prior to each series of calculations. Comparing the interaction energies between substituted (N-Me, N-EtF) and unsubstituted (N-H) pyrroles shows that the substituted compounds exist in a more narrow but deeper energy well than the unsubstituted analog, implying that the substituted pyrrole has less rotational freedom but makes more positive contacts with the receptor.
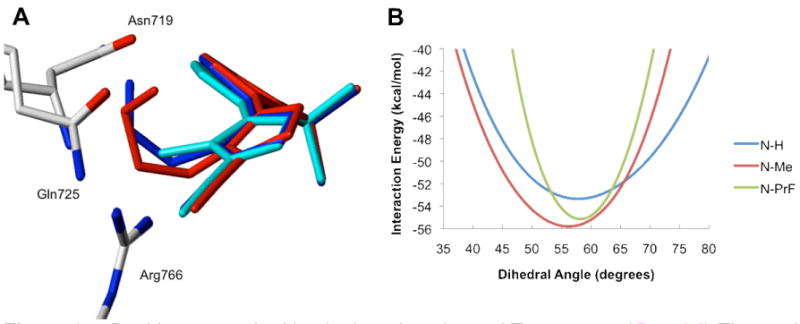
Shifting our focus to C4-substitution, we observed that while docked poses for the N-Me and N-H series of analogs show a conserved binding mode, the C4-substituent subtly affects ligand conformation and placement. Overlaying the docked poses for compounds in the N-Me (Figure 3) and N-H series (not shown) suggests that the gem-dimethyl substituted compounds adopt an intermediate position. As the size of the C4 substituent increases, the compound rotates to a degree related to steric bulk and in a direction that reflects the stereochemical configuration.
Figure 3.
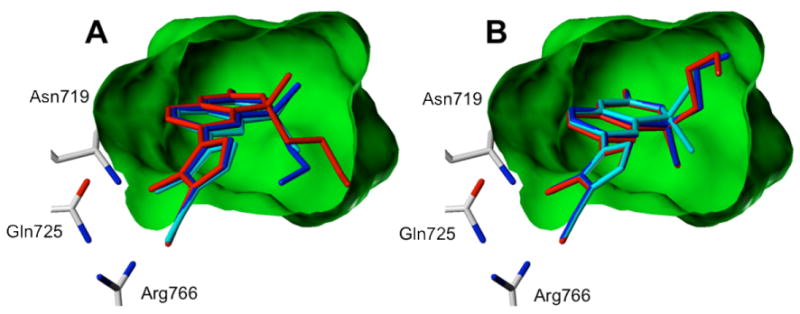
Effects of methyl (cyan), fluoroethyl (blue), and fluoropropyl (red) substitution at C4 of Tanaproget. As substituent bulk increases, the core of the R-enantiomer (Panel A) is forced upward to adopt a higher energy ligand conformation, while the S-enantiomer (Panel B) allows a downward rotation to a more energetically favorable dihedral angle.
The poses presented in Figure 3 imply that the stereochemical configuration at C4 should have a marked affect on ligand-protein interactions. The R-configuration directs the substituent down in the binding pocket, where steric constraints force the core to rotate up and adopt a relatively small dihedral angle (compare the dihedral angle of R/S-24 to 1 in Table 2). As clearly demonstrated in Figure 1B, the shift to a smaller dihedral angle represents a higher-energy ligand conformation. It is notable that N-H analogs follow a similar trend, although with a smaller energy difference that is obscured by the large energy range presented in Figure 1A. The S-configuration, in contrast, directs the substituent up in the pocket, causing the core to rotate downward and adopt a more energetically favorable dihedral angle. These qualitative observations are corroborated by energy calculations, whereby the total energy was calculated for each pose, and in all cases the S-stereoisomer for each chiral analog was lower in energy than the corresponding R-stereoisomer (Table 2).
Table 2.
Calculated Binding Metrics for Docked Poses of Tanaproget Analogs.
| Series | Ligand | Dihedral Angle | Energy (kcal/mol) | Meana H-Bonding Scoreb | |||
|---|---|---|---|---|---|---|---|
| Individual | Average | Individual | Average | RBA | |||
| NR | 5a | 59.6 | 488.9 | 488.9 | 0.396 | 0.396 | 18.5 |
| 5b | 64.8 | 498.7 | 498.7 | 0.311 | 0.311 | 0.99 | |
| NMe | Tanaproget (1) | 49.2 | 486.0 | 486.0 | 0.424 | 0.424 | 151 |
| R-16 | 47.6 | 490.3 | 488.1 | 0.419 | 0.425 | 151 | |
| S-16 | 52.1 | 485.9 | 0.430 | ||||
| R-24 | 45.3 | 500.5 | 496.7 | 0.411 | 0.421 | 90.9 | |
| S-24 | 52.5 | 493.0 | 0.431 | ||||
| NH | NH analog of 1 | 39 | 482.0 | 482.0 | 0.446 | 0.446 | 602 |
| R-17 | 35.6 | 485.8 | 484.0 | 0.439 | 0.445 | 198 | |
| S-17 | 43 | 482.2 | 0.451 | ||||
| R-25 | 33.7 | 496.7 | 493.3 | 0.434 | 0.440 | 189 | |
| S-25 | 46.3 | 490.0 | 0.446 | ||||
Geometric mean,
Calculated using the “ligand interactions” module in MOE
Finally, we set out to establish a model relating the molecular consequences for both N- and C4-substitution to the observed RBA values. Specifically we wanted to formalize the apparent reciprocity between the two substitution points, and to make predictions regarding the enantioselectivity for our chiral analogs, doing so in a simple yet functional manner. To this end we initially considered a number of models based on energy calculations. As illustrated by the calculated total energies in Table 2, these models were capable of predicting the relative order of the RBA values within each series (N-R, N-Me, and N-H); however, all failed in cross series comparisons (e.g. 5a vs 24 or 25). Ultimately, we developed a model focusing on hydrogen bonding contacts.
From experimental SAR and PR-ligand crystal structures, it has been well established that hydrogen bonding with residues Gln725, Arg766, and Asn719 is a requirement for tight binding. The remainder of the pocket is largely lipophilic and contributes to the binding affinity only through vdW interactions, a notoriously difficult energy contribution to calculate.39, 40 Based on the position and interdependence of the hydrogen bonding partners, we modeled the RBA values as a function of the geometric mean of calculated hydrogen bonding scores using the built-in “ligand interactions” module of MOE.41 Linearization of this function yielded a highly correlated relationship (Figure 4, slope=43.8, R2=0.96), and the mean hydrogen bonding scores correctly predict the relative order of binding affinity across all three series, including extremely low (e.g. 5a, 5b) and high (e.g. N-H analog of 1) affinity compounds.
Figure 4.
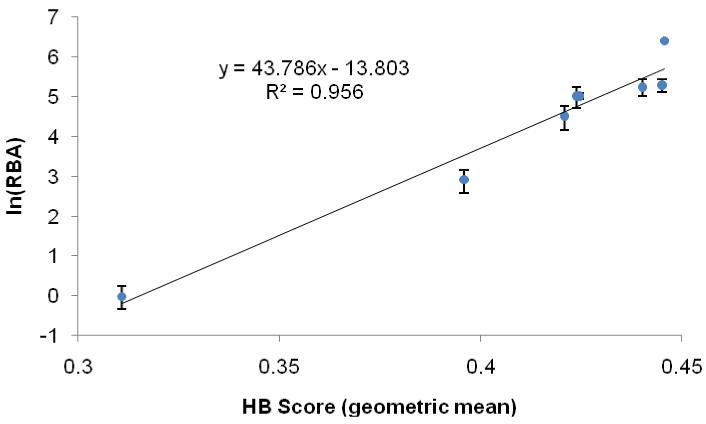
Linear Fit Relating the Calculated Hydrogen Bonding Score to RBA.
The application of this model to understand the observed RBA values for compounds 5a and 5b is rather straightforward. The rotation of the pyrrole away from Gln725, as highlighted in Figure 2, corresponds to the erosion of the mean hydrogen bonding score (Table 2, series N-R) with increased substituent size. The difference between RBA values for the remainder of the compounds in Table 1 are much smaller, and the analysis correspondingly more subtle. We propose that the ligand binds in a pose generally conserved for all analogs; the ligand conformation strikes a delicate balance between positioning the core to accommodate substitution at C4, maximizing hydrogen bonding contacts and positive interactions between the pyrrole and receptor, and minimizing internal ligand energy.
The interplay between these interactions is best illustrated by the marked decrease in RBA value for compound 24. The large substituent at C4 restricts the rotational freedom of the core, while N-Me substitution increases the internal vdW energy of the ligand, resulting in a high-energy conformation that precludes strong hydrogen bonding interactions. The reduction of C4-substituent size allowing the core to rotate to a more favorable position (ex. 15), or the removal of the N-substituent reducing the energy associated with internal torsion (ex. 25), results in increased hydrogen bonding scores that correspond well to the observed higher RBA values. This model also supports the prediction that the S-configuration of the C4-substitued systems will have higher affinity than the R-stereoisomer.
While our model is rather simplistic, we obtain with it a linear relationship between HB score and ln(RBA) having a correlation coefficient over 0.95. This relationship accurately predicts the relative order of binding affinity, and it gives predicted RBA values within an average factor of 1.4 (and always better than a factor of 2) relative to experimentally determined ones, comparable to the error in experimentally determined values (cf. Table 1). We suspect that the limitations in our model could be reduced through the development of a more complex model that includes a direct treatment of energy calculations, the evaluation of non-specific protein-ligand interactions, and multivariate analysis. However, our very simple approach of analyzing ligand binding affinity through the quantification of the hydrogen bonding contacts is suitable for explaining the binding trends of the compounds we have studied (Table 1), and it generates a valuable predictive model for exploring enantioselectivity.
