

Abstract
Increased lipoprotein-associated phospholipase A2 (Lp-PLA2) activity is associated with increased risk of cardiac events, but it is not known whether Lp-PLA2 is a causative agent. Here we show that selective inhibition of Lp-PLA2 with darapladib reduced development of advanced coronary atherosclerosis in diabetic and hypercholesterolemic swine. Darapladib markedly inhibited plasma and lesion Lp-PLA2 activity and reduced lesion lysophosphatidylcholine content. Analysis of coronary gene expression showed that darapladib exerted a general anti-inflammatory action, substantially reducing the expression of 24 genes associated with macrophage and T lymphocyte functioning. Darapladib treatment resulted in a considerable decrease in plaque area and, notably, a markedly reduced necrotic core area and reduced medial destruction, resulting in fewer lesions with an unstable phenotype. These data show that selective inhibition of Lp-PLA2 inhibits progression to advanced coronary atherosclerotic lesions and confirms a crucial role of vascular inflammation independent from hypercholesterolemia in the development of lesions implicated in the pathogenesis of myocardial infarction and stroke.
Atherosclerosis, the most common cause of myocardial infarction, stroke and cardiovascular death, is an inflammatory-immunomodulatory disease1,2. A key early step in its development is the accumulation and subsequent oxidation of low-density lipoproteins (LDLs) within the arterial intima. In vitro studies have shown that oxidized LDL (oxLDL) promotes leukocyte recruitment and activation, lipid accumulation and cell death3,4. Major clinical complications arise when atherosclerotic lesions evolve into complex, unstable forms characterized by a thin fibrous cap, a large lipid-filled necrotic core and an accumulation of macrophages5. These `vulnerable' plaques are prone to rupture, which can then trigger acute unstable coronary artery syndromes and ischemic strokes6–8.
Lp-PLA2, also known as platelet-activating factor acetylhydrolase or type VIIA PLA2, is a calcium-independent phospholipase A2. In humans, Lp-PLA2 is secreted by leukocytes and is associated with circulating LDL and macrophages in atherosclerotic plaques. Although some have hypothesized that Lp-PLA2 has a protective role in atherosclerotic lesion development9,10, the preponderance of recent data suggests that Lp-PLA2 has an active role in atherosclerotic development and progression11–13. Elevated circulating Lp-PLA2 activity predicts increased cardiovascular risk14. A proatherogenic role for Lp-PLA2 has been postulated on the basis of its ability to generate two key proinflammatory mediators, lysophosphatidylcholine (LPC) and oxidized nonesterified fatty acids (oxNEFAs), through the cleavage of oxidized or polar phospholipids generated during LDL oxidation15,16. Evidence exists for a regulatory role of these proinflammatory lipids, particularly of LPC12,13,17, in promoting atherosclerotic plaque development that can ultimately lead to the formation of a necrotic core. These steps include recruitment and activation of leukocytes12,18, induction of apoptosis12,19 and impaired removal of dead cells20,21. The demonstration that Lp-PLA2 is highly upregulated in macrophages undergoing apoptosis within the necrotic core and fibrous cap of vulnerable and ruptured plaques, but not within stable lesions22, supports the notion that Lp-PLA2 products may be crucial in determining plaque instability.
Taken together, these observations suggest a causative role for Lp-PLA2 in the development of atherosclerosis, and they also suggest that inhibition of Lp-PLA2 could decrease the phenotypic hallmarks of acute plaque instability, namely necrotic core development and smooth muscle cell and fibrous tissue destruction. To test this hypothesis, we treated pigs with induced diabetes and hypercholesterolemia (DM-HC) with the Lp-PLA2 inhibitor darapladib (SB-480848)23. The DM-HC porcine model of accelerated atherosclerosis is particularly relevant for studying the role of Lp-PLA2. Pigs have a plasma lipoprotein profile that is similar to that in humans, whereas Lp-PLA2 associates with different lipoprotein fractions in mice, rendering the mouse model inadequate for studying the effects of Lp-PLA2 inhibition. Diabetic pigs gain weight to a lesser degree than do nondiabetic pigs, and they show ketosis. Like humans, these animals develop advanced, human-like coronary lesions with variable phenotypes24,25. Moreover, the ability to investigate the coronary arteries in the DM-HC porcine model also provides information about a pathophysiological location known to be susceptible to enhanced oxidative stress, possessing similar dimensional and flow characteristics to human coronary disease. For example, branched, bifurcated and curved arteries such as the left descending coronary artery show disturbed flow conditions including oscillatory shear stress, which promotes oxidative stress26,27. These areas are particularly susceptible to atherosclerotic lesion development.
RESULTS
Selectivity of darapladib
Darapladib is a potent inhibitor of human Lp-PLA2, with a half-maximal inhibitory concentration of 270 pM when assayed at 20 μM substrate concentration, that is, at its approximate Km23. We initially examined its selectivity against other secretory PLA2s postulated to play a role in atherogenesis. When tested against secretory PLA2 IIA, PLA2 V and PLA2 X, the percentage by which 1 μM darapladib inhibited their activities was 0, 0 and 8.7%, respectively. The weak activity of darapladib against these three secretory PLA2s was expected, given that they have very different catalytic motifs and requirements for catalysis compared to Lp-PLA2 (ref. 28).
Plasma cholesterol, glucose and Lp-PLA2 activity
After 4 weeks of DM-HC induction, plasma glucose and cholesterol increased to levels that were maintained for the duration of the study (Fig. 1, glucose ~380 mg dl−1, cholesterol ~700 mg dl−1). Pigs not induced to develop diabetes and hypercholesterolemia maintained plasma glucose and cholesterol at ~70 mg dl−1 and 80 mg dl−1, respectively. There were no differences in plasma glucose and cholesterol levels (Fig. 1a,b) or in insulin units administered (data not shown) between induced groups treated with or without darapladib.
Figure 1.

Plasma glucose, cholesterol and Lp-PLA2 activity increase upon DM-HC induction, but only Lp-PLA2 activity is influenced by darapladib. (a–c) Pigs underwent DM-HC induction on day 0. Four weeks later, pigs were assigned to either a control or a treated group, and 10 mg kg−1 darapladib was orally administered daily to the treated group. Three age- and sex-matched pigs did not undergo DM-HC induction as a second control group. Plasma glucose (a), cholesterol (b) and Lp-PLA2 activity (c) levels were monitored throughout the 28-week study period. *P < 0.0001 darapladib-treated DM-HC pigs versus control DM-HC pigs. Data shown are means ± s.e.m. for glucose and cholesterol and means ± s.d. for Lp-PLA2 activity. n = 3 for control non–DM-HC pigs, n = 17 for control DM-HC pigs and n = 20 for darapladib-treated DM-HC pigs. (d,e) Distribution of Lp-PLA2 activity after gel filtration for lipoprotein fractionation of pooled plasma samples from DM-HC control pigs before induction (d) or at completion of the study at week 28 (e; n = 17 pigs in a pooled sample). (f) For comparison, a similar lipoprotein profile was obtained from pooled plasma from six double-transgenic mice expressing both human apolipoprotein B100 and CETP and fed a Western diet. HDL, high-density lipoprotein.
