

Abstract
Introduction
An HLA-DRA-DRB1*0101-restricted T-cell epitope in the factor VIII (FVIII) C2 domain occurred in a mild haemophilia A patient with missense substitution FVIII-A2201P. His T cells responded to synthetic peptides FVIII2186-2205 and FVIII2194-2213.
Aim
T cells from family members with genotype FVIII-A2201P were analyzed to determine if FVIII-specific T cells occur in individuals with a hemophilic mutation but no clinically significant inhibitor response.
Methods
Fluorescent MHC class II tetramers corresponding to subjects’ HLA-DRB1 types were loaded with 20-mer peptides and utilized to label antigen-specific CD4+ T cells. T-cell responses to peptides spanning the FVIII-C2 sequence were evaluated. T cells recognizing specific peptides were cloned, and antigen specificity was verified by proliferation assays. Plasma and/or purified IgG samples were tested for FVIII inhibitory activity.
Results
CD4+ T cells and T-cell clones from two brothers who shared the DRB1*0101 allele responded to FVIII2194-2213. A haemophilic cousin’s HLA-DRA-DRB1*1104-restricted response to FVIII2202-2221 was detected only when CD4+CD25+ cells were depleted. A great uncle and two obligate carriers had no detectable FVIII-C2-specific T cells. Concentrated IgG from the brother without a clinical inhibitor response showed a low-titer FVIII inhibitor.
Conclusion
FVIII-specific T cells and inhibitory IgG were found in a previously infused, haemophilic subject who had a sub-clinical FVIII inhibitor. CD4+CD25+ depleted T cells from a non-infused haemophilic cousin recognized an overlapping FVIII epitope, indicating a latent HLA-DRA-DRB1*1104-restricted T-cell response to FVIII. Specific T-cell responses to FVIII can occur without clinically significant inhibitors.
Keywords: haemophilia A, factor VIII inhibitors, HLA-DR, T-cell, epitope mapping
Introduction
Haemophilia A, a congenital bleeding disorder, is caused by a deficiency or functional defect of factor VIII (FVIII) and is treated by infusions of recombinant or plasma-derived FVIII [1]. Neutralizing antibodies to FVIII, clinically termed ‘inhibitors’, are a serious complication of FVIII infusion that develop in approximately one-third of severe patients, and they generally occur within the first five years of FVIII treatment [2-4]. FVIII inhibitors develop less frequently in mild/moderately severe patients [5-7], in whom they may occur later in life. Thus, the highest risk for inhibitor development is associated with mutations resulting in the absence or severe truncation of the FVIII protein [8, 9]. Inhibitors have been reported in mild or moderate haemophilia A patients with over 30 missense substitutions [7, 9-11]. Substitutions associated with an increased inhibitor risk occur predominantly in the FVIII A2 domain and light chain [12]. The assessment of possible risks posed by specific human leukocyte antigen (HLA) class II alleles in association with FVIII genotype has been limited [13-18]. Weak or no associations of HLA class II alleles with inhibitors have been described in subjects stratified according to the presence or absence of the FVIII intron 22 inversion [16, 17].
FVIII inhibitor development appears to depend on antigen-specific T-cell help. Evidence for this includes somatic hypermutations in the genes coding for the variable part of anti-FVIII antibodies [19], a large proportion of anti-FVIII antibodies belonging to the IgG1 and IgG4 subclasses, indicating isotype switching [20], and the presence of FVIII-specific memory B cells [21]. Direct evidence of the involvement of helper T cells in FVIII inhibitor responses came from a study of severe haemophilia A inhibitor subjects infected with human immunodeficiency virus type 1. Thirteen subjects with high-responder inhibitors lost their anamnestic response to FVIII infusions in the advanced stages of HIV-1 infection, indicating the virus impaired T cells necessary for anti-FVIII antibody production [22].
FVIII-specific T cells in the blood of haemophilia A subjects with inhibitors were suggested by testing peripheral blood mononuclear cells (PBMCs) depleted of B cells or CD8+ T cells for FVIII-specific proliferation [23, 24]. PBMCs depleted of CD8+ T cells from inhibitor subjects were shown to proliferate upon stimulation with various peptides corresponding to sequences in the FVIII A2 [25], A3 [26], and C2 domains [27]. Interestingly, proliferation of T cells from healthy subjects and haemophilia A subjects without inhibitors has also been observed when FVIII is added to cells in culture [23-29]. Some studies have noted that responses of CD4+ T cells from healthy controls and from haemophilia A subjects without inhibitors tend to be smaller in magnitude and transient [23, 24, 29], whereas stronger T-cell responses have been seen for healthy controls in other studies [26-28]. A recent report showed that PBMCs from healthy individuals proliferated or increased their proliferative response to FVIII when CD4+CD25+ cells expressing Foxp3 were depleted [30].
The relationships between T-cell responses to FVIII, FVIII genotypes, and HLA class II alleles are expected to be important in determining the immune outcome when FVIII is infused [31]. Jacquemin et al analyzed T cells from a mild haemophilia A inhibitor subject with missense substitution R2150H, isolating three T-cell clones that responded to wild-type FVIII and to a synthetic peptide containing the wild-type R2150 sequence, FVIII2144-2161 [32]. These clones were restricted by at least two of the subject’s HLA-DR allelic proteins. Jones et al. identified another C1 domain epitope(s) in a peptide corresponding to FVIII2089-2112. This peptide stimulated proliferation of a polyclonal T-cell line from a severe haemophilia A subject, and it bound to multiple HLA-DR allelic proteins [28]. We recently analyzed T cells from a mild haemophilia A subject with missense substitution A2201P [33], using the technique of tetramer guided epitope mapping (TGEM) [34], in serial blood samples obtained for over one year following initial detection of his inhibitor response. This epitope is within the FVIII C2 domain, FVIII2194-2205, which contains the wild-type sequence at the haemophilic missense site, and it was HLA-DRA-DRB1*0101-restricted. T-cell clones isolated using DRB1*0101 tetramers proliferated in response to peptides with the wild-type sequence but not to a peptide with the haemophilic P2201 sequence, indicating that these T cells could clearly distinguish self-versus wild-type FVIII.
In this report, we extend our study of HLA-DR-restricted FVIII T-cell epitopes in subjects with the A2201P substitution by analyzing the family members of the inhibitor subject described above [33]. Three additional family members had mild haemophilia A due to the A2201P missense substitution: two of these had received FVIII infusions, but none had a clinically significant inhibitor. T cells from two mothers who were obligate A2201P carriers were also analyzed.
Materials and Methods
Subjects
Blood samples from four related haemophilia A subjects and two carriers with the FVIII missense substitution A2201P (Fig. 1) were obtained following written, informed consent according to a protocol approved by the University of Washington’s Human Subjects Review Committee.
Figure 1. The A2201P mild haemophilia A family pedigree.
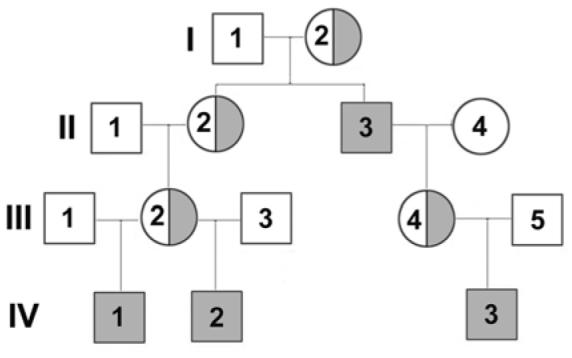
The high-titer inhibitor subject previously described [33] is depicted here as the first member of generation IV, or IV-1. Mild haemophilia A is indicated by shaded squares, and the half-shaded circles indicate the obligate carrier mothers.