Conclusion
We have synthesized a series of fluoroalkyl-substituted 6-aryl-1,4-dihydrobenzo-[d]-[1,3]oxazine-2-thiones, analogs of the high affinity non-steroidal progestin agonist 1 (Tanaproget), as potential diagnostic imaging agents using positron emission tomography (PET). Some of the synthesized compounds showed subnanomolar binding affinities, comparable to and even higher than that of the known high affinity PR ligand R5020 and of 1 itself. We have been able to rationalize the distinct structure-binding affinity relationships in this series by molecular modeling of ligand internal energy and ligand-receptor hydrogen bonding score, and our model yields an intriguing explanation for the interaction between substituents at the two ends of the Tanaproget core as well as offering predictions of enantioselectivity.
Compounds with high binding affinities are currently being evaluated as potential diagnostic imaging agents for PR in breast tumors, and further work on the radiolabeling of some of these compounds and studies of their biodistribution and in vivo PET imaging of mammary tumors in animals are currently underway and will be reported elsewhere. In addition, work on the asymmetric synthesis and separation of the racemates of the C4-substituted Tanaproget analogs are progressing, and the result of the binding selectivities will be reported in due course.
Experimental Procedures and Characterization Data
Materials and Methods
All reagents and solvents were obtained from Aldrich. Tetrahydrofuran, diethyl ether, toluene, and dichloromethane were obtained prior to use from a solvent-dispensing system.42 Glassware was oven-dried, assembled while hot, and cooled under an inert atmosphere. Unless otherwise noted, all reactions were conducted in an inert atmosphere. Reaction progress was monitored using analytical thin-layer chromatography (TLC) on 0.25 mm Merck F-254 silica gel glass plates. Visualization was achieved by either UV light (254 nm). Flash chromatography was performed with neutral aluminum (0.040-0.063 mm) packing. Compounds Tanaproget (1) and 2 were synthesized according to published procedures.24 The purity of all compounds for biological testing was determined by HPLC method (see Supporting Information), confirming >95% purity.
5-(4,4-Dimethyl-2-oxo-2,4-dihydro-1H-benzo[d][1,3]oxazin-6-yl)-1H-pyrrole-2-carbonitrile (2)
Compound 2 was prepared according to the previous paper.17 NMR (500 MHz, CDCl3) δ 1.62 (s, 6H), 6.36 (dd, 1H, J = 3.5 Hz, 2.5 Hz), 6.74 (dd, 1H, J = 3.5 Hz, 2.5 Hz), 6.86 (d, 1H, J = 9.0 Hz), 7.43-7.41 (m, 2H), 8.66 (brs, 1H), 11.08 (brs, 1H). Registry number: 304853-99-4.
2-Fluoroethyl trifluoromethanesulfonate (3a)
This compound 3a was prepared according to methods described in our prior publication.43 NMR (500 MHz, CDCl3) δ 4.70-4.66 (m, 2H), 4.77-4.74 (m, 2H).
Synthesis of N-fluoroalkyl derivatives 5
General Procedure for the Synthesis of N-fluoroalkyl derivatives 4
To a solution of 5-(4,4-dimethyl-2-oxo-1,4-dihydro-2H-3,1-benzoxazin-6-yl)-1H-pyrrole-2-carbonitrile 2 (61 mg, 0.23 mmol) in anhydrous DMF (5 mL) was added potassium carbonate (158 mg, 1.1 mmol) followed by fluoroalkyl triflate (0.21 mmol) and the reaction was stirred for 16 h at RT. The reaction mixture was poured into water (5 mL) and extracted with EtOAc (2 × 20 mL). The organic layers were combined, washed with brine (10 mL), dried over MgSO4, filtered and concentrated in vacuo. Purification of the concentrated filtrate by flash column chromatography on silica gel (40% EtOAc/hexane) or preparative thin-layer chromatography gave product; further purification was effected by recrystallization.
5-(4,4-Dimethyl-2-oxo-1,4-dihydro-2H-benzo[d][1,3]oxazin-6-yl)-1-(2-fluoro-ethyl)-1H-pyrro le-2-carbonitrile (4a)
Purified by preparative thin-layer chromatography (20% EtOAc/hexane) to give colorless needles (74% yield) that were recrystallized from EtOAc/hexane, mp 185-186 ºC. 1H NMR (500 MHz, acetone-d6) δ 1.71 (s, 6H), 4.45 (t, 1H, J = 5.0, 27.0 Hz), 4.51 (t, 1H, J = 5.0, 27.0 Hz), 4.59 (t, 1H, J = 5.0, 27.0 Hz), 4.69 (t, 1H, J = 5.0, 27.0 Hz), 6.29 (d, 1H, J = 4.0 Hz), 6.98 (d, 1H, J = 4.0 Hz), 7.15 (d, 1H, J = 8.0 Hz), 7.40 (d, 1H, J = 8.0 Hz), 7.44 (s, 1H), 9.40 (brs, 1H); 13C NMR (125 MHz, acetone-d6) δ 27.7, 45.83 (d, 1C, J = 20.4 Hz), 81.89 (d, 1C, J = 172.3 Hz), 82.46, 103.45, 111.83, 113.71, 114.38, 120.16, 125.01, 127.33, 127.48, 130.24, 132.67, 139.36, 153.78; MS m/e 313 (M+, 55%), HRMS (EI) calcd for C17H16O2N3F 313.1227 (M+), found 313.1225.
5-(4,4-Dimethyl-2-oxo-1,4-dihydro-2H-benzo[d][1,3]oxazin-6-yl)-1-(2-fluoro-propyl)-1H-pyrr ole-2-carbonitrile (4b)
Purified by flash chromatography (10-30% EtOAc/hexane) to give colorless needles (81% yield) that were recrystallized from EtOAc, mp 140-142 °C. 1H NMR (500 MHz, CDCl3) δ 1.75 (s, 6H), 2.08-2.18 (m, 2H), 4.21 (t, 2H, J = 7.2 Hz), 4.32 (t, 1H, J = 5.5, 47.0 Hz), 4.42 (t, 1H, J = 5.5, 47.0 Hz), 6.18 (d, 1H, J = 4.0 Hz), 6.88 (d, 1H, J = 4.0 Hz), 7.13 (d, 1H, J = 2.0 Hz), 7.24-7.26 (m, 1H), 9.60 (brs, 1H); 13C NMR (125 MHz, CDCl3) δ 28.3, 32.10 (d, J = 20.4 Hz), 43.13 (d, 1C, J =3.9 Hz) 80.66, (d, 1C, J = 166.3 Hz), 83.21, 104.61, 110.46, 114.35, 115.37, 120.65, 124.25, 126.67, 127.11, 130.13, 134.61, 139.51, 153.25. MS m/e 327 (M+, 65%), HRMS (EI) calcd for C18H18O2N3F 327.1383 (M+), found 327.1381.
General Procedure for the Synthesis of N-fluoroalkyl derivatives
To a solution of 4 (1.0 mmol) in toluene (6 mL) was added Lawesson's reagent (275 mg, 0.7 mmol) and the reaction was heated to 120 ºC for 2 h. The reaction was cooled to RT, poured into water (20 mL) and extracted with EtOAc (3 × 20 mL). The organic layers were combined, washed with brine (20 mL), dried over MgSO4, filtered and concentrated in vacuo gave the crude product (5).
5-(4,4-Dimethyl-2-thioxo-1,4-dihydro-2H-benzo[d][1,3]oxazin-6-yl)-1-(2-fluoro-ethyl)-1H-py rrole-2-carbonitrile (5a)
Purified by preparative thin-layer chromatography (5-30% EtOAc/hexane) to give light yellow needles (78% yield) that were recrystallized from EtOAc/hexane, mp 229-231 ºC. 1H NMR (500 MHz, acetone-d6) δ 1.75 (s, 6H), 4.47 (t, 1H, J = 5.0, 25.5 Hz), 4.52 (t, 1H, J = 5.0, 25.5 Hz), 4.60 (t, 1H, J = 4.8, 47.5 Hz), 4.69 (t, 1H, J = 4.8, 47.5 Hz), 6.33 (dd, 1H, J = 4.0, 0.5 Hz), 7.00 (dd, 1H, J = 4.0, 0.5 Hz), 7.25 (d, 1H, J = 8.0 Hz), 7.47 (d, 1H, J =8.0 Hz), 7.49 (s, 1H), 11.11 (brs, 1H); 13C NMR (125 MHz, acetone-d6) δ 26.8, 46.93 (d, 1C, J = 20.4 Hz), 82.49 (d, 1C, J = 170.1 Hz), 83.48, 105.35, 110.73, 113.76, 114.68, 120.20, 124.91, 127.68, 127.78, 130.34, 132.77, 139.81, 184.19. 19F NMR (470 MHz, acetone-d6) δ -88.56; MS m/e 329 (M+, 35%), HRMS (EI) calcd for C17H16ON3SF 329.0998 (M+), found 329.0997.