Plasma Lp-PLA2 activity increased by approximately 230% after 4 weeks of DM-HC (Fig. 1c). Subsequent initiation of darapladib treatment at 4 weeks resulted in a significant inhibition of plasma Lp-PLA2 activity (Fig. 1c), so that by study completion, elevated plasma Lp-PLA2 activities were reduced by 89% (P < 0.00001). In DM-HC control pigs with increased plasma Lp-PLA2 activity, this activity was associated with all lipoprotein species; however, as DM-HC induction led to a more dramatic increase in apolipoprotein B–containing lipoproteins (that is, more in very low density lipoprotein (VLDL) and LDL than in high-density lipoprotein (HDL)), the increase in Lp-PLA2 activity was predominantly associated with VLDL and LDL (Fig. 1d,e). This pattern of Lp-PLA2 association with lipoproteins is in contrast to that seen in mice, in which Lp-PLA2 binds exclusively high-density lipoprotein29; this is the case even when mice are engineered to have a more human-like plasma lipoprotein profile with elevated plasma VLDL and LDL abundance—that is, in double-transgenic mice expressing both human apolipoprotein B100 and cholesterol ester transfer protein (CETP) fed a Western-type diet (Fig. 1f). Lp-PLA2 in pigs, but not in mice, associates with VLDL and LDL because only porcine Lp-PLA2 (GenBank accession code NM_001113013) has two amino acid residues crucial for binding to the carboxy terminus of apoB100, which are also found in the human sequence (ref. 30).
Arterial Lp-PLA2 abundance
Consistent with observations made in human atherosclerosis22, DM-HC pigs had markedly upregulated iliac arterial Lp-PLA2 activity. At study completion, the mean (± s.e.m.) total PLA2 activities in the DM-HC control group (n = 17) and darapladib-treated group (n = 20) were 0.68 ± 0.21 nmol min−1 mg−1 and 0.14 ± 0.02 nmol min−1 mg−1 (P < 0.001), respectively. Darapladib treatment resulted in near normalization of arterial PLA2 activity to a level similar to that seen in age-matched pigs without DM-HC induction (0.10 ± 0.01 nmol min−1 mg−1). Because more than one type of PLA2 could contribute to the total PLA2 activity, we confirmed the contribution of Lp-PLA2 by adding a saturating concentration of a selective inhibitor. When iliac artery extracts were assayed in the presence of a saturating concentration of darapladib (1 μM), we observed the remaining activity to be uniform across the three study groups, averaging 0.047 ± 0.011 nmol min−1 mg−1. Thus, all of the increased arterial PLA2 activity in DM-HC–induced pigs was attributable to Lp-PLA2.
Arterial phosphatidylcholine molecular species composition
Diseased iliac arteries from DM-HC–induced pigs showed a 305% increase in arterial LPC content, as well as an altered phosphatidylcholine (PC) composition (Supplementary Table 1 online and Fig. 2a–c). Whereas iliac artery PC from normal noninduced pigs was comprised predominantly of monounsaturated species and species containing either linoleate (18:2) or arachidonate (20:4), PC from DM-HC–induced pigs had a decreased content of oleate (18:1)-containing species (PC 16:0/18:1, PC 18:0/18:1; see Fig. 2 legend for details on nomenclature) and increased content of 18:2-containing species (PC 16:0/18:2, PC 18:1a/18:2, PC 18:1/18:2 and PC 18:0/18:2). Notably, there was some evidence for modulation of the content of 20:4-containing species (for example, PC 18:0/20:4 and PC 16:1a/20:4), the chain elongation and desaturase product of 18:2-containing species. For example, whereas DM-HC induction decreased the content of both PC 16:1a/20:4 and PC 18:0/20:4, darapladib treatment significantly reduced the decrease in the latter species. Concentrations of PC species containing either of the n – 3 fatty acids docosahexaenoate (22:6) or eicospentanoate (20:5) were very low (<3 mole% of the total PC) in both control and DM-HC–induced pigs and did not change in the DM-HC group with Lp-PLA2 inhibitor treatment (data not shown). Notably, apart from ameliorating the decrease in PC 18:0/20:4 caused by DM-HC induction noted above, darapladib caused little alteration in iliac artery PC composition over and above that caused by DM-HC induction. Treatment with darapladib, however, resulted in a significant reduction in total arterial LPC content (Supplementary Table 1), an effect that was distributed across all of the major LPC species (Fig. 2b,c). Although the arterial LPC content varied with pathology and treatment, truncated oxidized PC species in the mass range m/z 594–666 were at low and similar abundances in all three groups (non–DM-HC control, DM-HC control and DM-HC–treated; Fig. 2d and Supplementary Table 1). As the P184 diagnostic precursor scan (Supplementary Methods online) would detect all oxidized PC species with an intact phosphocholine head group, this analysis suggests that oxidized PC comprised a very small proportion of total arterial PC (Supplementary Table 1).
Figure 2.

Influence of DM-HC induction and darapladib on arterial phospholipid composition. (a–d) Total lipid extracts from iliac arteries harvested at the end of the study were analyzed by mass spectrometry. All results were calculated relative to the total concentration of PC within each sample and are expressed as means ± s.e.m. Data shown are for individual PC species (a), a representative spectrum from each study group (b; top, no DM-HC; middle, DM-HC control; bottom, DM-HC darapladib), individual LPC species (c) and individual oxidized PC (oxPC) species (d). The relative contributions of LPC species with a m/z smaller than 670 are magnified by a factor of ten so that all species can be visualized on a single graph. The nomenclature used to denote PC molecular species is PC w:x/y:z, where PC is the phosphorylcholine headgroup and `w:z' and `y:z' denote the fatty acyl moieties esterified at the sn – 1 and sn – 2 positions, repsectively. For each fatty acyl group, `w' and `y' denote the number of carbon atoms, whereas `x' and `z' denote the number of unsaturated double bonds. PC species with fatty acids attached by an ether (alkyl) bond instead of an ester (acyl) bond are denoted by `a'. For a: +P < 0.0001 versus no DM-HC; *P < 0.05 versus no DM-HC; #P < 0.05 control versus darapladib. For c: *P < 0.01 no DM-HC versus DM-HC control; #P < 0.05 control versus darapladib. n = 3 no DM-HC, n = 16 DM-HC control and n = 18 DM-HC darapladib.