Genotyping
DNA was extracted from leukocytes in whole blood anti-coagulated with EDTA. HLADRB1 genotypes were determined using a micro-PCR-sequence-specific primers (SSP) method (Puget Sound Blood Center HLA Laboratory, Seattle, WA, USA). The f8-A2201P mutation was identified using heteroduplex screening of PCR-amplified FVIII exon fragments and DNA sequencing as described [35, 36], the latter using an ABI #3100 capillary sequencer.
Plasma analysis
FVIII inhibitor titers for plasma samples were determined by the Bethesda protocol [37]. IgG from subject IV-2 was purified from plasma on a Protein G affinity column (Pierce Biotechnology, Rockford, IL) according to the manufacturer’s instructions. The IgG eluate was dialyzed against phosphate buffered saline (0.05 M phosphate, 0.15M NaCl, pH 7.4) and concentrated to 10 mg/ml using Centricon-30 tubes (Amicon, Beverly, MA). Bethesda assays were carried out on serial dilutions of this IgG mixed with a normal human plasma pool.
FVIII peptides
A panel of 20-mer overlapping peptides (with a 12 amino-acid overlap) spanning the FVIII C2 domain sequence, plus two A2 domain peptides, was synthesized (Global Peptide Inc., Ft. Collins, CO, USA; SynPep, Dublin, CA, USA; Anaspec, San Jose, CA, USA). Peptide pools contained equal concentrations of five peptides with a total concentration of 10 mg/ml in DMSO/water. The sequences of these peptides and their division into five pools were described previously [33].
MHC class II tetramers
The proteins encoded by HLA-DR alleles, e.g. HLA-DRA-DRB1*0101, are referred to using the abbreviated DR convention, e.g. DR0101. All DR proteins in this study are encoded by DRB1 alleles. Fluorescent MHC class II tetramers were produced as described [38]. Briefly, soluble recombinant HLA-DR monomers were produced in Schneider S-2 insect cells, affinity-purified from cell supernatants, and biotinylated at a single site. These monomers were incubated with 0.2 mg/ml of either pooled or individual FVIII peptides in the presence of 0.25% n-octyl-β-D-glucopyranoside and 1 mM Pefabloc SC at 37°C for 72 h. Tetramers were formed by adding phycoerythrin (PE)-conjugated streptavidin (BioSource International, Camarillo, CA, USA) at a molar ratio of 8:1 to the following peptide-loaded HLA-DRA-DRB1 monomers: DR0101, DR0401, DR0404, DR0901, DR1104, and DR1501. The activities of all tetramer reagents were confirmed by loading the monomeric proteins with a reference peptide, adding streptavidin to form tetramers, and confirming their ability to stain a reference T-cell clone.
TGEM
As in our previous study [33], we used a TGEM strategy [34] to investigate T-cell responses in the extended family of an inhibitor subject with haemophilic missense substitution A2201P. CD4+ T cells were isolated from PBMCs by negative selection using a CD4 isolation kit (Miltenyi Biotec, Auburn, CA, USA). CD4+CD25+ T cells were then removed from half of the total CD4+ T-cell fraction by positive selection using CD25+ microbeads (Miltenyi Biotec). The non-CD4+ cell fraction was used to coat 48-well plates (3 million cells/well), which were incubated at 37°C for 1 h and washed, leaving adherent cells in the well. Total CD4+ or CD4+CD25+ depleted T cells (1.7 million cells/well) were added to the adherent cells and stimulated with 10 μg/ml pooled peptides in T-cell medium (RPMI 1640 with 25 mM HEPES, 15% human serum (MP Biomedicals, LLC, Solon, Ohio USA), 2 mM L-glutamine, 50 U/ml penicillin, 50 μg/ml streptomycin). The medium was supplemented with 40 U/ml IL-2 (Hemagen, Waltham, MD, USA) on day 7 and the cells were maintained with fresh medium and IL-2 for 13-19 days, at which point they were analyzed with tetramers. Approximately 0.75 × 105 cells were incubated with PE-tetramers loaded with pooled peptides (10 μg/ml final concentration in 75 μl T-cell medium) at 37°C for 1 h, then labeled with PerCP anti-human CD3 (BD Biosciences, San Jose, CA, USA), allophycocyanin (APC) anti-human CD4 (eBioscience, San Diego, CA, USA), and fluorescein isothiocyanate (FITC) anti-human CD25 (eBioscience) IgGs at 4°C for 20 min. (PE-Cy7 anti-human CD3 (eBioscience) was used instead of PerCP anti-human CD3 in one experiment). Stained cells were analyzed on a FACSCalibur (Becton Dickinson, San Jose, CA, USA). Tetramer-positive responses were then decoded by a second round of staining using tetramers carrying individual peptides. Flow cytometric data were gated for CD3+ lymphocytes using CD3 staining and forward/side scatter, than analyzed by plotting CD4-versus-tetramer staining and CD25-versus-tetramer staining. Antigen-specific tetramer staining produces similar patterns for these two plots, so both were analyzed in order to identify or rule out nonspecific, artifactual tetramer staining.
T-cell clone isolation
Tetramer- and FITC anti-human CD4 IgG (eBioscience)-stained cells were single-cell sorted using a FACS Vantage sorter (Becton Dickinson) into a 96-well plate containing T-cell medium. These single T cells were expanded by adding a mixture of 2 μg/ml phytohemagglutinin (PHA) and HLA-mismatched irradiated PBMCs. Twenty-four hours later, IL-2 was added at a final concentration of 40 U/ml IL-2. Cells that expanded after three weeks in culture were re-stimulated with PHA and HLA-mismatched irradiated PBMCs followed by the addition of 40 U/ml IL-2. Ten to 14 days later, the expanded cells were incubated with tetramer loaded with the relevant peptide, or with an irrelevant peptide as a negative control, followed by incubation with FITC or APC anti-human CD4 IgG (eBioscience). Staining of the cells was analyzed on the FACSCaliber (Becton Dickinson).
Antigen-specific T-cell proliferation assays
Proliferation assays of T-cell clones were performed as previously described [33]. Briefly, irradiated PBMCs from a non-haemophilic donor with a DRB1*0101 allele were plated into a 96-well plate at a concentration of 105 cells/well in 100 μl T-cell medium. Peptides diluted in DMSO were added in a 1 μl volume to give a final concentration of 10, 1, 0.1, and 0 μM in the well, and the plates were incubated at 37°C for 4 h. T-cell clones (104 cells/well) were then added in 100 μl T-cell medium and the co-cultures were incubated at 37°C. [3H]thymidine (1 μCi/well) was added at 48 h. Cells were harvested with a Tomtech Harvestor 96 (Hamden, CT USA) after 18 h of further incubation and [3H]thymidine uptake was measured with a Wallac Microbeta TriLux liquid scintillation counter (Waltham, MA USA).
Results
Haemophilic characteristics and DRB1 genotypes
Four members of this family had mild haemophilia A due to FVIII missense substitution A2201P; the inhibitor subject described previously (subject IV-1) [33], his brother (IV-2), his cousin (IV-3), and his great-uncle (II-3) (Fig. 1). Subject IV-1 had received only two FVIII infusions several years prior to receiving intensive FVIII treatment to support tonsillectomy/adenectomy, at which time he developed an inhibitor with a peak titer of 250 BU/ml. Subjects IV-2 and II-3 have each received over 50 FVIII infusions with cryoprecipitate and commercial concentrates but have not developed clinically significant inhibitors; IV-2 received prolonged therapy for major chest trauma and II-3 received support for a laminectomy. Subject IV-3 has not received FVIII infusions. The DRB1 genotypes of all of the haemophilic family members and of two obligate carriers were determined (Table 1). The inhibitor subject (IV-1), his brother (IV-2), and his mother (III-2) shared a DRB1*0101 allele. Subjects IV-3 and his mother, III-4 shared a DRB1*1104allele.