5-(4,4-Dimethyl-2-thioxo-1,4-dihydro-2H-benzo[d][1,3]oxazin-6-yl)-1-(2-fluoro-propyl)-1H-p yrrole-2-carbonitrile (5b)
Purified by flash chromatography (10-30% EtOAc/hexane) to give a yellow solid (82% yield) that was recrystallized from EtOAc, mp 219-221 ºC. 1H NMR (500 MHz, CDCl3) δ 1.78 (s, 6H), 2.09-2.20 (m, 2H), 4.21 (t, 2H, J = 7.2 Hz), 4.33 (t, 1H, J = 5.5, 47.0 Hz), 4.42 (t, 1H, J = 5.5, 47.0 Hz), 6.20 (d, 1H, J = 4.0 Hz), 6.89 (d, 1H, J = 4.0 Hz), 7.02-7.04 (m, 1H), 7.16 (d, 1H, J = 1.5 Hz), 7.33 (dd, 1H, J = 8.0, 1.5 Hz), 10.21 (brs, 1H); 13C NMR (125 MHz, CDCl3) δ 27.84, 32.09 (d, 1C, J = 19.4 Hz), 43.16 (d, 1C, J = 3.9 Hz), 80.61, (d, 1C, J = 166.1 Hz), 84.94, 104.98, 110.68, 114.21, 114.77, 120.72, 124.22, 127.55, 128.46, 130.23, 131.84, 139.05, 184.34. 19F NMR (470 MHz, CDCl3) δ -89.01; MS m/e 343 (M+, 85%), HRMS (EI) calcd for C18H18ON3SF 343.1155 (M+), found 343.1153.
Synthesis of compounds 16 and 17
4-Bromo-aminoacetophenone (6).44
To a solution of 2-aminoacetophenone (2.0 mg, 14.796 mmol) in CH2Cl2 (500 mL) was added slowly pyridine hydrobromide perbromide (5.26 mg, 14.796 mmol) in 0 °C. The reaction mixture was allowed warmed to RT for 24 h. The reaction mixture was added water (200 mL) and extracted with CH2Cl2 (50 mL × 3). Th combined organic layer was dried over Na2SO4 and concentrated by rotary evaporator. The residue, desired product 6 (2.97 g, 94%) as a yellow solid, was used in next step without further purification. 1H NMR (500 MHz, CDCl3) δ 7.80 (d, 1H, J = 2.5 Hz), 7.34 (dd, 1H, J = 11.0 Hz, 3.0 Hz), 6.56 (d, 1H, J = 11. 0 Hz), 6.29 (brs, 2H), 2.65 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 199.6, 149.0, 136.9, 134.0, 119.2, 118.9, 106.5, 27.7. Registry number: 29124-56-9
2-(2-Amino-5-bromophenyl)pent-4-en-2-ol (7)
To a solution of 4-bromo-aminoacetophenone 6 (2.97 g, 13.875 mmol) in THF (60 mL) was added allylmagnesium bromide (2.0 M, 20.81 mL, 41.624 mmol) at -78 °C under N2 atmosphere. The reaction mixture was stirred for 4 h. The reaction mixture was added water (300 mL) and extracted with EtOAc (50 mL × 3). The combined organic layer was washed with aq. NH4Cl (30 mL × 3) and dried over Na2SO4. The resulting solution was removed by reduced pressure. The residue was purified by flash column chromatography on silica gel (30% EtOAc/hexane) to give allylated product 7 (3.479 g, 98%) as a pale yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.27-7.23 (m, 2H), 6.62 (d, 1H, J = 10 Hz), 5.94-5.84 (m, 1H), 5.31-5.26 (m, 2H), 4.84 (brs, 2H), 2.94-2.89 (m, 1H), 2.74-2.66 (m, 1H), 1.70 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 27.2, 44.4, 75.0, 109.2, 118.9, 119.3, 129.0, 130.5, 131.2, 133.3 144.5; MS (EI): m/z 257 (M+2)+, 255 (M)+, 216, 214 (100), 198, 172, 158, 143, 136, 117. HRMS (EI) calcd for C11H14BrNO 255.0265, found 255.0259.
4-Allyl-6-bromo-4-methyl-1Hbenzo[d][1,3]oxazine-2(4H)-one (8)
To a solution of 7 (3.91 g, 15.25 mmol) in THF (150 mL) was added 1,1′-carbonyldiimidazole (2.72 g, 16.780 mmol) at RT and refluxed for 48 h. The reaction mixture was cooled to RT and added water (200 mL). The reaction solution was extracted with EtOAc (50 mL × 3). The combined organic layer was washed with aq. NH4Cl (30 mL × 3). The resulting solution was concentrated by reduced pressure and purified by flash column chromatography on silica gel (30% EtOAc/hexane) to provide benzo[d]oxaxines 8 (3.2 g, 74%) as a yellow oil. 1H NMR (500 MHz, CDCl3) δ 9.92 (s, 1H), 7.22 (td, J = 7.5 Hz, 1.5 Hz, 1H), 7.08-7.02 (m, 2H), 6.93 (d, J = 8.0 Hz, 1H), 5.78-5.69 (m, 1H), 5.13 (d, J =4.0 Hz, 1H), 5.10 (s, 1H), 2.76-2.66 (m, 2H), 1.72 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 153.3, 134.1, 131.4, 128.8, 124.2, 123.8, 123.2, 119.8, 114.7, 84.7, 45.4, 26.2; MS (EI): m/z 283 (M+2)+, 281 (M)+, 242, 240 (100), 224, 196, 161, 143, 115. HRMS (EI) calcd for C12H1281BrNO2 283.0030, found 283.0031.
2-(4-Allyl-4-methyl-2-oxo-1,4-dihydro-2H-benzo[d][1,3]oxazin-6-yl)-pyrrole-1-carboxylic acid tert-butyl ester (9)
Purified by flash chromatography (40-60% EtOAc/hexane) to give a light yellow solid (73%) that was recrystallized from EtOAc/hexane, mp 95-97 ºC. 1H NMR (500 MHz, CDCl3) δ 1.40 (s, 9H),1.72 (s, 3H), 2.66-2.73 (m, 2H), 5.12-5.15 (m, 2H), 5.73-5.80 (m, 1H), 6.15-6.16 (m, 1H), 6.21-6.22 (m, 1H), 6.89 (d, 1H, J = 8.0 Hz), 7.06 (d, 1H, J = 1.5 Hz), 7.22 (dd, 1H, J = 8.0, 1.5 Hz), 7.32-7.33 (m, 1H), 9.63 (brs, 1H); 13C NMR (125 MHz, CDCl3) δ 26.51, 27.96, 45.82, 83.92, 85.01, 110.81, 114.16, 114.67, 120.19, 122.78, 123.87, 125.06, 129.79, 130.20, 131.74, 133.52, 134.42, 149.38, 153.30. MS m/e 368 (M+, 30%), HRMS (EI) calcd for C21H24N2O4 368.1736 (M+), found 368.1737.
2-(4-Allyl-4-methyl-2-oxo-1,4-dihydro-2H-benzo[d][1,3]oxazin-6-yl)-5-cyano-pyrrole-1-carb oxylic acid tert-butyl ester (10)
To a solution of benzo[d]oxazines 9 (700 mg, 1.900 mmol) in dried THF (50 mL) was added chlorosulfonyl isocyanate (202 μL, 2.280 mmol) at -78 °C under N2 atmosphere and stirred for 2 h. DMF (1 mL) was added to the reaction mixture, which allowed warmed to RT for 1 h. The reaction mixture was added to water (200 mL) and extracted with EtOAc (30 mL × 3). The combined organic layer was washed with aq. NH4Cl (50 mL) and dried over Na2SO4. The resulting solution was concentrated by rotary evaporator. The residue was purified by flash column chromatography on silica gel (40% EtOAc/hexane) to obtain nitrilated benzo[d]oxazines 10 (478 mg, 64%) as a white solid. mp: 139.5-140.5 °C; 1H NMR (500 MHz, CDCl3) δ 9.38 (s, 1H), 7.22 (dd, J = 8.0 Hz, 2.0 Hz, 1H), 7.05 (d, J = 1.5 Hz, 1H), 6.97 (d, J = 3.5 Hz, 1H), 6.91 (d, J =8.5Hz, 1H), 6.23 (d, J = 3.5 Hz, 1H), 5.79-5.70 (m, 1H), 5.15 (s, 1H), 5.13 (d, J =2.0 Hz, 1H), 2.75-2.66 (m,2H), 1.72 (s,3H), 1.51 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 152.6, 147.1, 139.6, 134.4, 131.1, 129.7, 127.4, 124.8, 124.4, 124.1, 120.2, 114.2, 113.8, 113.3, 105.5, 87.1, 84.7, 45.5, 27.5, 26.2; MS (FAB): m/z 494.2 (M+H)+, 307.2, 298.1, 154.2 (100), 136.1, 106.8. EA calcd for C: 67.16, H: 5.89, N: 10.68 found C: 66.43, H: 5.82, N: 10.47.
2-Cyano-5-[4-methyl-2-oxo-4-(2-oxo-ethyl)-1,4-dihydro-2H-benzo[d][1,3]oxazin-6-yl]-pyrrol e-1-carboxylic acid tert-butyl ester (11)
To a solution of 2-(4-allyl-4-methyl-2-oxo-1,4-dihydro-2H-benzo[d][1,3]oxazin-6-yl)-5-cyano-pyrrole-1-carboxylic acid tert-butyl ester 10 (770 mg, 1.96 mmol) in a mixed solvent of THF/water (20 mL/2 mL) was added N-methylmorpholine-N-oxide (NMO, 276 mg, 2.35 mmol), followed by a solution of 4% aqueous OsO4 (0.25 mL, 0039mmol). After the solution was stirred at RT for 5 h, solid Na2SO3 was added. After the resulting mixture was stirred at RT for 1 h, it was worked up with EtOAc and water. The aqueous layer was extracted with EtOAc (3 × 50 mL). The combined organic layer was dried over Na2SO4. The solvent was evaporated under vacuum. The residue was purified by flash chromatography using pure diethyl ether to give 670 mg (80%) of diol.