Influence of darapladib on coronary artery gene expression
Analysis of coronary artery gene expression showed that darapladib exerted a general anti-inflammatory action, as evidenced by effects on the expression of various genes associated with leukocyte function (Table 1 for selected genes and Supplementary Table 2 online for all genes analyzed). Although it is unknown whether the changes in gene expression caused or were only reflective of the changes in arterial morphology, treatment with darapladib was significantly associated with the reduced expression of eight of the fourteen genes that showed the greatest (more than tenfold) upregulation with DM-HC induction (Table 1a). Furthermore, the expression of 24 of the 87 genes measured in coronary arteries evaluated at study completion was altered significantly after administration of darapladib (Table 1b). These data strongly suggest that darapladib was able to influence the recruitment or activation of the key inflammatory cell types in atheromas, namely monocytes/macrophages and T lymphocytes. For example, the increased coronary expression of the genes encoding CD68, Lp-PLA2 and cathepsin S resulting from DM-HC induction and reflecting mainly macrophage content was significantly reduced after darapladib treatment (Table 1a). A similar finding was observed for the genes encoding the inducible protein-10 (IP-10) chemokine receptor, CXCR3, a marker of T helper type 1 lymphocytes31 and the monocyte chemoattractant protein-1 (MCP-1) chemokine receptor, CCR2, a marker of a monocyte subset32. The effect of darapladib on the genes encoding CXCR3 and CCR2 was limited to their expression in coronary arteries, as their expression in circulating peripheral blood mononuclear cells (PBMCs) was not influenced by treatment (Figs. 3a–d). There was a strong and significant positive correlation between coronary expression of the genes encoding Lp-PLA2 and CD68 (r = 0.81; P < 0.0001) but not between coronary expression of the genes encoding Lp-PLA2 and CXCR3 (r = 0.33; not significant), indicating that macrophages were the probable primary source of Lp-PLA2 expression (Fig. 3e,f). Strong positive correlations were also observed between coronary expression of the genes encoding Lp-PLA2 and cathepsin S (r = 0.76; P < 0.0001), Lp-PLA2 and UCP2 (r = 0.82; P < 0.0001) and Lp-PLA2 and MMP-9 (r = 0.68; P < 0.0001).
Table 1.
Coronary gene expression at study completion: influence of darapladib
| (a) Influence of darapladib on the most upregulated (more than tenfold) genes. | |||
|---|---|---|---|
| Influence of darapladib |
|||
| Gene product | Upregulation with DM-HC (fold) | Percentage change | P value |
| MMP-9 | 70 | −72 | 0.090 |
| CD4 | 25 | −30 | 0.100 |
| IL-1a | 21 | −56 | 0.019 |
| gp91phox | 19 | −55 | 0.024 |
| CD48 | 19 | −53 | 0.068 |
| ApoE | 17 | −47 | 0.061 |
| CHI3L1 | 17 | −57 | 0.076 |
| CD68 | 17 | −48 | 0.040 |
| Cathepsin S | 16 | −53 | 0.020 |
| Lp-PLA2 | 15 | −55 | 0.012 |
| CD18 | 14 | −54 | 0.029 |
| CCR1 | 11 | −48 | 0.190 |
| NPL | 11 | −43 | 0.048 |
| p47phox | 10 | −54 | 0.025 |
| (b) All genes altered by darapladib | |||
|---|---|---|---|
| Influence of darapladib |
|||
| Gene product | Upregulated with DM-HC (fold) | Percentage change | P value |
| p-arrestin-2 | 3.0 | −47 | 0.004 |
| SLA-DMA | 3.6 | −38 | 0.007 |
| PTAFR | 5.8 | −49 | 0.009 |
| BIN2 | 7.1 | −53 | 0.009 |
| Lp-PLA2 | 14.6 | −55 | 0.012 |
| CXCR3 | 4.0 | −68 | 0.015 |
| GM2A | 5.4 | −50 | 0.016 |
| LAIR1 | 4.8 | −42 | 0.016 |
| IL-1a | 21.1 | −56 | 0.019 |
| Cathepsin S | 16.0 | −53 | 0.020 |
| gp91phox | 19.4 | −55 | 0.024 |
| p47phox | 10.4 | −54 | 0.025 |
| CCR2 | 1.9 | −86 | 0.026 |
| PLAUR | 6.7 | −39 | 0.026 |
| CD18 | 14.3 | −54 | 0.029 |
| UCP2 | 6.1 | −53 | 0.030 |
| HM0X1 | 6.4 | −44 | 0.033 |
| DENND2D | 6.9 | −45 | 0.034 |
| CD68 | 16.7 | −48 | 0.040 |
| EVI2A | 3.7 | −39 | 0.041 |
| EVI2B | 7.2 | −41 | 0.043 |
| CCL5 | 7.1 | −39 | 0.046 |
| SLC27A4 | 3.2 | −39 | 0.047 |
| NPL | 10.7 | −42 | 0.048 |
At study completion, coronary artery gene expression was evaluated by quantitative real-time PCR. The effect of DM-HC induction was first gauged by comparison with the age-matched control group receiving no induction; the influence of darapladib treatment was then calculated. MMP-9, matrix metalloproteinase-9; IL-1a, interleukin-1a; ApoE, apolipoprotein E; CHI3L1, chitinase-3-like 1 (cartilage glycoprotein-39); CCR1, CC chemokine receptor type 1; NPL, N-acetylneuraminate pyruvate lyase; SLA-DMA, porcine major histocompatibility complex, class II, DMa (human equivalent HLA-DMA); PTAFR, platelet-activating factor receptor; BIN2, bridging integrator-2; GM2A, GM2 ganglioside activator; LAIR1, leukocyte-associated Ig-like receptor-1; CCR2, CC chemokine receptor type 2; PLAUR, plasminogen activator, urokinase receptor; UCP2, uncoupling protein-2 (mitochondrial proton carrier); HMOX1, heme oxygenase; DENND2D, hypothetical protein FLJ22457; EVI2A, ecotropic viral integration site 2A; EVI2B, ecotropic viral integration site 2B; CCL5 is a chemokine; SLC27A4, solute carrier family 27 (fatty acid transporter), member 4. Please see Methods and Supplementary Methods for further details and Supplementary Table 2 for gene symbols.