Table 1.
DRB1 genotypes of the mild haemophilia A family with missense substitution A2201P
| Subject | Status | DRB1* genotypes |
|---|---|---|
| II-3 | haemophilic | 0404, 1501 |
| III-2 | carrier | 0101, 0901 |
| III-4 | carrier | 1104, 1501 |
| IV-1 | haemophilic | 0101, 1503 |
| IV-2 | haemophilic | 0101, 0401 |
| IV-3 | haemophilic | 0901, 1104 |
HLA-restricted T-cell epitopes in the FVIII C2 domain
Haemophilic subject IV-2 was screened for DR0101 and DR0401-restricted FVIII C2 T-cell epitopes using TGEM. The blood sample used for TGEM was obtained two years after his last FVIII exposure. A second sample was obtained recently, when he was receiving daily FVIII infusions as support after a minor sports injury. The tetramer-staining pattern was similar for the two blood samples; results of staining the first sample are shown in Fig. 2. T cells that bound DR0101 tetramers loaded with C2 peptide pools 1 and 2 were identified in total CD4+ T-cell cultures (Fig. 2A). A small population of tetramer-positive cells (0.6%) was observed when these CD4+ T cells were incubated with tetramers loaded with peptide pool 4 (Fig. 2A), but this was not observed for CD4+ cells from the more recent blood sample. Only a background level of tetramer-positive cells (0.3% or less) was observed when these CD4+ T cells were incubated with tetramers loaded with peptide pools 3 and 5. An aliquot of this subject’s CD4+ cells was depleted of CD4+CD25+ cells and TGEM was carried out as before (data not shown). An enhanced tetramer-positive response to peptide pool 1 was observed: 8.6% of cells incubated with tetramers carrying pool 1 peptides were tetramer-positive compared to 0.9% of total CD4+ cells. Tetramer-positive responses were observed (1.5%) but were not enhanced for peptide pool 2. Tetramer-positive responses were not observed for peptide pools 3-5. No DR0401-restricted T cells were detected in total CD4+ (Fig. 2B) or in CD4+CD25+-depleted CD4+ T-cell cultures (data not shown). Pool 1 and 2 tetramer-positive responses were decoded using both total CD4+ and CD4+CD25+-depleted CD4+ T-cell cultures. Fig. 2C presents results for the cultures that showed the strongest T-cell staining for pool 1 (CD4+CD25+-depleted T cells) and pool 2 (CD4+ T-cells) peptides, respectively. Three overlapping peptides contained DR0101-restricted T-cell epitopes: FVIII2187-2205 (peptide sequence: DAQITASSYFTNMFATWSP), FVIII2186-2205 (SDAQITASSYFTNMFATWSP) and FVIII2194-2213 (SYFTNMFATWSPSKARLHLQ). Tetramers loaded with these same three peptides also stained T cells from haemophilic inhibitor subject IV-1 [33].
Figure 2. T-cell epitope mapping for haemophilic subject IV-2.

CD4+ T cells were stimulated with pooled peptides spanning the FVIII C2 domain sequence. Eighteen days later, the cells were incubated with PE-labeled DR0101 tetramers loaded with FVIII C2 peptide pools (A) or with DR0401 tetramers loaded with FVIII C2 peptide pools (B) and antibodies as described in Methods. Decoding of positive CD4+ responses to DR0101 tetramers loaded with peptide pools 1 and 2 was carried out 22 days after stimulation of total CD4+ cells (top row) or CD4+CD25+-depleted CD4+ cells (bottom row), respectively (C). Decoding of DR0101-restricted responses to peptide pool 1 using tetramers loaded with individual peptides comprising the pool is shown in the top row. Decoding of DR0101-restricted responses to peptide pool 2 is shown in the bottom row.
Haemophilic subject IV-3, who had no prior exposure to wild-type FVIII, was screened for DR1104- and DR0901-restricted FVIII C2 T-cell epitopes using TGEM with peptide pools as above. No DR1104-restricted T cells were detected in total CD4+ T-cell cultures (Fig. 3A). However, tetramer-positive responses to peptide pool 2 were observed for 0.7% of the cells when the total CD4+ fraction was depleted of CD4+CD25+ T cells (Fig. 3B). This small but above-background T-cell response to peptide pool 2 was seen on analysis of two separate blood samples collected three months apart. Decoding identified FVIII2202-2221 (peptide sequence: TWSPSKARLHLQGRSNAWRP), which is adjacent to the A2201P missense substitution site, as the immunodominant DR1104-restricted T-cell epitope (Fig. 3C). The possibility of additional minor T-cell epitopes being present cannot be dismissed because of the above-background staining by tetramers loaded with FVIII2186-2205 and FVIII2194-2213. No DR0901-restricted T-cells were detected in total CD4+ or CD4+CD25+-depleted T-cell cultures (data not shown).
Figure 3. T-cell epitope mapping for haemophilic subject IV-3.

Total CD4+ cells (A) and CD4+ cells depleted of CD4+CD25+ cells (B) were stimulated with pooled peptides spanning the FVIII C2 domain sequence. Fifteen days later, the cells were incubated with PE-labeled DR1104 tetramers loaded with FVIII pooled peptides and antibodies as described in Methods. Decoding of DR1104-restricted responses to peptide pool 2 using tetramers loaded with individual peptides comprising the pool was carried out 22 days after stimulation of CD4+CD25+-depleted CD4+ cells with peptide pool 2 (C).
Subject II-3, the great-uncle of IV-1 and IV-2 and great-grandfather of IV-3 has HLA alleles DRB1*0404 and DRB1*1501; despite prior FVIII infusions, he has shown no evidence of an inhibitor. A blood sample was obtained from him several months after his last FVIII exposure and TGEM was carried out as above using his total CD4+ and CD4+CD25+-depleted T-cell fractions to screen for DR0404- and DR1501-restricted T-cell epitopes. Staining above background (0.9% tetramer-stained CD4 cells) was seen for total CD4 cells cultured with DR0404 tetramers loaded with pool 4 peptides. However, when tetramer binding was plotted versus CD25 staining the tetramer-positive cells did not form a distinct cell population, suggesting that this signal included significant non-specific binding, so we concluded that this staining did not indicate a legitimate epitope. Similar results were observed for the CD4+CD25+-depleted cultures (data not shown). No DR1501-restricted T cells were detected in total CD4+ or in CD4+CD25+-depleted T-cell cultures (data not shown).
Subject III-2, the obligate carrier mother of IV-1 and IV-2, is HLA-DRB1*0101, 0901, and subject III-4, the obligate carrier mother of subject IV-3, is HLA-DRB1*1104, 1501. Blood samples from each were screened for FVIII T-cell epitopes using TGEM with pooled peptides as above. Neither total CD4+ cells or CD4+CD25+-depleted CD4+ cells from the carrier mothers showed T-cell responses to the pooled peptides restricted by their respective HLA-DR allelic proteins (data not shown).
