

Abstract
Tumor metastasis is the dominant cause of death in cancer patients, including patients with oral tongue squamous cell carcinoma (TSCC). Previously, we reported that reduced miR-138 level is correlated with enhanced metastatic potential in TSCC cells. Here, we demonstrate that miR-138 suppresses TSCC cell migration and invasion by regulating two key genes in the Rho GTPase signaling pathway: RhoC and ROCK2. Direct targeting of miR-138 to specific sequences located in the 3′-untranslated regions of both RhoC and ROCK2 mRNAs were confirmed using luciferase reporter gene assays. Ectopic transfection of miR-138 reduced the expression of both RhoC and ROCK2 in TSCC cells. These reduced expressions, in consequence, led to the reorganization of the stress fibers and the subsequent cell morphology change to a round bleb-like shape, as well as the suppression of cell migration and invasion. In contrast, knockdown of miR-138 in TSCC cells enhanced the expression of RhoC and ROCK2, which resulted in an altered, elongated cell morphology, enhanced cell stress fiber formation, and accelerated cell migration and invasion. Taken together, our results suggest that miR-138 plays an important role in TSCC cell migration and invasion by concurrently targeting RhoC and ROCK2, and miR-138 may serve as a novel therapeutic target for TSCC patients at risk of metastatic disease.
Keywords: miR-138, RhoC, ROCK2, migration, invasion
Introduction
Oral cancer, predominately oral squamous cell carcinomas (OSCC), is one of the most devastating diseases. Oral tongue squamous cell carcinomas (TSCC), one of the major subtypes of OSCC, is significantly more aggressive than other forms of OSCCs, with a propensity for rapid local invasion, and spread,1 and a high recurrence rate.2 Despite improvements in surgery, radiotherapy and chemotherapy, the prognosis for TSCC patients has not significantly improved for the past 3 decades. Improvement in patient survival requires better understanding of tumor invasion and metastasis so that aggressive tumors can be detected early in the disease process, and so that targeted therapeutic interventions can be developed. While attempts have been made to identify genomic alterations that contribute to the aggressive phenotype of TSCC, most of these studies focus on protein coding genes. Our knowledge of genomic aberrations associated with non-coding genes (e.g., microRNA) and their contributions to cancer initiation and progression is relatively limited.
MicroRNAs (miRNAs) are non-coding small RNAs that control the target gene expression at the post-transcriptional level. It is currently estimated that the human genome may have over 1,000 miRNAs. Although they account for only a minor fraction of the expressed genome, microRNAs are essential regulators of diverse cellular processes including proliferation, differentiation, apoptosis, survival, motility and morphogenesis. Several microRNAs are believed to influence metastasis in various cancer types. These include: miR-23b which reduces cell migration by targeting urokinase and c-met in hepatocellular carcinoma cells,3 miR-222 which suppresses TSCC cell invasion by targeting MMP1,4 and miR-10b which stimulates cell invasion in breast cancers by suppressing HOXD10 expression which in turn leads to increased expression of RhoC, a well-characterized pro-metastatic gene.5
Our recent study demonstrated that reduced miR-138 level is associated with enhanced metastatic potential in OSCC.6 The decreased expression of miR-138 has been previously observed in several cancer types, including OSCC,7 TSCC,8 thyroid cancer,9 and lung cancer.10 Recently, two putative miR-138 precursor genes, termed pre-miR-138-1 and pre-miR-138-2, have been predicted in the mouse genome,11 and their human homologs have also been located on chromosome 3p21.33 and 16q13, respectively. Interestingly, loss of heterozygosity (LOH) at both chromosome loci have been frequently detected in OSCC and appears to correlate with tumor progression (i.e., cervical lymph node metastasis),12–14 which is in agreement with our previous observation that demonstrated miR-138’s contribute to the enhanced cell migration and invasion.6 However, the molecular mechanism(s) underlying the role of miR-138 in metastasis is poorly understood. This study seeks to investigate the molecular mechanism(s) that underlie the effect of miR-138 on metastasis.
Materials and Methods
Cell culture and transfection
Human OSSC cell lines used in this study were maintained in DMEM/F12 supplemented with 10% FBS, 100 U/mL penicillin and 100 μg/mL streptomycin (GIBCO) at 37°C in a humidified incubator containing 5% CO2. Primary normal human keratinocytes (NHOK) were prepared and cultured in OKM medium (ScienCell Research Laboratory) as previously described.16 For functional analysis, hsa-miR-138 mimics and non-targeting miRNA mimics (Dharmacon), anti-miR-138 peptide nucleic acid (PNA) and negative control PNA (Panagene), and gene specific siRNA (On-TargetPlus SMARTpool, Dharmacon) were transfected into cells using DharmaFECT Transfection Reagent 1 as described previously.4, 6
Real-time RT-PCR analysis
The relative expression level of miR-138 was determined using mirVana™ qRT-PCR microRNA Detection Kit (Ambion) as described previously.6 The relative mRNA levels of RhoC and ROCK2 were examined using a quantitative 2-step RT-PCR assay with gene specific primer sets (OriGene) as described previously.17 The relative expression level was determined using the 2−delta delta Ct analysis method,18 where actin was used as an internal reference.