To the above diol (341 mg, 0.805 mmol) was added THF (8 mL), water (2 mL) and NaIO4 (343 mg, 1.61 mmol). After the resulting mixture was stirred at RT for 1 h, it was worked up with diethyl ether and water. The aqueous layer was extracted with diethyl ether (3 × 50 mL). The combined organic layer was washed with brine and dried over Na2SO4. The solvent was evaporated under vacuum. The residue was purified by flash chromatography using 20% diethyl ether/petroleum ether to give aldehyde 11 as viscous oil (65% yield). 1H NMR (500 MHz, CDCl3) δ 1.52 (s, 9H), 1.82 (s, 3H), 3.04-3.13 (m, 2H), 6.22 (d, 1H, J =4.0 Hz), 6.94-6.97 (m, 2H), 7.08 (d, 1H, J = 1.5 Hz), 7.25-7.27 (m, 1H), 9.44 (brs, 1H), 9.76 (s, 1H); 13C NMR (125 MHz, CDCl3) δ 18.92, 27.74, 52.71, 82.74, 87.64, 105.93, 113.50, 114.34, 114.89, 123.40, 124.71, 124.75, 128.17, 130.60, 134.36, 147.30, 150.97, 152.01, 198.53. MS m/e 395 (M+, 60%), HRMS (EI) calcd for C21H21O5N3 395.1481 (M+), found 395.1480.
2-Cyano-5-[4-(2-hydroxy-ethyl)-4-methyl-2-oxo-1,4-dihydro-2H-benzo[d][1,3]oxazin-6-yl]-pyrrole-1-carboxylic acid tert-butyl ester (12)
To a solution of aldehyde 11 (138 mg, 0.35 mmol) in MeOH (3 mL) at RT was added NaBH4 (39 mg, 1.05 mmol). The reaction mixture was stirred for 2 h at RT. The mixture was added 5 mL water and extracted with EtOAc several times. The combined organic layer was washed with brine, dried MgSO4, and concentrated in vacuo. Purified by flash chromatography (40-60% EtOAc/hexane) to give 12 as a white solid (88% yield) that was recrystallized from EtOAc/hexane, mp 145-147 ºC. 1H NMR (500 MHz, CDCl3) δ 1.52 (s, 9H), 1.75 (s, 3H), 2.22-2.33 (m, 2H), 3.80 (m, 2H), 6.23 (d, 1H, J = 4.0 Hz), 6.91 (d, 1H, J = 8.0 Hz), 6.97 (d, 1H, J = 4.0 Hz), 7.09 (d, 1H, J = 1.5 Hz), 7.22 (dd, 1H, J = 8.0, 1.5 Hz), 9.40 (s, br, 1H); 13C NMR (125 MHz, CDCl3) δ 27.59, 27.82, 43.05, 58.47, 85.08, 87.62, 105.80, 113.55, 114.21, 124.43, 124.72, 124.81, 127.76, 130.00, 134.55, 139.78, 147.43, 152.69. MS m/e 397 (M+, 70%), HRMS (EI) calcd for C21H23O5N3 397.1638 (M+), found 397.1635.
5-[4-(2-Hydroxy-ethyl)-4-methyl-2-oxo-1,4-dihydro-2H-benzo[d][1,3]oxazin-6-yl]-1H-pyrrole -2-carbonitrile(13)
2-Cyano-5-[4-(2-hydroxy-ethyl)-4-methyl-2-oxo-1,4-dihydro-2H-benzo[d][1,3]oxazin-6-yl]-pyrrole -1-carboxylic acid tert-butyl ester (12) (397 mg, 1.0 mmol) was placed in a 25 mL round bottomed flask stopped with a rubber septum and equipped with nitrogen inlet and a needle to allow gaseous outflow. A vigorous flow of nitrogen was maintained as the flask was placed in an oil bath and heated to 160 ºC. After 20 min at this temperature, the flask was removed from the oil bath and allowed to cool. The yellow residue was purified by flash chromatography (40-60% EtOAc/hexane) to give 13 as a yellow powder (169 mg, 57%) that was recrystallized from EtOAc/hexane, mp 241-242 ºC. 1H NMR (500 MHz, acetone-d6) δ 1.74 (s, 3H), 2.19-2.24 (m, 1H), 2.31-2.33 (m, 1H), 3.63-3.70 (m, 2H, 6.66-6.67 (m, 1H), 6.94-6.95 (m, 1H), 7.05 (d, 1H, J = 8.5 Hz), 7.66 (dd, 1H, J = 8.5, 2.0 Hz), 7.71-7.72 (m, 1H), 9.26 (brs, 1H), 11.62 (brs, 1H); 13C NMR (125 MHz, acetone-d6) δ 26.74, 43.15, 57.33, 83.52, 101.24, 106.96, 114.45, 114.92, 114.99, 120.90, 120.99, 125.84, 126.11, 135.24, 137.31, 150.23. MS m/e 297 (M+, 90%), HRMS (EI) calcd for C16H15O3N3 297.1113 (M+), found 297.1112.
5-[4-(2-Fluoro-ethyl)-4-methyl-2-oxo-1,4-dihydro-2H-benzo[d][1,3]oxazin-6-yl]-1H-pyrrole-2-carbonitrile (14)
In a one-necked round-bottom flask, which contained 5-[4-(2-hydroxy-ethyl)-4-methyl-2-oxo-1,4-dihydro-2H-benzo[d][1,3]oxazin-6-yl]-1H-pyrrole-2-car bonitrile (13) (297 mg, 1 mmol) in CH2Cl2 (5 mL) was cooled to -78 ºC under nitrogen atmosphere.. Diethylaminosulfur trifluoride (232 mg, 1.2 mmol) was dropped very slowly by syringe at -78 ºC. The cooling was removed, and the reaction mixture was stirred for 1 h at RT. The reaction mixture was cooled again to -78 ºC, and MeOH (200 μl) was added. The solution was additionally stirred for 30 min after removal of the dry ice bath, and the reaction was quenched by saturated NaHCO3 solution and extracted with excess EtOAc. The organic layer was dried over Na2SO4 and concentrated. Purified by flash chromatography (40-60% EtOAc/hexane) to give 14 as a yellow solid (57% yield) that was recrystallized from EtOAc/hexane, mp 96-98 ºC. 1H NMR (500 MHz, acetone-d6) δ 1.76 (s, 3H), 2.41-2.56 (m, 2H), 4.50-4.72 (m, 2H), 6.68 (d, 1H, J = 4.0 Hz), 6.95 (d, 1H, J = 4.0 Hz), 7.07 (d, 1H, J = 8.5 Hz), 7.69 (dd, 1H, J = 8.5, 2.0 Hz), 7.76 (d, 1H, J = 2.0 Hz), 9.32 (brs, 1H), 11.60 (brs, 1H); 13C NMR (125 MHz, acetone-d6) δ 26.58, 40.45 (d, 1C, J = 20.4 Hz), 79.82 (d,1C, J = 163.3 Hz), 82.71, 101.29, 107.04, 114.43, 115.11, 119.55, 120.89, 120.99, 125.63, 126.06, 126.25, 135.16, 155.82. 19F NMR (470 MHz, acetone-d6) δ -89.10. MS m/e 299 (M+, 80%), HRMS (EI) calcd for C16H14O2N3F 299.1070, found 299.1070.
5-[4-(2-Fluoro-ethyl)-4-methyl-2-oxo-1,4-dihydro-2H-benzo[d][1,3]oxazin-6-yl]-1-methyl-1H-pyrrole-2-carbonitrile (15)
To a solution of 5-[4-(2-fluoro-ethyl)-4-methyl-2-oxo-1,4-dihydro-2H-benzo[d][1,3]oxazin-6-yl]-1H-pyrrole-2-carbo nitrile (14) (68 mg, 0.23 mmol) in anhydrous DMF (5 mL) was added potassium carbonate (158 mg, 1.1 mol) followed by methyl iodide (0.0129 mL, 0.21 mmol). The reaction mixture was stirred for 16 h at RT. The reaction mixture was poured into water (10 mL) and extracted with EtOAc (2 × 20 mL). The combined organic layer was washed with brine (10 mL), dried over MgSO4 and concentrated in vacuo. Purification by flash chromatography (20-40% EtOAc/hexane) gave 15 as a white solid (90% yield) that was recrystallized from EtOAc/hexane, mp 160-162 ºC. 1H NMR (500 MHz, CDCl3) δ 1.79 (s, 3H), 2.38-2.51 (m, 2H), 3.71 (s, 3H), 4.51-4.74 (m, 2H), 6.19 (d, 1H, J = 4.0 Hz), 6.85 (d, 1H, J = 4.0 Hz), 6.90 (d, 1H, J = 8.0 Hz), 7.17 (m, 1H),7.29 (dd, 1H, J =8.0, 2.0 Hz), 8.06 (brs, 1H); 13C NMR (125 MHz, CDCl3) δ 26.58, 37.05, 41.33 (d,1C, J = 20.4 Hz), 79.67 (d, 1C, J = 163.6 Hz), 83.01, 101.56, 106.98, 114.41, 115.17, 119.67, 121.03, 121.10, 125.37, 125.98, 126.12, 134.98, 155.62. 19F NMR (470 MHz, acetone-d6) δ -90.00. MS m/e 313 (M+, 65%), HRMS (EI) calcd for C17H16O2N3F 313.1227 (M+), found 313.1225.