Figure 3.

Inhibition of Lp-PLA2 reduces leukocyte subset marker abundance in coronary arteries in the absence of an effect in blood PBMCs. (a–d) Expression of the genes encoding the MCP-1 chemokine receptor CCR2 (a,b) and the IP-10 chemokine receptor CXCR3 (c,d) at the end of study in coronary arteries (a,c) and circulating PMBCs (b,d). (e,f) Correlation between coronary expression of the genes encoding Lp-PLA2 and CD68 (e, r = 0.81, P < 0.0001, n = 34) and between coronary expression of the genes encoding Lp-PLA2 and CXCR3 (f, r = 0.33, not significant, n = 29).
An effect on mRNA levels of CD18 and BIN2, two proteins highly expressed in monocytes and T cells, confirms a general influence of darapladib on monocyte and T cell recruitment (Table 1). Significant reduction in the expression of the genes encoding two of the subunits of NADPH oxidase, gp91phox and p47phox (Table 1) suggests a possible reduction of oxidative stress upon treatment with darapladib. Several of the genes whose expression in coronary arteries was influenced by darapladib treatment have been previously identified as markers of unstable human atheroma, namely those encoding Lp-PLA2, CD68, PTAFR, GM2A, LAIR1, cathepsin S, PLAUR, UCP2, HMOX1 and NPL33.
Darapladib reduces complex coronary lesion development
Atherosclerotic lesions in the darapladib-treated group appeared to be less severe and less complex, with noticeably better preservation of the medial layer, compared with lesions from the control group (Fig. 4a). This observation was confirmed by quantification of the left anterior descending coronary artery lesion area (Fig. 4b) and maximal necrotic core area (Fig. 4c) and the use of a scoring system to evaluate destruction of the medial layer (see Supplementary Methods).
Figure 4.

Darapladib treatment reduces complex coronary lesion development. (a) Photomicrograph of the two largest lesions observed in each study group (Movat's pentachrome stains). Arrows point to areas of medial destruction observed primarily in the control arteries. NC, necrotic core. Scale bar, 0.5 mm. (b) Mean plaque size in coronary arteries of DM-HC pigs left untreated or treated with darapladib. The bars represent median values. (c) Coronary artery necrotic core size in DM-HC pigs left untreated or treated with darapladib. (d) For each study group, a cross-section of the coronary artery adjacent to the lesion with the largest area (shown in a) was stained for the presence of macrophages (cathepsin S, FITC, green), collagens I and III (Texas Red) and smooth muscle α-actin (7-amino-4-methyl-3-coumarinylacetic acid (AMCA), blue). Yellow staining represents autofluorescence in the area of a focal calcification. The bottom images are higher magnifications of the top images. I, intima; L, lumen; M, medial layer. Scale bar, 100 μM.
Plaque area in the left anterior descending coronary artery was significantly reduced in the treated group compared to the control group (mean ± s.e.m. 0.178 ± 0.046 mm2 versus 0.636 ± 0.212 mm2), with the median plaque area reduced from 0.222 mm2 to 0.086 mm2 (P < 0.05, Fig 4b). The corresponding mean intimal area for the non–DM-HC pigs was 0.068 ± 0.027 mm2. There was no significant correlation between total plasma cholesterol levels and coronary artery lesion areas in both the DM-HC–induced control pigs and the DM-HC–induced treated pigs (Spearman coefficient, 0.094; P = 0.579). Coronary lesions in the control group were more advanced than those in the darapladib-treated group on the basis of the modified American Heart Association criteria7, with seven of seventeen control pigs showing a fibrous or thin fibrous cap atheroma (41%) compared to two of twenty (10%) in the treated group (neither with a thin fibrous cap). This difference approached statistical significance (P = 0.05). Of note, the mean necrotic core area (± s.e.m.) from the arterial section with the greatest plaque area was significantly reduced from 0.87 ± 0.33 mm2 to 0.03 ± 0.003 mm2 (P = 0.015) with treatment (Fig. 4c). Immunohistochemical staining for the presence of macrophages, smooth muscle cells and collagen showed that lesions in the treated group had a greater amount of smooth muscle cells, whereas the DM-HC control lesions were more macrophage rich (an example is shown in Fig. 4d).
Lp-PLA2 inhibition also resulted in reduced destruction of the tunica media: the mean medial destruction score for control coronary vessels was 2.4 ± 0.027 (range 1.0–4.0), whereas the score was significantly reduced in the treatment group (1.2 ± 0.24, range 0.0–3.0, P = 0.003).
Lesion macrophage content
Lesion macrophage content was assessed through the use of cathepsin S immunohistochemistry (Fig. 5)34. The mean ratio of macrophages to total intimal and medial area was 1.78 ± 0.44% in the DM-HC control pigs, 0.71 ± 0.18% in the DM-HC–treated group (59% reduction, P = 0.036) and 0.007 ± 0.003% in the non–DM-HC group. The lower macrophage content in the darapladib-treated pigs is consistent with the effects seen on macrophage marker gene expression (Table 1).
Figure 5.
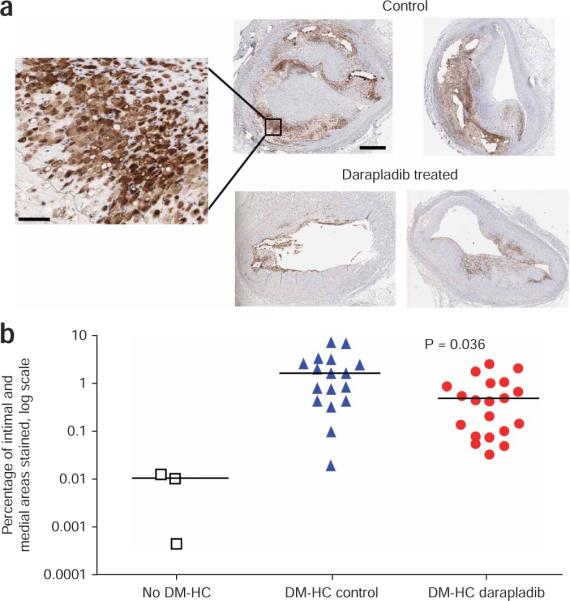
Inhibition of Lp-PLA2 results in fewer lesion macrophages in coronary arteries. Cathepsin S immunohistochemical staining was used as a marker of lesion macrophages and quantified in both the intima and media. (a) Staining of the two largest lesions observed in each study group (the same lesions as shown in Figure 4a); the inset clearly shows staining in cells. Scale bars, 0.5 mm and 25 μm (inset). (b) Quantification of lesion macrophage areas in the three study groups. The bars represent median values. The groups being compared for the P value are DM-HC control and DM-HC darapladib.