Isolation of T-cell clones specific for DR0101 and DR1104 epitopes
T cells from the first blood draw of haemophilic subject IV-2 that were stimulated with peptide pool 2 were next stained using DR0101 tetramers carrying FVIII2194-2213 and then were single-cell sorted into 96-well plates as described above. Cells in 21 wells expanded sufficiently to be tested for tetramer binding, and 20/21 wells contained expanded T cells that were 99-100% tetramer-positive (data not shown). Six of these clones were cryo-preserved. After thawing and multiple rounds of expansion with HLA-mismatched PBMCs and PHA, greater than 96% of the cells expanded from each clone were stained by the DR0101 tetramer loaded with FVIII2194-2213 (Fig. 4A). When the DR0101 tetramer was loaded with FVIII2218-2237, staining of the T-cell clones was at background levels, demonstrating the specificity of the clones for the DR0101-FVIII2194-2213 peptide complex (Fig. 4B). The antigen specificity of the T-cell clones was further tested by proliferation assays. Each T-cell clone proliferated in response to FVIII2194-2213 presented by DR0101 (Fig. 5). The amount of proliferation was dose-dependent over the range of peptide concentrations tested (0.1-10 μM). The stimulation indices (ratios of measured proliferation/background proliferation) were highly significant, ranging from 62 to 248 at 0.1 μM peptide concentration for the six clones. The T-cell clones also proliferated in response to a peptide with the haemophilic sequence, FVIII2194-2213, 2201P, but at significantly reduced levels. However, this proliferation was above the background levels established using an irrelevant peptide, FVIII519-538. This stimulation by the haemophilic (missense) sequence was in contrast to the behavior of clones isolated from the clinical inhibitor subject IV-1, which did not proliferate in response to the haemophilic peptide [33].
Figure 4. Tetramer staining of T-cell clones isolated from haemophilic subject IV-2.
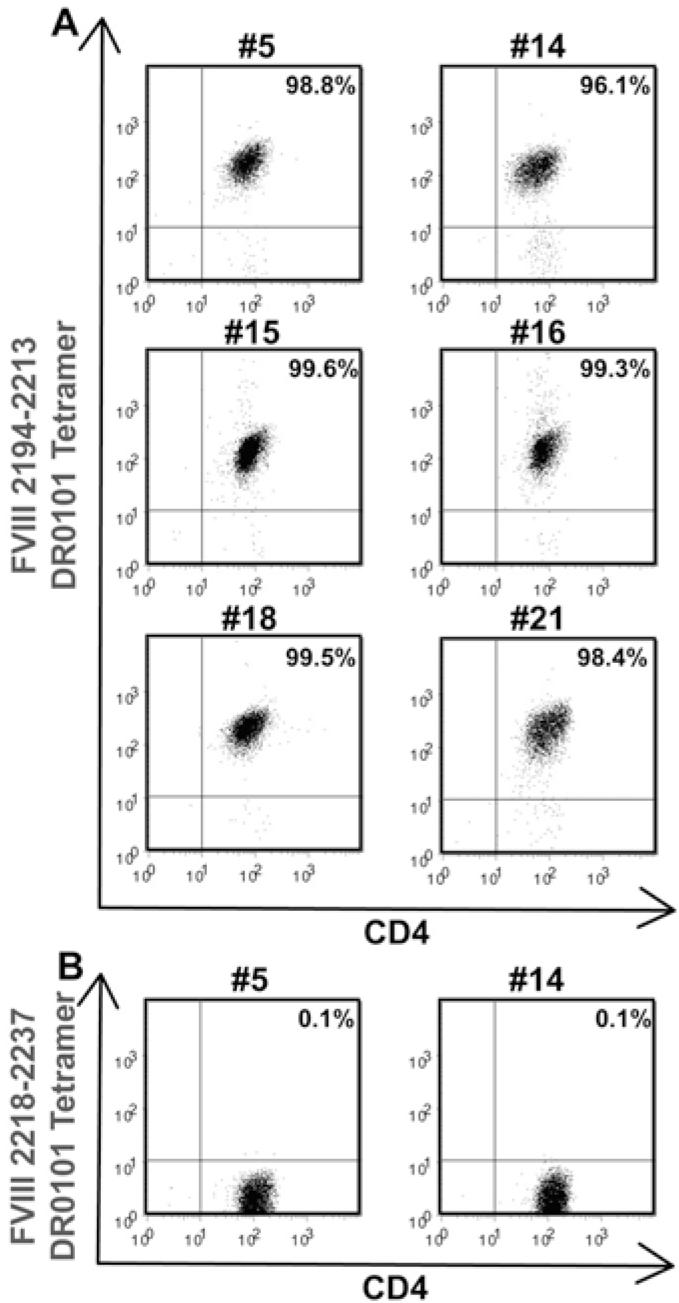
A. T-cell clones #5, 14, 15, 16, 18, and 21 were stimulated with HLA-mismatched PBMCs and PHA. Fourteen days later, the clones were incubated with PE-labeled DR0101 tetramers loaded with peptide FVIII2194-2213 and FITC-labeled anti-human CD4 IgG. B. Two of the clones were incubated with DR0101 tetramers loaded with an irrelevant peptide (FVIII2218-2237) as a negative control.
Figure 5. Antigen-specific proliferation of T-cell clones from haemophilic subject IV-2.
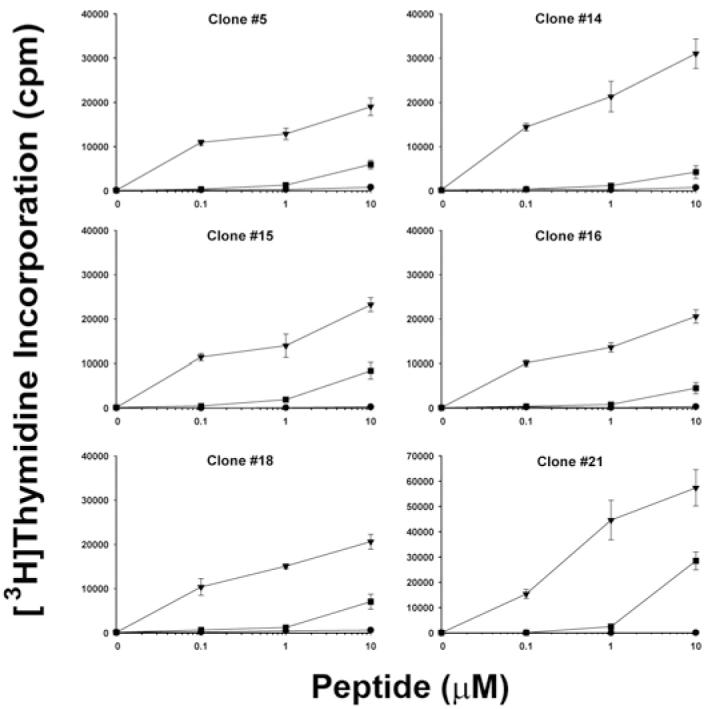
Resting T-cell clones #5, 14, 15, 16, 18, and 21 were stimulated with PBMCs from a healthy DRB1*0101 donor plus wild-type peptide FVIII2194-2213 (triangle symbols), haemophilic peptide FVIII2194-2213 2201P (square symbols), or irrelevant peptide FVIII519-538 (circle symbols) at 0, 0.1, 1.0, and 10 μM final concentration. [3H]thymidine uptake was measured. Data show mean ± SD of triplicate determinations.
The T cells from haemophilic subject IV-3 that were stimulated with peptide pool 2 were next stained using DR1104 tetramers carrying FVIII2202-2221 and then single-cell sorted into 96-well plates as described above. Cells in 13 wells expanded sufficiently to be tested for tetramer binding, and cells in four of these wells were tetramer-positive (Fig. 6). The binding avidity of these T-cell clones for the DR1104 tetramer loaded with FVIII2202-2221 was low (Fig. 6A) compared with that of the clones shown in Fig. 4A. These T-cell clones did not bind to the DR1104 tetramer loaded with FVIII2186-2205 (Fig. 6B), verifying the specificity of these T-cell clones for a peptide adjacent to the missense substitution A2201P. None of these T-cell clones expanded well under conditions routinely used for expansion of human T-cell clones, so they could not be further analyzed or cryo-preserved.
Figure 6. Tetramer staining of T-cell clones isolated from haemophilic subject IV-3.