5-[4-(2-Fluoro-ethyl)-4-methyl-2-thioxo-1,4-dihydro-2H-benzo[d][1,3]oxazin-6-yl]-1-methyl-1H-pyrrole-2-carbonitrile (16)
Compound 16 was synthesized using the procedure similar to that described for compounds 5. Purification by preparative thin-layer chromatography (30% EtOAc/hexane) gave a light yellow solid (65% yield), mp 172-174 ºC. 1H NMR (500 MHz, acetone-d6) δ 2.36-2.56 (m, 2H), 3.73 (s, 3H), 4.49-4.78 (m, 2H), 6.21 (d, 1H, J = 4.0 Hz), 6.86 (d, 1H, J = 4.0 Hz), 6.91 (d, 1H, J =8.0 Hz), 7.20 (brs, 1H), 7.33-7.34 (m, 1H), 9.13 (brs, 1H); 13C NMR (125 MHz, acetone-d6) δ 27.89, 37.45, 41.24 (d, 1C, J = 20.4 Hz), 80.66 (d, 1C, J = 163.6 Hz), 83.03, 101.55, 107.01, 114.43, 115.13, 119.78, 119.89, 121.56, 125.56, 126.12, 126.23, 135.00, 159.62. 19F NMR (470 MHz, acetone-d6) δ -88.90. MS m/e 329 (M+, 55%), HRMS (EI) calcd for C17H16ON3SF 329.0998 (M+), found 329.0996.
5-[4-(2-Fluoro-ethyl)-4-methyl-2-thioxo-1,4-dihydro-2H-benzo[d][1,3]oxazin-6-yl]-1H-pyrrol e-2-carbonitrile (17)
Compound 17 was synthesized using the procedure similar to that described for compounds 5. Purified by preparative thin-layer chromatography (10-35% EtOAc/hexane) to give a light yellow solid (75% yield), mp 104-106 ºC. 1H NMR (500 MHz, acetone-d6) δ 1.82 (s, 3H), 2.40-2.58 (m, 2H), 4.51-4.76 (m, 2H), 6.73 (dd, 1H, J = 4.0, 2.0 Hz), 6.96 (dd, 1H, J = 4.0, 2.0 Hz), 7.75 (dd, 1H, J = 8.0, 2.0 Hz), 7.81 (d, 1H, J = 2.0 Hz), 10.96 (brs, 1H), 11.66 (brs, 1H); 13C NMR (125 MHz, acetone-d6) δ 25.98, 40.42 (d,1C, J = 20.4 Hz), 80.32 (d, 1C, J = 163.3 Hz), 82.45, 101.12, 106.98, 114.22, 115.19, 119.95, 120.56, 121.09, 125.56, 126.09, 126.20, 134.16, 161.12. 19F NMR (470 MHz, acetone-d6) δ -89.10. MS m/e 315 (M+, 20%), HRMS (EI) calcd for C16H14ON3SF 315.0842 (M+), found 315.0842.
Synthesis of Fluorine Compounds 24 and 25
6-Bromo-4-(3-hydroxy-propyl)-4-methyl-1,4-dihydro-benzo[d][1,3]oxazin-2-one (18)
To a solution of 4-allyl-6-bromo-4-methyl-1,4-dihydro-benzo[d][1,3]oxazin-2-one (8) (983.5 mg, 3.50 mmol) in THF (30 mL) at RT was added 9-borabicyclo[3.3.1]nonane (9-BBN; 21 mL, 0.5 M solution in hexane, 10.50 mmol). The reaction mixture was stirred for 20 h. To the mixture were added, successively, EtOH (0.64 mL), 6 N NaOH (0.215 mL), and 30% H2O2 (0.426 mL). The mixture was heated at 50 ºC for 1 h, cooled to RT, and extracted with EtOAc several times. The combined organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo. Compound 18 was purified by flash chromatography (40-60% EtOAc/hexane) to give light yellow oil (89% yield). 1H NMR (500 MHz, CDCl3) δ 1.63 (s, 3H), 1.75-2.01 (m, 4H), 3.51-3.52 (m, 2H), 6.75 (d, 1H, J =8.0 Hz), 7.16 (d, 1H, J =2.0 Hz), 7.28 (dd, 1H, J =8.0, 2.0 Hz); 13C NMR (125 MHz, CDCl3) δ 26.99, 27.50, 35.78, 56.77, 84.93, 115.43, 116.67, 127.06, 132.45, 133.67, 152.77. MS m/e 299 (M+, 30%), HRMS (EI) calcd for C12H14O3NBr 299.0157 (M+), found 299.0153.
6-Bromo-4-(3-fluoro-propyl)-4-methyl-1,4-dihydro-benzo[d][1,3]oxazin-2-one (19)
Compound 19 was synthesized using the procedure similar to that described for compound 14. Purification by flash chromatography (10-30% EtOAc/hexane) gave a light yellow oil (70% yield). 1H NMR (500 MHz, CDCl3) δ 1.70 (s, 3H), 1.73-1.86 (m, 2H), 2.07-2.18 (m, 2H), 4.40 (t, 1H, J = 5.8, 47.5 Hz), 4.50 (t, 1H, J = 5.8, 47.5 Hz), 6.77 (d, 1H, J = 8.0 Hz), 7.20 (d, 1H, J = 2.0 Hz), 7.35 (dd, 1H, J = 8.0, 2.0 Hz), 9.46 (brs, 1H); 13C NMR (125 MHz, CDCl3) δ 25.08 (d,1C, J = 20.4 Hz), 27.50, 36.78 (d, 1C, J = 3.9 Hz), 83.77 (d, 1C, J = 165.3 Hz), 84.93, 116.23, 116.66, 126.96, 132.25, 133.57, 152.71 (one carbon missing as a result of overlap). MS m/e 301 (M+, 25%), HRMS (EI) calcd for C12H13O2NBrF 301.0114, found 301.0115.
2-[4-(3-Fluoro-propyl)-4-methyl-2-oxo-1,4-dihydro-2H-benzo[d][1,3]oxazin-6-yl]-pyrrole-1-carboxylic acid tert-butyl ester (20)
A solution of 6-bromo-4-(3-fluoro-propyl)-4-methyl-1,4-dihydro-benzo[d][1,3]oxazin-2-one (19) (602 mg, 2 mmol) and tetrakis(triphenylphosphine)palladium(0) (58 mg, 0.05 mmol) in toluene (20 mL) was stirred under a flow of nitrogen for 25 min. To the solution was added sequentially 1-tert-butoxycarbonylpyrrole-2-boronic acid (824 mg, 3.9 mmol) in absolute EtOH (5 mL) and potassium carbonate (539 mg, 3.9 mmol) in water (5 mL). The mixture was heated to 80 ºC for 16 h and allowed to cool. The reaction mixture was poured into aqueous saturated sodium bicarbonate solution (20 mL) and extracted with EtOAc (3 × 20 mL). The organic layers were combined, washed with water (20 mL) and brine (10 mL) and dried over MgSO4. The solution was filtered, concentrated in vacuo, and the residue was purified by flash chromatography (10-30% EtOAc/hexane) to give 20 as a light yellow solid (72% yield) that was recrystallized from EtOAc/hexane, mp 87-89 ºC. 1H NMR (500 MHz, CDCl3) δ 1.41 (s, 9H), 1.72 (s, 3H),1.81-1.90 (m, 2H), 2.08-2.21 (m, 2H), 4.39 (t, 1H, J = 7.5, 47.5 Hz), 4.48 (t, 1H, J = 7.5, 47.5 Hz), 6.15-6.15 (m, 1H), 6.19-6.22 (m, 1H), 6.90 (d, 1H, J = 8.0 Hz), 7.08-7.10 (m, 1H), 7.22 (d, 1H, J = 8.0 Hz), 7.31-7.32 (m, 1H), 9.69 (brs, 1H); 13C NMR (125 MHz, CDCl3) δ 25.75 (d, 1C, J = 20.4 Hz), 27.77, 27.96, 36.85 (d, 1C, J = 4.9 Hz), 83.97 (d, 1C, J = 164.3 Hz), 83.93, 85.51, 110.84, 114.32, 114.74, 122.83, 123.57, 124.88, 129.97, 130.18, 133.58, 134.37, 149.33, 153.28. MS m/e 388 (M+, 20%), HRMS (EI) calcd for C21H25O4N2F 388.1798 (M+), found 388.1802.
2-Cyano-5-[4-(3-fluoro-propyl)-4-methyl-2-oxo-1,4-dihydro-2H-benzo[d][1,3]oxazin-6-yl]-pyrrole-1-carboxylic acid tert-butyl ester (21)
To a solution of 2-[4-(3-fluoro-propyl)-4-methyl-2-oxo-1,4-dihydro-2H-benzo[d][1,3]oxazin-6-yl]-pyrrole-1-carboxylic acid tert-butyl ester (20) (225 mg, 0.58 mmol) in THF (anhydrous, 5 mL) at -78 ºC was added chlorosulfonyl isocyanate (0.066 mL, 0.67 mmol). After 90 min, DMF (0.9 mL, 11.6 mmol) was added, and the reaction was allowed to warm to RT. The reaction mixture was poured into water (10 mL) and extracted with EtOAc (2 × 20 mL). The organic layers were combined, washed with brine (10 mL), dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by flash chromatography (10-30% EtOAc/hexane) to give 21 as a light yellow solid (76% yield) that was recrystallized from EtOAc/hexane, mp 87-89 ºC. 1H NMR (500 MHz, CDCl3) δ 1.53 (s, 9H), 1.75 (s, 3H),1.78-1.88 (m,2H), 2.07-2.13 (m, 1H), 2.16-2.22 (m, 1H), 4.40 (t, 1H, J =5.75, 47.0 Hz), 4.49 (t, 1H, J = 5.75, 47.0 Hz), 6.24 (d, 1H, J = 3.5 Hz), 6.90 (d, 1H, J = 8.0 Hz), 6.97 (d, 1H, J = 3.5 Hz), 7.07 (d, 1H, J = 1.5 Hz), 7.23 (dd, 1H, J = 8.0, 1.5 Hz), 9.05 (brs, 1H); 13C NMR (125 MHz, CDCl3) δ 25.14 (d, 1C, J = 20.4 Hz), 27.66, 27.80, 36.81 (d, 1C, J = 4.0 Hz), 83.74 (d, 1C, J = 165.3 Hz), 85.38 87.53, 105.85, 113.55, 114.19, 114.53, 124.21, 124.67, 124.88, 127.86, 129.97, 134.66, 139.74, 147.34, 152.49. 19F NMR (470 MHz, acetone-d6) δ -89.10. MS m/e 413 (M+, 75%), HRMS (EI) calcd for C22H24O4N3F 413.1551 (M+), found 413.1750.