DISCUSSION
Selective inhibition of Lp-PLA2 with darapladib reduced development of coronary atherosclerosis and, more notably, inhibited the subsequent progression to advanced lesions, resulting in a more stable plaque phenotype. As such, the major finding of this study was the marked reduction in necrotic core development and the change in arterial lesion composition resulting from Lp-PLA2 inhibition. To our knowledge, this is the first study in a large-animal model of advanced, human-like coronary artery disease to show that inhibition of vascular inflammation in the absence of an effect on cholesterol abundance reduces development of coronary lesions with high-risk phenotypic characteristics. This antiatherogenic effect was associated with a marked decrease in macrophage content and the expression of various proinflammatory genes, the majority of which are crucial for macrophage and T lymphocyte recruitment and functioning. Hence, darapladib decoupled the effect of hypercholesterolemia on inflammation, resulting in stabilization of the potential vulnerable coronary lesions thought to be responsible for unstable ischemic syndromes and myocardial infarction.
Individuals with both diabetes mellitus and hypercholesterolemia have an increased risk of macrovascular atherosclerotic complications35, an observation replicated in the DM-HC porcine model24,25. Such individuals have an increased percentage of coronary artery lesion area occupied by lipid-rich atheroma, macrophages and T lymphocytes and have larger necrotic cores compared to individuals without diabetes36,37. These differences are noteworthy, as inflammation and necrotic core size are indicative of atherosclerotic plaque progression to a more vulnerable phenotype. Increased oxidative stress seems to be important for diabetic cardiovascular disease38. Lp-PLA2 hydrolyzes oxidatively-modified PC, such that Lp-PLA2 inhibitors would be expected to reduce the generation of two proinflammatory lipids, LPC and oxNEFA11. Thus, conditions that are associated with high oxidative stress, as seen in the DM-HC–induced pig with greatly upregulated coronary NADPH oxidase expression, represent an ideal scenario for studying the involvement of Lp-PLA2 in the development of complex coronary atherosclerosis. The finding that Lp-PLA2 expression was elevated in both circulating apolipoprotein B–containing lipoproteins and in complex coronary lesions in DM-HC–induced pigs provides further support for the clinical relevance of this animal model, as both observations mirror what has been reported in humans14,22.
Darapladib treatment was associated with a decrease in elevated arterial LPC abundance together with a marked decrease in the coronary expression of proinflammatory genes. The LPC species influenced by darapladib treatment in the current in vivo study bore close resemblance to those identified during the in vitro oxidation of LDL, namely LPC 16:0, LPC 18:1 and LPC 18:0 (ref. 16). It should be noted that our analysis of LPC abundance does not distinguish between species generated intracellularly and extracellularly, with the latter more likely to be associated with Lp-PLA2 activity. Additionally, this static analysis provides no information on the relative turnover rates of these different intracellular and extracellular pools of LPC in arterial samples, which is likely to be a key determinant of the effectiveness of Lp-PLA2 inhibition in reducing LPC abundance. Nonetheless, these data provide mechanistic support for the proposal that Lp-PLA2, generated predominately by intimal macrophages, contributes to atherosclerosis through the generation of proinflammatory mediators, such as LPC12,13,17, that activate pathways promoting leukocyte infiltration and activation. Of note was the striking near normalization of arterial CCR2 expression by darapladib, consistent with recent findings showing that the subset of circulating monocytes that express the MCP-1 chemokine receptor CCR2 preferentially accumulate within atherosclerotic lesions32,39. A similar decrease in arterial expression was observed for the gene encoding the IP-10 chemokine receptor, CXCR3, which is a recognized marker of T helper type 1 lymphocytes, the principal T cell type detected within atheroma31. Taken together, these data show that darapladib was able to influence the recruitment, activation or both of the principal inflammatory cell types present within atheromas, namely monocyte-derived macrophages and T lymphocytes.
The observation that the arterial oxidized PC content was not influenced by inhibition of Lp-PLA2, nor were oxidized PC species elevated in atherosclerotic lesions, is of considerable interest. The low values detected of these species were not due to methodological limitations, as mass spectrometry analyses performed at the same time readily detected a considerably higher abundance of oxidized PC species in lipid extracts of human carotid artery plaques16. Blockade of Lp-PLA2 activity would be predicted to result in the accumulation of short chain oxidized PC substrates (molecular mass 594–666 kDa), as was shown with in vitro oxidation of LDL16. The observation that these substrates did not accumulate suggests they are alternatively metabolized. In addition, the observation that the arterial concentration of oxidized PC did not change upon DM-HC induction casts doubt on previous suggestions that these phospholipids are centrally involved in promoting disease progression10,40. A limitation of our findings is that we measured the abundances of oxidized PC species at a single time point, which may overlook their potential role in the initiation of the disease process and does not provide insight into the dynamics of their clearance. As these are reactive molecules, it is probable that they could form a number of protein adducts41. Nevertheless, DM-HC induction provides favorable circumstances to address the importance of these molecules because the fat-feeding regimen used led to a decreased content of oleate (18:1)-containing PC species and an increased content of 18:2-containing PC species, the net effect of which results in a greater percentage of PC species prone to oxidative modification.
The diet used in this study contained sodium cholate to increase cholesterol levels and the development of atherosclerotic lesions. In mouse models, the addition of sodium cholate to the diet is associated with pleiotropic effects that may lead to chronic inflammation42. No such effects have been reported in swine models of atherosclerosis, although it is possible that some of the effects observed on inflammatory gene expression may have resulted from an exaggerated effect of sodium cholate on inflammation rather than from coronary atherosclerosis per se. However, both the control and treated DM-HC–induced groups were subjected to similar cholate doses, and, therefore, the results reflect the impact of Lp-PLA2 inhibition. Also, lesion area did not correlate significantly with cumulative cholesterol or cholate doses. Of note, the lesions shown in Figures 4 and 5 were obtained from pigs that had received the same diet.
In summary, the results of this study support the hypothesis that Lp-PLA2 is causally involved in the development of coronary atherosclerosis and formation of an unstable lesion phenotype. Selective inhibition of Lp-PLA2 resulted in a marked decrease in progression to complex coronary artery lesions. The treated lesions were less severe, contained fewer macrophages and showed smaller necrotic cores. These morphological observations were paralleled by a reduction in inflammatory gene expression. As this effect was independent of cholesterol abundance or the severity of dysglycemia, these results demonstrate the crucial and independent role for vascular inflammation in the development of complex coronary artery disease. Ongoing and planned clinical studies will test the hypothesis that selective inhibition of Lp-PLA2 can reduce necrotic core formation and the incidence of myocardial infarction and death in humans.