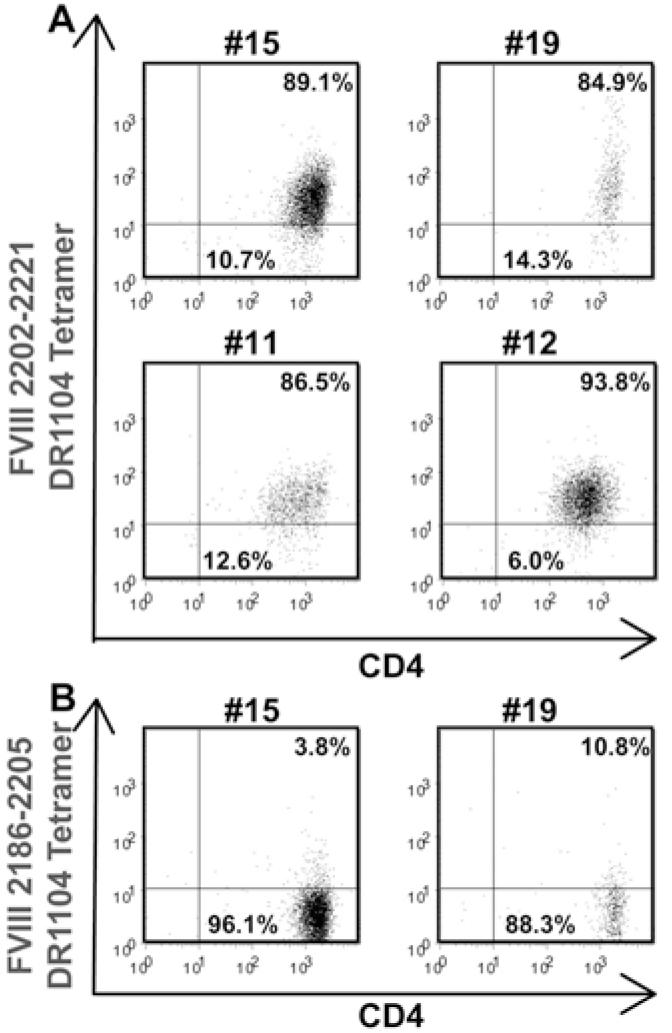
A. T-cell clones #15, 19, 11, and 12 were stimulated with HLA-mismatched PBMCs and PHA. Thirteen days later, the clones were incubated with PE-labeled DR1104 tetramers loaded with peptide FVIII2202-2221 and APC-labeled anti-human CD4 IgG. B. Two of the clones were incubated with DR1104 tetramers loaded with an irrelevant peptide (FVIII2186-2205) as a negative control.
Sub-clinical inhibitor response
The presence of FVIII-responsive T cells in subject IV-2, who did not have an inhibitor but whose T-cell response was highly similar to that of his brother raised the possibility that he might have circulating inhibitory antibodies, albeit at a sub-clinical level. To address this possibility, IgG was Protein G-purified from his plasma and Bethesda assays were carried out for serial dilutions of the concentrated IgG samples. At 10 mg/ml, his IgG sample had an inhibitor titer of 1 BU/ml, demonstrating a trace level of circulating inhibitory antibody.
Discussion and Conclusions
Our previous study of T-cell responses to FVIII peptides in a mild haemophilia A inhibitor subject with missense substitution FVIII-A2201P established a powerful approach to identify HLA-DR-restricted T-cell epitopes in FVIII [33]. We applied this same approach to study FVIII-specific T-cell responses in family members of the original subject (IV-1). DR0101 tetramer staining of T cells from his haemophilic brother, subject IV-2, was quite similar, as a comparable number of T cells recognized the same HLA-DR-FVIII peptide complexes. In addition, the avidities of the T-cell clones isolated from both brothers for DR0101 tetramer loaded with synthetic peptide FVIII2194-2213 were similar, and both sets of clones showed strong, dose-dependent proliferation when stimulated with FVIII2194-2213.
An intriguing difference between the T-cell responses of IV-1 and IV-2 was noted, in that low-level proliferation of T-cell clones from subject IV-2 was elicited by a peptide with the haemophilic missense sequence, FVIII2194-2213 2201P, but this was not seen for any clones isolated from inhibitor subject IV-1. Staining of polyclonal T cells from IV-1 using DR0101 tetramers loaded with the haemophilic peptide was seen only during analysis of the sample obtained three days following initial detection of his inhibitor response. Staining of T cells from this time point was consistent with clinical evidence for an immune response to his self (haemophilic) FVIII protein, as well as against the wild-type FVIII that he received in infusions to support surgery. At this time point, his peak inhibitor titer of 250 Bethesda units/ml coincided with a clotting activity (FVIII:C) of 3%, which was well below his pre-inhibitor baseline FVIII activity of 8-10%. His FVIII activity corrected to his normal baseline level as his inhibitor titer fell to 30 Bethesda units/ml over the ensuing four weeks. A possible explanation is that under the inflamed conditions at the time of his surgery and FVIII infusions, a subset of his T cells, primed by “danger signals” accompanying this inflammatory response, recognized the lower-avidity self-sequence with P2201 as well as the “non-self” FVIII containing A2201. Signaling from T cells stimulated by wild-type and/or haemophilic FVIII fragments may have contributed to the transient production of antibodies inhibiting the function of the haemophilic FVIII.
The presence of T cells recognizing the haemophilic peptide in subject IV-2 is interesting, as he had only very low levels of circulating IgG that inhibited FVIII activity, and this blood sample was not obtained at a time of trauma or major inflammation. Because our sample size is small, it is not yet known how frequently T cells from individuals with mild haemophilia A will recognize their haemophilic “self-FVIII” as well as wild-type “non-self” FVIII.
Our documentation of T cells from haemophilia A subjects without a clinically significant inhibitor responding to a specific epitope in FVIII is consistent with several previous reports of T-cell responses in inhibitor-negative haemophilia A subjects or in nonhaemophilic subjects. Singer et al. reported proliferation of B-cell depleted PBMCs from haemophilia A subjects without inhibitors and from nonhaemophilic controls in response to recombinant human FVIII; these cells proliferated with stimulation index (SI) values from 1.6 to 6.0 [23]. Hu et al. reported that CD8+ depleted PBMCs from four non-inhibitor haemophilic subjects proliferated when stimulated by FVIII or by several A2 domain peptides, [25]. Reding et al. did not consistently observe proliferation of T cells from non-inhibitor haemophilic subjects in response to A3 domain peptides [26], while C2 domain peptides spanning FVIII residues 2190-2210 and 2291-2330 elicited proliferation in some subjects with and without haemophilia or inhibitors [27]. We cloned T cells from an individual with haemophilia A but no clinically significant inhibitor (IV-2); these clones retained their antigen specificity for FVIII2194-2213 presented by DR0101, as demonstrated by both tetramer binding and proliferation assays. The SI values for proliferation assays utilizing six of these T-cell clones were highly significant, ranging between 62 and 248 at a low peptide concentration of 0.1 μM. To our knowledge, this study is the first to demonstrate conclusively an HLA-DR-restricted T-cell response, validated by isolation of relevant epitope-specific T-cell clones, to a FVIII epitope in a haemophilia A subject without a clinically significant inhibitor.
We isolated FVIII-responsive T cells from two haemophilia A subjects who did not have a clinically significant inhibitor, one of whom had received infusions of wild-type FVIII to achieve hemostasis. T cells from these subjects may well have different immune characteristics compared to those from subjects with an established antibody response to FVIII. A recent study demonstrated that cytokine production by FVIII-stimulated polyclonal CD4+ T cells differs between healthy subjects, haemophilia A subjects without inhibitors, and haemophilia A subjects with inhibitors [39]. T cells from haemophilia A subjects with inhibitors produced higher levels of IFN-γ and IL-4, whereas T cells from controls and haemophilia A subjects without inhibitors secreted higher levels of TGF-β but did not produce IL-4. We are utilizing the T-cell clones isolated from the A2201P missense substitution subjects, which are restricted by the same HLA-DR-FVIII peptide complex, to investigate additional features of T-cell immune responses to FVIII, including cytokine secretion and T-cell receptor variations. As noted above, T-cell responses to the haemophilic peptide differed in these individuals. Their T cells may also have different thresholds of responsiveness to haemophilic and/or wild-type peptides.