5-[4-(3-Fluoro-propyl)-4-methyl-2-oxo-1,4-dihydro-2H-benzo[d][1,3]oxazin-6-yl]-1H-pyrrole -2-carbonitrile (22)
Compound 22 was synthesized using a procedure similar to that described for compound 13. The desired compound was purified by flash chromatography (40-60% EtOAc/hexane) to give 22 as a light yellow solid (68% yield) that was recrystallized from EtOAc/hexane, mp 150-152 ºC. 1H NMR (400 MHz, acetone-d6) δ 1.72 (s, 3H),1.72-1.82 (m, 2H), 2.02-2.10 (m, 1H), 2.19-2.24 (m, 1H), 4.40 (t, 1H, J = 7.5, 48.0 Hz), 4.52 (t, 1H, J = 7.5, 48.0 Hz), 6.66 (dd, 1H, J = 4.0, 2.0 Hz), 6.94 (dd, 1H, J = 4.0, 2.0 Hz), 7.06 (d, 1H, J = 8.0 Hz), 7.66-7.71 (m, 2H),9.31 (brs, 1H), 11.58 (brs, 1H); 13C NMR (100 MHz, acetone-d6) δ 25.12 (d, 1C, J = 24.8 Hz), 26.58, 36.42 (d, 1C, J =5.6 Hz), 83.50 (d, 1C, J = 163.1 Hz), 84.47, 101.25, 107.01, 114.47, 115.06, 120.90, 121.02, 125.68, 125.89, 126.22, 135.38, 137.25, 150.26. MS m/e 313 (M+, 70%), HRMS (EI) calcd for C17H16O2N3F 313.1227 (M+), found 313.1225.
5-[4-(3-Fluoro-propyl)-4-methyl-2-oxo-1,4-dihydro-2H-benzo[d][1,3]oxazin-6-yl]-1-methyl-1 H-pyrrole-2-carbonitrile (23)
Compound 23 was synthesized using a procedure similar to that described for compound 15. The desired product was purified by preparative thin-layer chromatography (10-35% EtOAc/hexane) to give 23 as a light yellow solid (95% yield) that was recrystallized from EtOAc/hexane, mp 150-152 ºC. 1H NMR (500 MHz, CDCl3) δ 1.74 (s, 3H),1.81-1.90 (m, 2H), 2.09-2.17 (m, 1H), 2.21-2.24 (m, 1H), 3.72 (s, 3H), 4.40-4.43 (m, 1H), 4.50-4.52 (m, 1H), 6.19 (d, 1H, J = 4.0 Hz), 6.85 (d, 1H, J = 4.0 Hz), 6.97 (d, 1H, J = 8.0 Hz), 7.10 (d, 1H, J =1.5 Hz), 7.27-7.28 (m, 1H), 9.36 (brs, 1H); 13C NMR (125 MHz, CDCl3) δ 25.23 (d, 1C, J = 24.8 Hz), 27.77, 33.94, 36.72 (d, 1C, J = 5.6 Hz), 83.64 (d, 1C, J = 164.0 Hz), 85.42, 105.96, 110.01, 114.07, 114.34, 115.33, 124.79, 125.02, 126.95, 129.96, 134.75, 139.23, 161.46. 19F NMR (470 MHz, acetone-d6) δ -90.07. MS m/e 327 (M+, 65%), HRMS (EI) calcd for C18H18O2N3F 327.1383 (M+), found 327.1381.
5-[4-(3-Fluoro-propyl)-4-methyl-2-thioxo-1,4-dihydro-2H-benzo[d][1,3]oxazin-6-yl]-1-methy l-1H-pyrrole-2-carbonitrile (24)
Compound 24 was synthesized using the procedure similar to that described for compounds 5. The desired product was purified by preparative thin-layer chromatography (30% EtOAc/hexane) to give a light yellow solid (71% yield) that was recrystallized from EtOAc/hexane, mp 72-74 ºC. 1H NMR (500 MHz, CDCl3) δ 1.77 (s, 3H),1.77-1.90 (m, 2H), 2.14-2.19 (m, 1H), 2.22-2.27 (m, 1H), 3.73 (s, 3H), 4.40-4.43 (m, 1H), 4.49-4.52 (m, 1H), 6.21 (d, 1H, J = 4.0 Hz), 6.86 (d, 1H, J = 4.0 Hz), 6.99-7.03 (m, 1H), 7.12-7.13 (m, 1H), 7.31-7.33 (m, 1H), 9.81 (brs, 1H); 13C NMR (125 MHz, CDCl3) δ 25.28 (d, 1C, J = 20.4 Hz), 27.23, 34.03, 36.64 (d, 1C, J = 3.9 Hz), 83. 51 (d, 1C, J = 165.3 Hz), 87.08, 106.41, 110.30, 114.17, 114.38, 115.21, 119.85, 124.77, 128.72, 130.03, 134.39, 138.76, 161.28. 19F NMR (470 MHz, acetone-d6) δ -89.10. MS m/e 343 (M+, 100%), HRMS (EI) calcd for C18H18ON3SF 343.1155 (M+), found 343.1155.
5-[4-(3-Fluoro-propyl)-4-methyl-2-thioxo-1,4-dihydro-2H-benzo[d][1,3]oxazin-6-yl]-1H-pyrr ole-2-carbonitrile (25)
Compound 25 was synthesized using a procedure similar to that described for compounds 5. The desired product was purified by preparative thin-layer chromatography (30% EtOAc/hexane) to give a light yellow solid (78% yield) that was recrystallized from EtOAc/hexane, mp 103-105 ºC. 1H NMR (500 MHz, acetone-d6) δ 1.78 (s, 3H), 2.01-2.25 (m, 4H), 4.41-4.44 (m, 1H), 4.50-4.53 (m, 1H), 6.71 (dd, 1H, J = 4.0, 1.0 Hz), 6.96 (dd, 1H, J = 4.0, 1.0 Hz), 7.21 (d, 1H, J = 8.5 Hz), 7.73-7.76 (m, 2H), 9.60 (brs, 1H), 11.62 brs, 1H); 13C NMR (125 MHz, CDCl3) δ 25.08 (d, 1C, J = 20.3 Hz), 25.97, 36.33 (d, 1C, J = 4.0 Hz), 83.57 (d, 1C, J = 162.3 Hz), 85.85, 107.49, 114.67, 115.08, 120.76, 121.04, 125.99, 126.21, 127.88, 132.21, 135.21, 136.91, 160.56. 19F NMR (470 MHz, acetone-d6) δ -90.11. MS m/e 329 (M+, 15%), HRMS (EI) calcd for C17H16ON3SF 329.0998 (M+), found 329.0998.
Receptor Binding Affinity Assays
Relative binding affinities for PR were determined by a competitive radiometric binding assay as previously described,19, 30 using 10 nM [3H]R5020 as tracer ([17α-methyl-3H]-promegestone) (Perkin-Elmer, Boston, MA), unlabeled R5020 as standard, and purified full length progesterone receptor B from PanVera/Invitrogen (Carlsbad, CA). Comparable experiments with AR30, 45 used [3H]R1881 ([17α methyl-3H] methyltrienolone) (Perkin-Elmer, Boston, MA), unlabeled R1881, and purified recombinant rat androgen receptor ligand binding domain (Pan Vera/Invitrogen, Carlsbad, CA), and with GR,46 3H Dexamethasone ([1,2,4-3H] dexamethasone (Amersham Biosciences, Piscataway, NJ), unlabeled dexamethasone, and adrenalectomized male rat liver cytosol. The protein was incubated for buffer or several concentrations of competitor for 18-24 h at 0 °C. Hydroxyapatite (BioRad, Hercules, CA) was used to absorb the receptor-ligand complexes, and free ligand was removed by washing with cold buffer. The binding affinities are expressed as relative binding affinity values with the RBA of the standard set to 100%. The values given are the average ± range or SD of two or more independent determinations. R5020 binds to PR with a KD of 0.4 nM.; R1881 to AR with a KD of 0.6nM; and Dexamethasone to GR with a KD of 19nM..
Incubations were for 18-24 h at 0 °C. Hydroxyapatite (BioRad, Hercules, CA) was used to absorb the receptor-ligand complexes, and free ligand was removed by washing with cold buffer. The binding affinities are expressed as relative binding affinity values with the RBA of R5020 set to 100%. The values given are the average ± range or SD of two or more independent determinations. R5020 binds to PR with a KD of 0.4 nM.