METHODS
Pig model
We induced diabetes in male Yorkshire domestic farm pigs (~25–35 kg; Archer Farms) with a single intravenous injection of 125 mg kg−1 streptozotocin25. We closely monitored blood glucose and cholesterol (Supplementary Methods). Three days after diabetes induction, we used a hyperlipidemic diet to achieve hypercholesterolemia with serum cholesterol concentrations clamped between 400 and 800 mg dl−1. Because pigs have a variable serum cholesterol response to a high cholesterol diet, we checked cholesterol concentrations every two months. One month after DM-HC induction, we randomly assigned pigs into either a control group or a treatment group receiving 10 mg kg−1 d−1 orally of the selective Lp-PLA2 inhibitor darapladib (SB480848, GlaxoSmithKline). We killed the pigs 28 weeks after induction (that is, 24 weeks after initiation of treatment). At the time of completion of the study, there were 17 pigs in the control group and 20 in the darapladib-treated group, as three pigs in the control group were excluded from analysis because of persistently elevated cholesterol levels (Supplementary Methods). Three pigs did not undergo DM-HC induction and acted as age-matched controls. Investigators were blinded to the group allocation. All experiments were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania.
Arterial harvest
At baseline and before killing, we collected whole blood for PBMC isolation (Supplementary Methods). We killed pigs with Eutasol (Virbac AH) and harvested the tissues under sterile conditions. After thoracotomy, we quickly removed the heart. We evaluated gene expression from the isolated left circumflex and right coronary arteries by quantitative real-time PCR. We used the left anterior descending artery for histology and immunohistochemistry (Supplementary Methods). We harvested the iliac arteries from origin at the aortic bifurcation to the femoral artery and used them for analysis of Lp-PLA2 activity and quantification of LPC abundance by mass spectrometry. In this model, iliac arteries develop high-grade atherosclerotic lesions, similar in severity, incidence and composition to those in coronary arteries.
Measurement of plasma and lipoprotein Lp-PLA2 activity
We measured plasma Lp-PLA2 activity by adding 50 μl of plasma to tubes containing 150 μl of [3H]-platelet-activating factor in buffer (20 mM HEPES, 150 mM NaCl, 50 μM [3H]-platelet-activating factor final concentration, pH 7.4) and incubating them at 37 °C for 30 s as described previously43. We used lipoprotein fractionation to determine the lipoprotein distribution of plasma Lp-PLA2 activity. We pooled plasma samples from control pigs at the start (before induction) and the end (week 28) of the study (17 pigs total, 30 μl plasma from each), and we fractionated 50 μl of this pooled plasma by HPLC43. For the purpose of comparison with the pig model, we drew approximately 100 μl of blood from double-transgenic mice (hemizygous for the genes encoding human apolipoprotein B100 and CETP Taconic) expressing both human apolipoprotein B100 and CETP fed a Western-type diet by tail bleeding, and we fractionated plasma as described previously44. We assayed fractions for cholesterol content and Lp-PLA2 activity (Supplementary Methods). Mouse studies were approved by the Institutional Animal Care and Use Committee of GlaxoSmithKline.
Measurement of arterial Lp-PLA2 activity and phospholipids
Because we had insufficient coronary artery tissue available for all planned measurements, we analyzed sections of a similarly bifurcated and atherosclerotic iliac artery for their Lp-PLA2 activity and phospholipid composition (Supplementary Methods).
Determining selectivity of darapladib
We studied the selectivity of darapladib for the three different human secretory PLA2s (types GIIA, GV and GX) exactly as described previously45. We assayed inhibition of secretory PLA2s with the fluorometric substrate 1-palmitoyl-2-(10-pyrenedecanoyl)-phosphatidylglycerol in a microtiter plate assay as described previously46.
Gene expression determination by quantitative real-time PCR
To evaluate gene expression, we built a Taqman plate with selected genes that had been shown to be involved in the human atherosclerotic process33 and for which a pig ortholog had been identified (Supplementary Table 3 online). We homogenized minced coronary arterial tissue (<0.5 cm thick) or pelleted PBMCs in Trizol reagent (Sigma) on ice. We extracted total RNA by chloroform and purified it twice through RNeasy (Qiagen) minicolumns. After on-column DNase treatment, we eluted RNA with RNase-free water. We removed any genomic DNA contamination in the RNA with DNase I (Ambion). We judged RNA samples to be free of genomic DNA if they showed no amplification in a standard TaqMan assay with 10 ng of RNA and ACTB (the gene encoding β-actin) primer-probe oligonucleotides. We then quantified the RNA with Ribogreen RNA quantification reagent (Molecular Probes) and converted it to cDNA by reverse transcription with the High-Capacity cDNA Archive Kit (Applied Biosystems, Supplementary Methods). We analyzed the Taqman gene expression data on the basis of normalized expression values, using scaled geometric mean of the selected reference genes for the normalization factor calculations (Supplementary Methods).
Statistical analyses
We applied the nonparametric exact two-sided Wilcoxon test to find differences with most continuous variables. The normal distributions of the arterial phospholipid PC species values and of the transformed and normalized Taqman gene expression values allowed these comparisons to use contrasts based on one-way analysis of variance. We used the Cochran-Mantel-Haenszel row mean difference test for comparison of the categorical media destruction score values. We used the Spearman rank test to find correlations between expressions of different genes. Except for the analyses of the coronary tissue Taqman gene expression data, we used Statistical Analysis System software (SAS release 8.02).
For statistical tests of gene expression differences, we used log-transformed values of the normalized expression values. For comparisons of the study groups' (induced control versus uninduced and induced control versus induced with treatment) gene expression, we applied one-way analysis of variance tests. We applied Dunnett's P value correction to the comparisons for each gene, with a P value ≤ 0.05 used as a cutoff for selecting genes that were significantly differentially expressed in the comparison groups. We expressed the fold change estimates as ratios of the geometric mean normalized expression levels with positive fold change values indicating upregulation of the treatment group versus the control group. We calculated the relative treatment effect with untransformed normalized expression values using the following formula on mean normalized intensity of each comparison group: [(treatment group – induced group) / (induced group – uninduced group)] × 100. Results are presented as the means ± s.e.m. unless otherwise noted. A P value of < 0.05 was considered significant.