DRB1 proteins corresponding to the second allele (DRB1*1503) of inhibitor subject IV-1 were not available, but his brother’s second allele was DRB1*0401. This subject, IV-2, did not have DR0401-restricted T cells that recognized FVIII C2 domain peptides, although he did have a pronounced DR0101-restricted T-cell response to one of these FVIII peptides. This is compelling evidence that an individual’s MHC class II allelic proteins are essential contributors to a T-cell immune response against FVIII. Mechanistic differences in T-cell epitope selection between DR0101 and DR0401 are explained by polymorphisms in the MHC Class II-DRB1 peptide binding groove that alter the dimensions of peptide-binding pockets 4, 6, and 9 (these numbers correspond to pockets in the canonical Class II peptide binding groove) [40, 41]. The lack of FVIII-responsive T cells restricted by DR0404 and DR1501 in haemophilic subject II-3 suggests that these particular MHC class II allelic proteins do not bind epitopes in wild-type FVIII. However, other genetic and non-genetic factors are known to affect the risk of inhibitor development [42-49]. Sibling studies are valuable in attempting to discern genetic factors that may predispose some individuals to developing FVIII inhibitors. The Malmö International Brother Study (MIBS) [50-52] has identified polymorphisms associated with inhibitor development in the IL10 [53], TNFA [54] and CTLA4 [55] genes. These risk factors are currently under investigation for the subjects of the present study.
It was previously noted that FVIII-specific T-cell responses can be enhanced or uncovered when CD4+/CD25high regulatory T cells (Tregs) are depleted from PBMCs of healthy subjects [30]. Our experiments directly demonstrate the presence of DR1104-restricted FVIII2202-2221-responsive T cells in a haemophilia A subject who has never been infused with FVIII, but these responses were only apparent in CD4+CD25+-depleted CD4+ cultures. The presence of auto-reactive T cells directed against other auto-antigens in the blood of healthy individuals has previously been noted using similar experimental conditions [56, 57]. The results of these studies suggest that auto-reactive T cells, including T cells specific for FVIII, escape thymic deletion and are under the control of Tregs in the periphery. The CD4+CD25+ cell subset contains Tregs, which play a key role in the maintenance of peripheral tolerance [58].
T cells from obligate female carrier subjects III-2 and III-4 were not stained by tetramers loaded with pooled FVIII C2 domain peptides. This suggests that one copy of the wild-type FVIII DNA sequence resulting in at least low-level wild-type FVIII expression, as found in heterozygous carriers of haemophilic mutations, is sufficient to promote central tolerance despite their sharing the DRB1-reactive allele with their sons. The mechanism by which the A2201P missense substitution alters presentation of the T-cell epitope identified within the FVIII2202-2221 peptide is not yet clear. A predicted DR1104 binding motif is present within the immunogenic peptide FVIII2202-2221 between amino acids 2210 and 2218 [59], making it unlikely that alanine 2201 interacts directly with the DR1104 peptide-binding groove or with its cognate T-cell receptor. However, the missense substitution could influence antigen processing and presentation in more subtle ways.
In conclusion, this study demonstrates the presence of HLA-DR-restricted FVIII T cells in the blood of two haemophilia A subjects without clinically recognized inhibitors. The FVIII2194-2213 peptide contains a dominant DR0101-restricted T-cell epitope that was recognized by CD4+ T cells from two mild haemophilia A subjects with the A2201P missense substitution. We suggest that modification of this FVIII epitope could facilitate efforts to engineer versions of FVIII that would be less immunogenic for individuals who are DRB1*0101. The promiscuity of this epitope across other DRB1 types is under investigation.
Acknowledgement
We thank Mr. Charles Cooper, RN, for help with protocols, Ms. Shelley Nakaya for carrying out FVIII genotyping assays, Ms. Laura Stewart for carrying out Bethesda assays, and all subjects for their voluntary blood donations. This work was supported by a Bayer Haemophilia Award (K.P. Pratt), a CSL Behring Haemophilia Research Award (K.P. Pratt), NIH R01-HL 071093-01 (A.R. Thompson), and NIH contract HHSN266200400028C (W. W. Kwok).
Footnotes
Disclosure of Conflicts of Interest The authors state that they have no conflict of interest.
Dedication
It is an honor to dedicate this manuscript, with great respect, to Professor Hans-Hermann Brackmann.
References
- 1.Hoyer LW. Hemophilia. N Engl J Med. 1994;330:38–47. doi: 10.1056/NEJM199401063300108. [DOI] [PubMed] [Google Scholar]
- 2.Ehrenfort S, Kreuz W, Scharrer I, Linde R, Funk M, Gungor T, et al. Incidence of development of factor VIII and factor IX inhibitors in haemophiliacs. Lancet. 1992;339:594–8. doi: 10.1016/0140-6736(92)90874-3. [DOI] [PubMed] [Google Scholar]
- 3.Lusher JM, Arkin S, Abildgaard CF, Schwartz RS, The Kogenate Previously Untreated Patient Study Group Recombinant Factor VIII for the treatment of previously untreated patients with hemophilia A — safety, efficacy, and development of inhibitor. N Engl J Med. 1993;328:453–9. doi: 10.1056/NEJM199302183280701. [DOI] [PubMed] [Google Scholar]
- 4.Darby SC, Keeling DM, Spooner RJ, Kan SW, Giangrande PL, Collins PW, et al. The incidence of factor VIII and factor IX inhibitors in the hemophilia population of the UK and their effect on subsequent mortality, 1977-99. J Thromb Haemost. 2004;2:1047–54. doi: 10.1046/j.1538-7836.2004.00710.x. [DOI] [PubMed] [Google Scholar]
- 5.Thompson AR, Murphy ME, Liu ML, Saenko EL, Healey JF, Lollar P, et al. Loss of tolerance to exogenous and endogenous factor VIII in a mild hemophilia A patient with an Arg 593 to Cys mutation. Blood. 1997;90:1902–10. [PubMed] [Google Scholar]
- 6.Fijnvandraat K, Turenhout EA, van den Brink EN, ten Cate JW, van Mourik JA, Peters M, et al. The missense mutation Arg593→Cys is related to antibody formation in a patient with mild hemophilia A. Blood. 1997;89:4371–7. [PubMed] [Google Scholar]
- 7.Hay CR. Factor VIII inhibitors in mild and moderate-severity haemophilia A. Haemophilia. 1998;4:558–63. doi: 10.1046/j.1365-2516.1998.440558.x. [DOI] [PubMed] [Google Scholar]