Molecular Modeling
Procedure for docking Tanaproget (1) and analogs
The PR structure was obtained from the PDB databank (1zuc) and prepared using the Molecular Operating Environment (MOE). Explicit hydrogen atoms were added, partial charges were computed using the MMFF94x force field, and the receptor-ligand complex was minimized with a termination gradient of 0.5. All water molecules were then deleted except the single water molecule hydrogen bound to Gln725 and Arg766. Finally, the ligand was removed and the receptor structure was processed using AutoDock Tools47 to define the AD4 atom types and calculate Gasteiger charges.
All compounds in Table 1 were constructed in Sybyl 8.1.1, cleaned up using the built-in Concord module,48 and minimized using the Powell method with a termination gradient of 0.5kcal/(mol*A), 100K maximum iterations, and MMFF94 force fields and charges. The ligands were prepared for docking using AutoDock Tools to assign AD4 atom types, calculate Gasteiger charges, and set all rotatable bonds as active torsions. Each ligand was docked into the receptor using AutoDock Vina. The grid box was centered on the ligand in the original crystal structure, and measured 18 Å by 18 Å by 22 Å. To ensure that the proper binding conformation was found, the exhaustiveness parameter was set to 100 (default=8, linear scale); all other default settings were used.
The top five poses for each ligand were visually inspected in MOE. Unreasonable poses were discarded and the lowest energy conformation of the remaining poses was selected for further use. The selected pose and receptor structure were merged into a single file, all hydrogen atoms were explicitly added, the partial charges were calculated using the MMFF94x force field, and the protein-ligand complex was minimized. The minimization was conducted in four stages: stage 1 minimized the hydrogen atoms that were added to the structure in the previous step; stage 2 minimized the ligand; stage 3 minimized any residues within 4.5 Å of the ligand; and stage 4 minimized both the ligand and any residues within 4.5 Å. This worked up structure was used for all other calculations and analyses.
Procedure for calculating the torsion energetics of N-substituted analogs (Figure 1)
The worked up structure was opened in MOE and all atoms deleted except for the ligand. The dihedral angle relating the pyrrole to the benzoxazin-2-thione core was rotated from 0-90 degrees, measuring each energy term in 0.5-degree increments. The relative value of each term was calculated by subtracting the minimum measured value.
Procedure for calculating the interaction energy of the protein-ligand complex (Figure 2B)
Similar to the above process, the pyrrole was rotated with respect to the core by setting the dihedral angle from 0-90 degrees, measuring the interaction energy in 0.5-degree increments. To ensure that any observed change in the interaction energy was due solely to a difference in the N-substitution, we started from the worked up pose of compound 5a, deleting atoms as necessary to obtain N-Me (1) and N-H (analog of 1). The partial charges were recalculated using MMFF94x force field prior to beginning the rotation.
Procedure for calculating the mean hydrogen bond score (Table 2)
The hydrogen bonds formed between the ligand and protein were scored using the “dock_HydrogenBonds” module found in the standard scientific vector language (SVL) library for MOE. This module forms the basis for the “ligand interactions” feature accessible from MOE's graphical user interface. The mean score was subsequently calculated as the geometric mean of the individual scores.
Supplementary Material
Acknowledgments
Supported by grants from the NIH (PHS 5R37 DK015556). Funding for NMR and MS instrumentation is from the Keck Foundation, NIH and NSF We are grateful to Dr. Sung Hoon Kim for helpful comments.
Abbreviations
- AR
androgen receptor
- 9-BBN
9-borabicyclo[3.3.1]nonage
- CDI
carbonyl diimidazole
- CSI
chlorosulfonyl isocynate
- DAST
diethylaminosulfur trifluoride
- ER
estrogen receptor
- FES
16α-[18F]fluoroestradiol
- FFNP
fluoro furanyl norprogesterone
- GR
glucocorticoid receptor
- PET
positron emission tomography
- RBA
relative binding affinity
- PR
progesterone receptor
- vdW
van der Waals
Footnotes
Supporting Information Available: Normal and reversed-phase HPLC results and chromatograms of the fluoro-substituted compounds 5a-b, 16, 17, 24 and 25. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Benagiano G, Bastianelli C, Farris M. Selective progesterone receptor modulators 3: use in oncology, endocrinology and psychiatry. Expert Opin Pharmacother. 2008;9:2487–2496. doi: 10.1517/14656566.9.14.2487. [DOI] [PubMed] [Google Scholar]
- 2.Lanari C, Molinolo AA. Progesterone receptors--animal models and cell signalling in breast cancer. Diverse activation pathways for the progesterone receptor: possible implications for breast biology and cancer. Breast Cancer Res. 2002;4:240–243. doi: 10.1186/bcr539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klijn JG, Setyono-Han B, Foekens JA. Progesterone antagonists and progesterone receptor modulators in the treatment of breast cancer. Steroids. 2000;65:825–830. doi: 10.1016/s0039-128x(00)00195-1. [DOI] [PubMed] [Google Scholar]
- 4.Hughes-Davies L, Caldas C, Wishart GC. Tamoxifen: the drug that came in from the cold. Br J Cancer. 2009;101:875–878. doi: 10.1038/sj.bjc.6605231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Normanno N, Morabito A, De Luca A, Piccirillo MC, Gallo M, Maiello MR, Perrone F. Target-based therapies in breast cancer: current status and future perspectives. Endocr Relat Cancer. 2009;16:675–702. doi: 10.1677/ERC-08-0208. [DOI] [PubMed] [Google Scholar]
- 6.Peng J, Sengupta S, Jordan VC. Potential of selective estrogen receptor modulators as treatments and preventives of breast cancer. Anticancer Agents Med Chem. 2009;9:481–499. doi: 10.2174/187152009788451833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dehdashti F, Flanagan FL, Mortimer JE, Katzenellenbogen JA, Welch MJ, Siegel BA. Positron emission tomographic assessment of “metabolic flare” to predict response of metastatic breast cancer to antiestrogen therapy. Eur J Nucl Med. 1999;26:51–56. doi: 10.1007/s002590050359. [DOI] [PubMed] [Google Scholar]
- 8.Mortimer JE, Dehdashti F, Siegel BA, Trinkaus K, Katzenellenbogen JA, Welch MJ. Metabolic flare: indicator of hormone responsiveness in advanced breast cancer. J Clin Oncol. 2001;19:2797–2803. doi: 10.1200/JCO.2001.19.11.2797. [DOI] [PubMed] [Google Scholar]
- 9.Mortimer JE, Dehdashti F, Siegel BA, Katzenellenbogen JA, Fracasso P, Welch MJ. Positron emission tomography with 2-[18F]Fluoro-2-deoxy-D-glucose and 16alpha-[18F]fluoro-17beta-estradiol in breast cancer: correlation with estrogen receptor status and response to systemic therapy. Clin Cancer Res. 1996;2:933–939. [PubMed] [Google Scholar]
- 10.Dehdashti F, Mortimer JE, Trinkaus K, Naughton MJ, Ellis M, Katzenellenbogen JA, Welch MJ, Siegel BA. PET-based estradiol challenge as a predictive biomarker of response to endocrine therapy in women with estrogen-receptor-positive breast cancer. Breast Cancer Res Treat. 2009;113:509–517. doi: 10.1007/s10549-008-9953-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Santen R, Manni A, Harvey H, Redmond C. Endocrine treatment of breast cancer in women. Endocrine Rev. 1990;11:221–265. doi: 10.1210/edrv-11-2-221. [DOI] [PubMed] [Google Scholar]
- 12.Bardou VJ, Arpino G, Elledge RM, Osborne CK, Clark GM. Progesterone receptor status significantly improves outcome prediction over estrogen receptor status alone for adjuvant endocrine therapy in two large breast cancer databases. J Clin Oncol. 2003;21:1973–1979. doi: 10.1200/JCO.2003.09.099. [DOI] [PubMed] [Google Scholar]
- 13.Osborne C, Tripathy D. Aromatase inhibitors: rationale and use in breast cancer. Annu Rev Med. 2005;56:103–116. doi: 10.1146/annurev.med.56.062804.103324. [DOI] [PubMed] [Google Scholar]
- 14.Furr BJ, Jordan VC. The pharmacology and clinical uses of tamoxifen. Pharmacol Ther. 1984;25:127–205. doi: 10.1016/0163-7258(84)90043-3. [DOI] [PubMed] [Google Scholar]
- 15.Noguchi S, Miyauchi K, Nishizawa Y, Koyama H. Induction of progesterone receptor with tamoxifen in human breast cancer with special reference to its behavior over time. Cancer. 1988;61:1345–1349. doi: 10.1002/1097-0142(19880401)61:7<1345::aid-cncr2820610712>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 16.Howell A, Harland RN, Barnes DM, Baildam AD, Wilkinson MJ, Hayward E, Swindell R, Sellwood RA. Endocrine therapy for advanced carcinoma of the breast: relationship between the effect of tamoxifen upon concentrations of progesterone receptor and subsequent response to treatment. Cancer Res. 1987;47:300–304. [Google Scholar]