Supplementary Material
ACKNOWLEDGMENTS
The superb animal husbandry of H. Profka is gratefully acknowledged. We greatly appreciate the input by M. Hurle into the design and confirmation of the Taqman probes and the advice and support related to immunofluorescence procedures of P. Crino. These studies were supported by funding from GlaxoSmithKline through an industry-academic alliance via the Alternative Drug Discovery Initiative with the University of Pennsylvania School of Medicine. M.H.G. is funded by US National Institutes of Health grant R37HL036235.
Footnotes
Note: Supplementary information is available on the Nature Medicine website.
COMPETING INTERESTS STATEMENT The authors declare competing financial interests: details accompany the full-text HTML version of the paper at http://www.nature.com/naturemedicine/.
References
- 1.Ross R. Atherosclerosis—an inflammatory disease. N. Engl. J. Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 2.Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat. Rev. Immunol. 2006;6:508–519. doi: 10.1038/nri1882. [DOI] [PubMed] [Google Scholar]
- 3.Steinberg D. Thematic review series: the pathogenesis of atherosclerosis. An interpretive history of the cholesterol controversy, part V: the discovery of the statins and the end of the controversy. J. Lipid Res. 2006;47:1339–1351. doi: 10.1194/jlr.R600009-JLR200. [DOI] [PubMed] [Google Scholar]
- 4.Witztum JL. The oxidation hypothesis of atherosclerosis. Lancet. 1994;344:793–795. doi: 10.1016/s0140-6736(94)92346-9. [DOI] [PubMed] [Google Scholar]
- 5.Shah PK. Molecular mechanisms of plaque instability. Curr. Opin. Lipidol. 2007;18:492–499. doi: 10.1097/MOL.0b013e3282efa326. [DOI] [PubMed] [Google Scholar]
- 6.Kolodgie FD, et al. Intraplaque hemorrhage and progression of coronary atheroma. N. Engl. J. Med. 2003;349:2316–2325. doi: 10.1056/NEJMoa035655. [DOI] [PubMed] [Google Scholar]
- 7.Virmani R, et al. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 2000;20:1262–1275. doi: 10.1161/01.atv.20.5.1262. [DOI] [PubMed] [Google Scholar]
- 8.Spagnoli LG, et al. Extracranial thrombotically active carotid plaque as a risk factor for ischemic stroke. J. Am. Med. Assoc. 2004;292:1845–1852. doi: 10.1001/jama.292.15.1845. [DOI] [PubMed] [Google Scholar]
- 9.McIntyre TM, et al. Biologically active oxidized phospholipids. J. Biol. Chem. 1999;274:25189–25192. doi: 10.1074/jbc.274.36.25189. [DOI] [PubMed] [Google Scholar]
- 10.Prescott SM, et al. Platelet-activating factor and related lipid mediators. Annu. Rev. Biochem. 2000;69:419–445. doi: 10.1146/annurev.biochem.69.1.419. [DOI] [PubMed] [Google Scholar]
- 11.Macphee CH, Nelson JJ, Zalewski A. Lipoprotein-associated phospholipase A2 as a target of therapy. Curr. Opin. Lipidol. 2005;16:442–446. doi: 10.1097/01.mol.0000174155.61307.5f. [DOI] [PubMed] [Google Scholar]
- 12.Zalewski A, Macphee C. Role of lipoprotein-associated phospholipase A2 in atherosclerosis: biology, epidemiology, and possible therapeutic target. Arterioscler. Thromb. Vasc. Biol. 2005;25:923–931. doi: 10.1161/01.ATV.0000160551.21962.a7. [DOI] [PubMed] [Google Scholar]
- 13.Tsimikas S, Tsironis LD, Tselepis AD. New insights into the role of lipoprotein(a)-associated lipoprotein-associated phospholipase A2 in atherosclerosis and cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2007;27:2094–2099. doi: 10.1161/01.ATV.0000280571.28102.d4. [DOI] [PubMed] [Google Scholar]
- 14.Garza CA, et al. Association between lipoprotein-associated phospholipase A2 and cardiovascular disease: a systematic review. Mayo Clin. Proc. 2007;82:159–165. doi: 10.4065/82.2.159. [DOI] [PubMed] [Google Scholar]
- 15.Macphee CH, et al. Lipoprotein-associated phospholipase A2, platelet-activating factor acetylhydrolase, generates two bioactive products during the oxidation of low-density lipoprotein: use of a novel inhibitor. Biochem. J. 1999;338:479–487. [PMC free article] [PubMed] [Google Scholar]
- 16.Davis B, et al. Electrospray ionization mass spectrometry identifies substrates and products of lipoprotein-associated phospholipase A2 in oxidized human low density lipoprotein. J. Biol. Chem. 2008;283:6428–6437. doi: 10.1074/jbc.M709970200. [DOI] [PubMed] [Google Scholar]
- 17.Kougias P, et al. Lysophosphatidylcholine and secretory phospholipase A2 in vascular disease: mediators of endothelial dysfunction and atherosclerosis. Med. Sci. Monit. 2006;12:RA5–RA16. [PubMed] [Google Scholar]
- 18.Shi Y, et al. Role of lipoprotein-associated phospholipase A2 in leukocyte activation and inflammatory responses. Atherosclerosis. 2007;191:54–62. doi: 10.1016/j.atherosclerosis.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 19.Carpenter KL, et al. Mildly oxidised LDL induces more macrophage death than moderately oxidised LDL: roles of peroxidation, lipoprotein-associated phospholipase A2 and PPARg. FEBS Lett. 2003;553:145–150. doi: 10.1016/s0014-5793(03)01007-x. [DOI] [PubMed] [Google Scholar]
- 20.Perez R, et al. Involvement of group VIA calcium-independent phospholipase A2 in macrophage engulfment of hydrogen peroxide–treated U937 cells. J. Immunol. 2006;176:2555–2561. doi: 10.4049/jimmunol.176.4.2555. [DOI] [PubMed] [Google Scholar]
- 21.Aprahamian T, et al. Impaired clearance of apoptotic cells promotes synergy between atherogenesis and autoimmune disease. J. Exp. Med. 2004;199:1121–1131. doi: 10.1084/jem.20031557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kolodgie FD, et al. Lipoprotein-associated phospholipase A2 protein expression in the natural progression of human coronary atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2006;26:2523–2529. doi: 10.1161/01.ATV.0000244681.72738.bc. [DOI] [PubMed] [Google Scholar]