- 8.Schwaab R, Brackmann HH, Meyer C, Seehafer J, Kirchgesser M, Haack A, et al. Haemophilia A: mutation type determines risk of inhibitor formation. Thromb Haemost. 1995;74:1402–6. [PubMed] [Google Scholar]
- 9.Goodeve A. The incidence of inhibitor development according to specific mutations – and treatment? Blood Coagul Fibrinolysis. 2003;14(Suppl 1):S17–21. doi: 10.1097/00001721-200306001-00005. [DOI] [PubMed] [Google Scholar]
- 10.Schwaab R, Oldenburg J, Schwaab U, Johnson DJ, Schmidt W, Olek K, et al. Characterization of mutations within the factor VIII gene of 73 unrelated mild and moderate haemophiliacs. Br J Haematol. 1995;91:458–64. doi: 10.1111/j.1365-2141.1995.tb05322.x. [DOI] [PubMed] [Google Scholar]
- 11.Haemostasis Research Group at the MRC clinical sciences centre, Imperial College Medical School Haemophilia A mutation database. http://europiu.mmrc.rpms.ac.uk/HAMSterS
- 12.Scandella DH, Nakai H, Felch M, Mondorf W, Scharrer I, Hoyer LW, et al. In hemophilia A and autoantibody inhibitor patients: the factor VIII A2 domain and light chains are most immunogenic. Thromb Res. 2001;101:377–85. doi: 10.1016/s0049-3848(00)00418-7. [DOI] [PubMed] [Google Scholar]
- 13.Mayr WR, Lechner K, Niessner H, Pabinger-Fasching I. HLA-DR and factor VIII antibodies in hemophilia A. Thromb Haemost. 1984;30:293. [PubMed] [Google Scholar]
- 14.Lippert LE, Fisher LM, Schook LB. Relationship of major histocompatibility complex class II genes to inhibitor antibody formation in hemophilia A. Thromb Haemost. 1990;28:564–8. [PubMed] [Google Scholar]
- 15.Aly AM, Aledort LM, Lee TD, Hoyer LW. Histocompatibility antigen patterns in haemophilic patients with factor VIII antibodies. Br J Haematol. 1990;76:238–41. doi: 10.1111/j.1365-2141.1990.tb07878.x. [DOI] [PubMed] [Google Scholar]
- 16.Oldenburg J, Picard JK, Schwaab R, Brackmann HH, Tuddenham EG, Simpson E. HLA genotype of patients with severe haemophilia A due to intron 22 inversion with and without inhibitors of factor VIII. Thromb Haemost. 1997;77:238–42. [PubMed] [Google Scholar]
- 17.Hay CR, Ollier W, Pepper L, Cumming A, Keeney S, Goodeve AC, et al. HLA class II profile: a weak determinant of factor VIII inhibitor development in severe haemophilia A. Thromb Haemost. 1997;77:234–7. [PubMed] [Google Scholar]
- 18.Bril WS, Maclean PE, Kaijen PH, van den Brink EN, Lardy NM, Fijnvandraat K, et al. HLA class II genotype and factor VIII inhibitors in mild haemophilia A patients with an Arg593 to Cys mutation. Haemophilia. 2004;10:509–14. doi: 10.1111/j.1365-2516.2004.01011.x. [DOI] [PubMed] [Google Scholar]
- 19.van den Brink EN, Turenhout EA, Davies J, Bovenschen N, Fijnvandraat K, Ouwehand WH, et al. Human antibodies with specificity for the C2 domain of factor VIII are derived from VH1 germline genes. Blood. 2000;95:558–63. [PubMed] [Google Scholar]
- 20.Fulcher CA, de Graaf Mahoney S, Zimmerman TS. FVIII inhibitor IgG subclass and FVIII polypeptide specificity determined by immunoblotting. Blood. 1987;69:1475–1480. [PubMed] [Google Scholar]
- 21.van Helden PM, Kaijen PH, Fijnvandraat K, van den Berg HM, Voorberg J. Factor VIII-specific memory B cells in patients with hemophilia A. J Thromb Haemost. 2007;5:2306–8. doi: 10.1111/j.1538-7836.2007.02736.x. [DOI] [PubMed] [Google Scholar]
- 22.Bray GL, Kroner BL, Arkin S, Aledort LW, Hilgartner MW, Eyster ME, et al. Loss of high-responder inhibitors in patients with severe hemophilia A and human immunodeficiency virus type 1 infection: A report from the multi-center hemophilia cohort study. Am J Hematol. 1993;42:375–79. doi: 10.1002/ajh.2830420408. [DOI] [PubMed] [Google Scholar]
- 23.Singer ST, Addiego JE, Reason DC, Lucas AH. T lymphocyte proliferative responses induced by recombinant factor VIII in hemophilia A patients with inhibitors. Thromb Haemost. 1996;76:17–22. [PubMed] [Google Scholar]
- 24.Reding MT, Wu H, Krampf M, Okita DK, Diethelm-Okita BM, Christie BA, et al. Sensitization of CD4+ T cells to coagulation factor VIII: response in congenital and acquired hemophilia patients and in healthy subjects. Thromb Haemost. 2000;84:643–52. [PubMed] [Google Scholar]
- 25.Hu GL, Okita DK, Conti-Fine BM. T cell recognition of the A2 domain of coagulation factor VIII in hemophilia patients and healthy subjects. J Thromb Haemost. 2004;2:1908–17. doi: 10.1111/j.1538-7836.2004.00918.x. [DOI] [PubMed] [Google Scholar]
- 26.Reding MT, Okita DK, Diethelm-Okita BM, Anderson TA, Conti-Fine BM. Epitope repertoire of human CD4+ T cells on the A3 domain of coagulation factor VIII. J Thromb Haemost. 2004;2:1385–94. doi: 10.1111/j.1538-7836.2004.00850.x. [DOI] [PubMed] [Google Scholar]
- 27.Reding MT, Okita DK, Diethelm-Okita BM, Anderson TA, Conti-Fine BM. Human CD4+ T-cell epitope repertoire on the C2 domain of coagulation factor VIII. J Thromb Haemost. 2003;1:1777–84. doi: 10.1046/j.1538-7836.2003.00251.x. [DOI] [PubMed] [Google Scholar]
- 28.Jones TD, Phillips WJ, Smith BJ, Bamford CA, Nayee PD, Baglin TP, et al. Identification and removal of a promiscuous CD4+ T cell epitope from the C1 domain of factor VIII. J Thromb Haemost. 2005;3:991–1000. doi: 10.1111/j.1538-7836.2005.01309.x. [DOI] [PubMed] [Google Scholar]
- 29.Hu GL, Okita DK, Diethelm-Okita BM, Conti-Fine BM. Recognition of coagulation factor VIII by CD4+ T cells of healthy humans. J Thromb Haemost. 2003;1:2159–66. doi: 10.1046/j.1538-7836.2003.00366.x. [DOI] [PubMed] [Google Scholar]
- 30.Kamaté C, Lenting PJ, van den Berg HM, Mutis T. Depletion of CD4+/CD25high regulatory T cells may enhance or uncover factor VIII-specific T-cell responses in healthy individuals. J Thromb Haemost. 2007;5:611–3. doi: 10.1111/j.1538-7836.2007.02336.x. [DOI] [PubMed] [Google Scholar]
- 31.White GC, Kempton CL, Grimsley A, Nielsen B, Roberts HR. Cellular immune responses in hemophilia: why do inhibitors develop in some, but not all hemophiliacs? J Thromb Haemost. 2005;3:1676–81. doi: 10.1111/j.1538-7836.2005.01375.x. [DOI] [PubMed] [Google Scholar]
- 32.Jacquemin M, Vantomme V, Buhot C, Lavend’homme R, Burny W, Demotte N, et al. CD4+ T-cell clones specific for wild-type factor VIII: a molecular mechanism responsible for a higher incidence of inhibitor formation in mild/moderate hemophilia A. Blood. 2003;101:1351–58. doi: 10.1182/blood-2002-05-1369. [DOI] [PubMed] [Google Scholar]