- 17.Namer M, Lalanne C, Baulieu EE. Increase of progesterone receptor by tamoxifen as a hormonal challenge test in breast cancer. Cancer Res. 1980;40:1750–1752. [PubMed] [Google Scholar]
- 18.Buckman BO, Bonasera TA, Kirschbaum KS, Welch MJ, Katzenellenbogen JA. Fluorine-18-labeled progestin 16 alpha, 17 alpha-dioxolanes: development of high-affinity ligands for the progesterone receptor with high in vivo target site selectivity. J Med Chem. 1995;38:328–337. doi: 10.1021/jm00002a014. [DOI] [PubMed] [Google Scholar]
- 19.Zhou D, Carlson KE, Katzenellenbogen JA, Welch MJ. Bromine- and iodine-substituted 16alpha,17alpha-dioxolane progestins for breast tumor imaging and radiotherapy: synthesis and receptor binding affinity. J Med Chem. 2006;49:4737–4744. doi: 10.1021/jm060348q. [DOI] [PubMed] [Google Scholar]
- 20.Zhou D, Sharp TL, Fettig NM, Lee H, Lewis JS, Katzenellenbogen JA, Welch MJ. Evaluation of a bromine-76-labeled progestin 16alpha, 17alpha-dioxolane for breast tumor imaging and radiotherapy: in vivo biodistribution and metabolic stability studies. Nucl Med Biol. 2008;35:655–663. doi: 10.1016/j.nucmedbio.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Verhagen A, Studeny M, Luurtsema G, Visser GM, De Goeij CC, Sluyser M, Nieweg OE, Van der Ploeg E, Go KG, Vaalburg W. Metabolism of a [18F]fluorine labeled progestin (21-[18F]fluoro-16 alpha-ethyl-19-norprogesterone) in humans: a clue for future investigations. Nucl Med Biol. 1994;21:941–952. doi: 10.1016/0969-8051(94)90083-3. [DOI] [PubMed] [Google Scholar]
- 22.Kochanny MJ, VanBrocklin HF, Kym PR, Carlson KE, O'Neil JP, Bonasera TA, Welch MJ, Katzenellenbogen JA. Fluorine-18-labeled progestin ketals: synthesis and target tissue uptake selectivity of potential imaging agents for receptor-positive breast tumors. J Med Chem. 1993;36:1120–1127. doi: 10.1021/jm00061a002. [DOI] [PubMed] [Google Scholar]
- 23.Katzenellenbogen JA. Receptor Imaging of Tumors (Non-Peptide) In: Welch MJ, Redvanly C, editors. Handbook of Radiopharmaceuticals: Radiochemistry and Applications. John Wiley & Sons, Ltd; London: 2003. pp. 715–750. [Google Scholar]
- 24.Fensome A, Bender R, Chopra R, Cohen J, Collins MA, Hudak V, Malakian K, Lockhead S, Olland A, Svenson K, Terefenko EA, Unwalla RJ, Wilhelm JM, Wolfrom S, Zhu Y, Zhang Z, Zhang P, Winneker RC, Wrobel J. Synthesis and structure-activity relationship of novel 6-aryl-1,4-dihydrobenzo[d][1,3]oxazine-2-thiones as progesterone receptor modulators leading to the potent and selective nonsteroidal progesterone receptor agonist tanaproget. J Med Chem. 2005;48:5092–5095. doi: 10.1021/jm050358b. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Z, Olland AM, Zhu Y, Cohen J, Berrodin T, Chippari S, Appavu C, Li S, Wilhem J, Chopra R, Fensome A, Zhang P, Wrobel J, Unwalla RJ, Lyttle CR, Winneker RC. Molecular and pharmacological properties of a potent and selective novel nonsteroidal progesterone receptor agonist tanaproget. J Biol Chem. 2005;280:28468–28475. doi: 10.1074/jbc.M504144200. [DOI] [PubMed] [Google Scholar]
- 26.Winneker RC, Fensome A, Zhang P, Yudt MR, McComas CC, Unwalla RJ. A new generation of progesterone receptor modulators. Steroids. 2008;73:689–701. doi: 10.1016/j.steroids.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 27.Zhang P, Terefenko EA, Fensome A, Wrobel J, Winneker R, Zhang Z. Novel 6-aryl-1,4-dihydrobenzo[d]oxazine-2-thiones as potent, selective, and orally active nonsteroidal progesterone receptor agonists. Bioorg Med Chem Lett. 2003;13:1313–1316. doi: 10.1016/s0960-894x(03)00128-8. [DOI] [PubMed] [Google Scholar]
- 28.Trost BM, Tang W. Migratory hydroamination: a facile enantioselective synthesis of benzomorphans. J Am Chem Soc. 2003;125:8744–8745. doi: 10.1021/ja0360539. [DOI] [PubMed] [Google Scholar]
- 29.Sakaitani M, Ohfune Y. Syntheses and reactions of silyl carbamates. 2. A new mode of cyclic carbamate formation from tert-butyldimethylsilyl carbamate. J Am Chem Soc. 1990;112:1150–1158. [Google Scholar]
- 30.Brandes SJ, Katzenellenbogen JA. Fluorinated androgens and progestins: molecular probes for androgen and progesterone receptors with potential use in positron emission tomography. Mol Pharmacol. 1987;32:391–403. [PubMed] [Google Scholar]
- 31.Dong Y, Roberge JY, Wang Z, Wang X, Tamasi J, Dell V, Golla R, Corte JR, Liu Y, Fang T, Anthony MN, Schnur DM, Agler ML, Dickson JK, Jr, Lawrence RM, Prack MM, Seethala R, Feyen JH. Characterization of a new class of selective nonsteroidal progesterone receptor agonists. Steroids. 2004;69:201–217. doi: 10.1016/j.steroids.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 32.Dore JC, Gilbert J, Ojasoo T, Raynaud JP. Correspondence analysis applied to steroid receptor binding. J Med Chem. 1986;29:54–60. doi: 10.1021/jm00151a009. [DOI] [PubMed] [Google Scholar]
- 33.Hoppe C, Steinbeck C, Wohlfahrt G. Classification and comparison of ligand-binding sites derived from grid-mapped knowledge-based potentials. J Mol Graph Model. 2006;24:328–340. doi: 10.1016/j.jmgm.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 34.Ojasoo T, Delettre J, Mornon JP, Turpin-VanDycke C, Raynaud JP. Towards the mapping of the progesterone and androgen receptors. J Steroid Biochem. 1987;27:255–269. doi: 10.1016/0022-4731(87)90317-7. [DOI] [PubMed] [Google Scholar]
- 35.Ojasoo T, Dore JC, Gilbert J, Raynaud JP. Binding of steroids to the progestin and glucocorticoid receptors analyzed by correspondence analysis. J Med Chem. 1988;31:1160–1169. doi: 10.1021/jm00401a015. [DOI] [PubMed] [Google Scholar]
- 36.Palmer S, Campen CA, Allan GF, Rybczynski P, Haynes-Johnson D, Hutchins A, Kraft P, Kiddoe M, Lai M, Lombardi E, Pedersen P, Hodgen G, Combs DW. Nonsteroidal progesterone receptor ligands with unprecedented receptor selectivity. J Steroid Biochem Mol Biol. 2000;75:33–42. doi: 10.1016/s0960-0760(00)00134-5. [DOI] [PubMed] [Google Scholar]
- 37.Wilkinson JM, Hayes S, Thompson D, Whitney P, Bi K. Compound profiling using a panel of steroid hormone receptor cell-based assays. J Biomol Screen. 2008;13:755–765. doi: 10.1177/1087057108322155. [DOI] [PubMed] [Google Scholar]
- 38.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mordasini T, Curioni A, Bursi R, Andreoni W. The Binding Mode of Progesterone to Its Receptor Deduced from Molecular Dynamics Simulations. ChemBioChem. 2003;4:155–161. doi: 10.1002/cbic.200390026. [DOI] [PubMed] [Google Scholar]
- 40.Harada T, Yamagishi K, Nakano T, Kitaura K, Tokiwa H. Ab initio fragment molecular orbital study of ligand binding to human progesterone receptor ligand-binding domain. Naunyn-Schmiedeberg's Arch Pharmacol. 2008;377:607–615. doi: 10.1007/s00210-008-0268-9. [DOI] [PubMed] [Google Scholar]
- 41.Molecular Operating Environment (MOE), Chemical Computing Group. http://www.chemcomp.com/
- 42.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Safe and Convenient Procedure for Solvent Purification. Organometallics. 1996;15:1518–1520. [Google Scholar]
- 43.Chi DY, Kilbourn MR, Katzenellenbogen JA, Welch MJ. A Rapid and Efficient Method for the Fluoroalkylation of Amines and Amides. Development of a Method Suitable for Incorporation of the Short-Lived Positron Emitting Radionuclide Fluorine- 18. J Org Chem. 1987:658–664. [Google Scholar]
- 44.Baker LJ, Copp BR, Rickard CEF. 2′-Amino-5′-bromoacetophenone. Acta crystallographica. 2001;57:o540–o541. [Google Scholar]
- 45.Parent EE, Dence CS, Jenks C, Sharp TL, Welch MJ, Katzenellenbogen JA. Synthesis and biological evaluation of [18F]bicalutamide, 4-[76Br]bromobicalutamide, and 4-[76Br]bromo-thiobicalutamide as non-steroidal androgens for prostate cancer imaging. J Med Chem. 2007;50:1028–1040. doi: 10.1021/jm060847r. [DOI] [PubMed] [Google Scholar]
- 46.Pomper MG, Kochanny MJ, Thieme AM, Carlson KE, VanBrocklin HF, Mathias CJ, Welch MJ, Katzenellenbogen JA. Fluorine-substituted corticosteroids: synthesis and evaluation as potential receptor-based imaging agents for positron emission tomography of the brain. Int J Rad Appl Instrum B. 1992;19:461–480. doi: 10.1016/0883-2897(92)90161-q. [DOI] [PubMed] [Google Scholar]
- 47.Sanner MF. Python: a programming language for software integration and development. J Mol Graph Model. 1999;17:57–61. [PubMed] [Google Scholar]
- 48.Pearlman RS. Concord. St. Louis, Missouri, 63144, USA: distributed by Tripos International. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.