- 23.Blackie JA, et al. The identification of clinical candidate SB-480848: a potent inhibitor of lipoprotein-associated phospholipase A2. Bioorg. Med. Chem. Lett. 2003;13:1067–1070. doi: 10.1016/s0960-894x(03)00058-1. [DOI] [PubMed] [Google Scholar]
- 24.Gerrity RG, et al. Diabetes-induced accelerated atherosclerosis in swine. Diabetes. 2001;50:1654–1665. doi: 10.2337/diabetes.50.7.1654. [DOI] [PubMed] [Google Scholar]
- 25.Mohler ER, III, et al. Site-specific atherogenic gene expression correlates with subsequent variable lesion development in coronary and peripheral vasculature. Arterioscler. Thromb. Vasc. Biol. 2008;28:850–855. doi: 10.1161/ATVBAHA.107.154534. [DOI] [PubMed] [Google Scholar]
- 26.Chatzizisis YS, et al. Role of endothelial shear stress in the natural history of coronary atherosclerosis and vascular remodeling: molecular, cellular and vascular behavior. J. Am. Coll. Cardiol. 2007;49:2379–2393. doi: 10.1016/j.jacc.2007.02.059. [DOI] [PubMed] [Google Scholar]
- 27.Jo H, et al. Role of NADPH oxidases in disturbed flow- and BMP4- induced inflammation and atherosclerosis. Antioxid. Redox Signal. 2006;8:1609–1619. doi: 10.1089/ars.2006.8.1609. [DOI] [PubMed] [Google Scholar]
- 28.Schaloske RH, Dennis EA. The phospholipase A2 superfamily and its group numbering system. Biochim. Biophys. Acta. 2006;1761:1246–1259. doi: 10.1016/j.bbalip.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 29.Gardner AA, Reichert EC, Topham MK, Stafforini DM. Identification of a domain that mediates association of platelet-activating factor acetylhydrolase with high-density lipoprotein. J. Biol. Chem. 2008;283:17099–17106. doi: 10.1074/jbc.M802394200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stafforini DM, et al. Molecular basis of the interaction between plasma platelet-activating factor acetylhydrolase and low density lipoprotein. J. Biol. Chem. 1999;274:7018–7024. doi: 10.1074/jbc.274.11.7018. [DOI] [PubMed] [Google Scholar]
- 31.Heller EA, et al. Chemokine CXCL10 promotes atherogenesis by modulating the local balance of effector and regulatory T cells. Circulation. 2006;113:2301–2312. doi: 10.1161/CIRCULATIONAHA.105.605121. [DOI] [PubMed] [Google Scholar]
- 32.Swirski FK, et al. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J. Clin. Invest. 2007;117:195–205. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Papaspyridonos M, et al. Novel candidate genes in unstable areas of human atherosclerotic plaques. Arterioscler. Thromb. Vasc. Biol. 2006;26:1837–1844. doi: 10.1161/01.ATV.0000229695.68416.76. [DOI] [PubMed] [Google Scholar]
- 34.Reddy V.y., Zhang Z-Y, Weiss SJ. Pericellular mobilization of the tissue-destructive cysteine proteinases, cathepsins B, L and S by human monocyte-derived macrophages. Proc. Natl. Acad. Sci. USA. 1995;92:3849–3853. doi: 10.1073/pnas.92.9.3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heart Protection Study Collaborative Group MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5693 people with diabetes: a randomized placebo-controlled trial. Lancet. 2003;361:2005–2013. doi: 10.1016/S0140-6736(11)61125-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moreno PR, et al. Coronary composition and macrophage infiltration in atherectomy specimens from patients with diabetes mellitus. Circulation. 2000;102:2180–2184. doi: 10.1161/01.cir.102.18.2180. [DOI] [PubMed] [Google Scholar]
- 37.Burke AP, et al. Morphologic findings of coronary atherosclerotic plaques in diabetics: a postmortem study. Arterioscler. Thromb. Vasc. Biol. 2004;24:1266–1271. doi: 10.1161/01.ATV.0000131783.74034.97. [DOI] [PubMed] [Google Scholar]
- 38.Pennathur S, Heinecke JW. Mechanisms for oxidative stress in diabetic cardiovascular disease. Antioxid. Redox Signal. 2007;9:955–969. doi: 10.1089/ars.2007.1595. [DOI] [PubMed] [Google Scholar]
- 39.Tacke F, et al. Monocyte subsets differentially employ CCR2, CCR5 and CX3CR1 to accumulate within atherosclerotic plaques. J. Clin. Invest. 2007;117:185–194. doi: 10.1172/JCI28549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Navab M, et al. The oxidation hypothesis of atherogenesis: the role of oxidized phospholipids and HDL. J. Lipid Res. 2004;45:993–1007. doi: 10.1194/jlr.R400001-JLR200. [DOI] [PubMed] [Google Scholar]
- 41.Sangvanich P, Mackness B, Gaskell SJ, Durrington P, Mackness M. The effect of high-density lipoproteins on the formation of lipid/protein conjugates during in vitro oxidation of low-density lipoprotein. Biochem. Biophys. Res. Commun. 2003;300:501–506. doi: 10.1016/s0006-291x(02)02849-8. [DOI] [PubMed] [Google Scholar]
- 42.Getz GS, Reardon CA. Diet and murine atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2006;26:242–249. doi: 10.1161/01.ATV.0000201071.49029.17. [DOI] [PubMed] [Google Scholar]
- 43.Caslake MJ, et al. Lipoprotein-associated phospholipase A2, platelet-activating factor acetylhydrolase: a potential new risk factor for coronary artery disease. Atherosclerosis. 2000;150:413–419. doi: 10.1016/s0021-9150(99)00406-2. [DOI] [PubMed] [Google Scholar]
- 44.Van Eck M, et al. Bone marrow transplantation in apolipoprotein E-deficient mice. Effect of ApoE gene dosage on serum lipid concentrations, bVLDL catabolism, and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 1997;17:3117–3126. doi: 10.1161/01.atv.17.11.3117. [DOI] [PubMed] [Google Scholar]
- 45.Singer AG, et al. Interfacial kinetic and binding properties of the complete set of human and mouse groups I, II, V, X and XII secreted phospholipases A2. J. Biol. Chem. 2002;277:48535–48549. doi: 10.1074/jbc.M205855200. [DOI] [PubMed] [Google Scholar]
- 46.Smart BP, et al. Inhibition of the complete set of mammalian secreted phospholipases A2 by indole analogues: a structure-guided study. Bioorg. Med. Chem. 2004;12:1737–1749. doi: 10.1016/j.bmc.2004.01.022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.