- 33.James EA, Kwok WW, Ettinger RA, Thompson AR, Pratt KP. T-cell responses over time in a mild hemophilia A inhibitor subject: epitope identification and transient immunogenicity of the corresponding self-peptide. J Thromb Haemost. 2007;5:2399–407. doi: 10.1111/j.1538-7836.2007.02762.x. [DOI] [PubMed] [Google Scholar]
- 34.Novak EJ, Liu AW, Gebe JA, Falk BA, Nepom GT, Koelle DM, et al. Tetramer-guided epitope mapping: rapid identification and characterization of immunodominant CD4+ T cell epitopes from complex antigens. J Immunol. 2001;166:6665–70. doi: 10.4049/jimmunol.166.11.6665. [DOI] [PubMed] [Google Scholar]
- 35.Liu ML, Shen BW, Nakaya S, Pratt KP, Fujikawa K, Davie EW, et al. Hemophilic factor VIII, C1- and C2-domain missense mutations and their modeling to the 1.5-angstrom human C2 domain crystal structure. Blood. 2000;96:979–87. [PubMed] [Google Scholar]
- 36.Liu ML, Nakaya S, Thompson AR. Non-inversion factor VIII mutations in 80 hemophilia A families including 24 with alloimmune responses. Thromb Haemost. 2002;87:273–6. [PubMed] [Google Scholar]
- 37.Kasper CK, Aledort L, Aronson D, Counts R, Edson JR, van Eys J, et al. Proceedings: A more uniform measurement of factor VIII inhibitors. Thromb Diath Haemorrh. 1975;34:612. [PubMed] [Google Scholar]
- 38.Novak EJ, Liu AW, Nepom GT, Kwok WW. MHC class II tetramers identify peptide-specific human CD4+ T cells proliferating in response to influenza A antigen. J Clin Invest. 1999;104:R63–7. doi: 10.1172/JCI8476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu GL, Guo D, Key NS, Conti-Fine BM. Cytokine production by CD4+ T cells specific for coagulation factor VIII in healthy subjects and haemophilia A patients. Thromb Haemost. 2007;97:788–94. [PubMed] [Google Scholar]
- 40.Stern LJ, Brown JH, Jardetzky TS, Gorga JC, Urban RG, Strominger JL, et al. Crystal structure of the human class II MHC protein HLA-DR1 complexed with an influenza virus peptide. Nature. 1994;368:215–21. doi: 10.1038/368215a0. [DOI] [PubMed] [Google Scholar]
- 41.Dessen A, Lawrence CM, Cupo S, Zaller DM, Wiley DC. X-Ray crystal structure of HLA-DR4 (DRA*0101, DRB1*0401) complexed with a peptide from human collagen II. Immunity. 1997;7:473–81. doi: 10.1016/s1074-7613(00)80369-6. [DOI] [PubMed] [Google Scholar]
- 42.Oldenburg J, Brackmann HH, Schwaab R. Risk factors for inhibitor development in hemophilia A. Haematologica. 2000;85(10 Suppl):7–13. [PubMed] [Google Scholar]
- 43.Oldenburg J, El-Maarri O, Schwaab R. Inhibitor development in correlation to factor VIII genotypes. Haemophilia. 2002;8(Suppl 2):23–9. doi: 10.1046/j.1351-8216.2001.00134.x. [DOI] [PubMed] [Google Scholar]
- 44.Brackmann HH, Albert T, Graw J, Oldenburg J, Schramm W, Schwaab R. [Gathering and evaluation of phenotype data of haemophilia A patients for correlation with genotype data] Hamostaseologie. 2003;23:24–7. [PubMed] [Google Scholar]
- 45.Oldenburg J, Schröder J, Brackmann HH, Müller-Reible C, Schwaab R, Tuddenham E. Environmental and genetic factors influencing inhibitor development. Semin Hematol. 2004;41(1 Suppl 1):82–8. doi: 10.1053/j.seminhematol.2003.11.016. [DOI] [PubMed] [Google Scholar]
- 46.Astermark J. Basic aspects of inhibitors to factors VIII and IX and the influence of non-genetic risk factors. Haemophilia. 2006;12(Suppl 6):8–13. doi: 10.1111/j.1365-2516.2006.01360.x. [DOI] [PubMed] [Google Scholar]
- 47.Lee CA, Lillicrap D, Astermark J. Inhibitor development in hemophiliacs: the roles of genetic versus environmental factors. Semin Thromb Hemost. 2006;32(Suppl 2):10–4. doi: 10.1055/s-2006-946909. [DOI] [PubMed] [Google Scholar]
- 48.Oldenburg J, Pavlova A. Genetic risk factors for inhibitors to factors VIII and IX. Haemophilia. 2006;12(Suppl 6):15–22. doi: 10.1111/j.1365-2516.2006.01361.x. [DOI] [PubMed] [Google Scholar]
- 49.Dasgupta S, Navarrete A-M, Delignat S, Wootla B, Andre S, Nagaraja V, et al. Immune response against therapeutic factor VIII in hemophilia A patients – A survey of probable risk factors. Immunol Letters. 2007;110:23–8. doi: 10.1016/j.imlet.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 50.Astermark J, Berntorp E. Malmö International Brother Study (MIBS): an international survey of brother pairs with haemophilia. Vox Sang. 1999;77(Suppl 1):80–2. doi: 10.1159/000056723. [DOI] [PubMed] [Google Scholar]
- 51.Astermark J, Berntorp E, White GC, Kroner BL, MIBS Study Group The Malmö International Brother Study (MIBS): further support for genetic predisposition to inhibitor development in hemophilia patients. Haemophilia. 2001;7:267–72. doi: 10.1046/j.1365-2516.2001.00510.x. [DOI] [PubMed] [Google Scholar]
- 52.Astermark J, Oldenburg J, Escobar M, White GC, 2nd, Berntorp E, Malmö International Brother study group The Malmö International Brother Study (MIBS). Genetic defects and inhibitor development in siblings with severe hemophilia A. Haematologica. 2005;90:924–31. [PubMed] [Google Scholar]
- 53.Astermark J, Oldenburg J, Pavlova A, Berntorp E, Lefvert A-K, MIBS Study Group Polymorphisms in the IL10 but not in the IL1beta and IL4 genes are associated with inhibitor development in patients with hemophilia A. Blood. 2006;107:3167–72. doi: 10.1182/blood-2005-09-3918. [DOI] [PubMed] [Google Scholar]
- 54.Astermark J, Oldenburg J, Carlson J, Pavlova A, Kavakli K, Berntorp E, et al. Polymorphisms in the TNFA gene and the risk of inhibitor development in patients with hemophilia A. Blood. 2006;108:3739–45. doi: 10.1182/blood-2006-05-024711. [DOI] [PubMed] [Google Scholar]
- 55.Astermark J, Wang X, Oldenburg J, Berntorp E, Lefvert A-K, MIBS Study Group Polymorphism in the CTLA-4 gene and inhibitor development in patients with severe hemophilia A. J Thromb Haemost. 2007;5:263–5. doi: 10.1111/j.1538-7836.2007.02290.x., on behalf of the
- 56.Danke NA, Koelle DM, Yee C, Beheray S, Kwok WW. Autoreactive T cells in healthy individuals. J Immunol. 2004;172:5967–72. doi: 10.4049/jimmunol.172.10.5967. [DOI] [PubMed] [Google Scholar]
- 57.Yang J, Danke NA, Berger D, Reichstetter S, Reijonen H, Greenbaum C, et al. Islet-specific glucose-6-phosphatase catalytic subunit-related protein-reactive CD4+ T cells in human subjects. J Immunol. 2006;176:2781–9. doi: 10.4049/jimmunol.176.5.2781. [DOI] [PubMed] [Google Scholar]
- 58.Sakaguchi S. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol. 2005;6:345–52. doi: 10.1038/ni1178. [DOI] [PubMed] [Google Scholar]
- 59.Verreck FA, Termijteien A, Koning F. HLA-DR beta chain residue 86 controls DR alpha beta stability. Eur J Immunol. 1993;23:1346–50. doi: 10.1002/eji.1830230624. [DOI] [PubMed] [Google Scholar]