Western Blotting Analysis
Western blots were performed as described previously 4 using antibodies specific against RhoA (Cell Signaling), RhoB (Cell Signaling), RhoC (Cell Signaling), ROCK1 (Santa Cluz), ROCK2 (Santa Cluz), and beta-actin (Sigma).
Dual luciferase reporter assay
An 62-bp fragment from the 3′ untranslated region (3′-UTR) of RhoC (position 1210 to 1271, NM_175744) and a 75-bp fragment from the 3′-UTR of ROCK2 (position 4932 to 5006, NM_175744) containing the miRNA-138 binding sites were cloned into the Xba I site of the pGL3-Control firefly luciferase reporter vector (Promega). The corresponding mutant constructs were created by mutating the seed regions of the miR-138 binding sites. The constructs were then verified by sequencing. Using lipofectamine 2000 (Invitrogen), cells were transfected with the reporter constructs containing either the targeting sequence from the RhoC 3′-UTR (named pGL3-RhoC) or its mutant (named pGL3-RhoCm), the targeting sequence from the ROCK2 3′-UTR (named pGL3-ROCK2) or its mutant (named pGL3-ROCK2m). The pRL-TK vector (Promega) was co-transfected as internal control for normalization of the transfection efficiency. The luciferase activities were then determined as described previously 4 using a Lumat LB 9507 Luminometer (Berthold Technologies).
In vitro cell migration assay and invasion assay
The in vitro cell migration was measured using the Oris™ 96-well cell migration assay kit (Platypus Technologies) following the manufacturer’s instructions. In brief, on the first day, 5 × 104 cells were seeded in each well. On day 2, cells were washed and transfected with appropriate miRNA (or siRNA) reagents. On day 3, 24 hrs after transfection, the stopper was removed to allow cells to migrate into the detection zone. On day 4, 24 hrs after initiation of the migration, cells were fixed with 75% ethyl alcohol and nucleus-stained with hematoxilin (Sigma). The cells that migrated into the detection zone were counted using ImageJ analysis software (version 1.421) following the manufacturer’s protocol. The in vitro cell invasion assay was performed using a Cultrex 96-well membrane invasion assay kit (R&D Systems) as described previously.4, 6
Cell stress fiber visualization
To visualize the stress fiber, cells were cultured on Lab-Tek slide chambers, and fixed using 3.7% formaldehyde solution (Thermo Scientific). The F-actin was stained with rhodamine phalloidin (Invitrogen) and mounted with ProLong Gold antifade reagent containing DAPI (Invitrogen) following the manufacturer’s protocol. The slides were then examined with a fluorescence microscope (Carl Zeiss). Representative images of cells at the migration fronts were captured at a magnification of x400.
Statistical analysis
Statistical analysis was done with Student’s t-test. Differences with P < 0.05 were considered statistically significant.
Results and Discussion
The UM1 and UM2 are paired TSCC cell lines with different metastatic potential that were previously established from a single patient.15 Significantly lower miR-138 level was observed in the highly invasive cell line (UM1) as compared to UM2.6 As shown in Figure 1A, ectopic transfection of the miR-138 mimic to the UM1 cells led to an increase of the miR-138 level as measured by quantitative RT-PCR. Introduction of the anti-miR-138 peptide nucleic acid (PNA) to the UM2 cells led to specific knockdown of miR-138. While the increased miR-138 level in UM1 resulted in reduced cell migration and cell invasion, the reduced miR-138 level in UM2 led to enhanced cell migration and invasion (Figure 1B & C). These findings confirmed our early observations.6 Accompanied with the changes in the cell migration and invasion, apparent differences in cell morphology and stress-fiber formation (F-actin filament) were also observed. As shown in Figures 1D & E, strikingly different morphologies were observed when UM1 cells were transfected with the miR-138 mimic. The cells switched from an elongated morphology to a rounded bleb-like morphology. Moreover, the stress-fiber was found to be significantly reduced and less organized in UM1 cells transfected with the miR-138 mimic as compared to UM1 cells transfected with the control mimic. For UM2 (Figures 1F and G), when cells were treated with anti-miR-138 PNA, they switched from the rounded morphology to the elongated morphology. Enhanced stress-fiber formation was observed in UM2 cells after treatment with anti-miR-138 PNA (Figure 1G). Small F-actin-rich protrusions were also observed. Similar results were observed in additional OSCC cell line 1386Ln and 686Ln (Supplementary Figure 1). These observations are in agreement with the notion that coordinated regulation of the actin cytoskeleton is central to cell motility, invasion and metastasis.
Figure 1. The effects of miR-138 on cell migration, invasion and stress-fiber formation in TSCC cells.
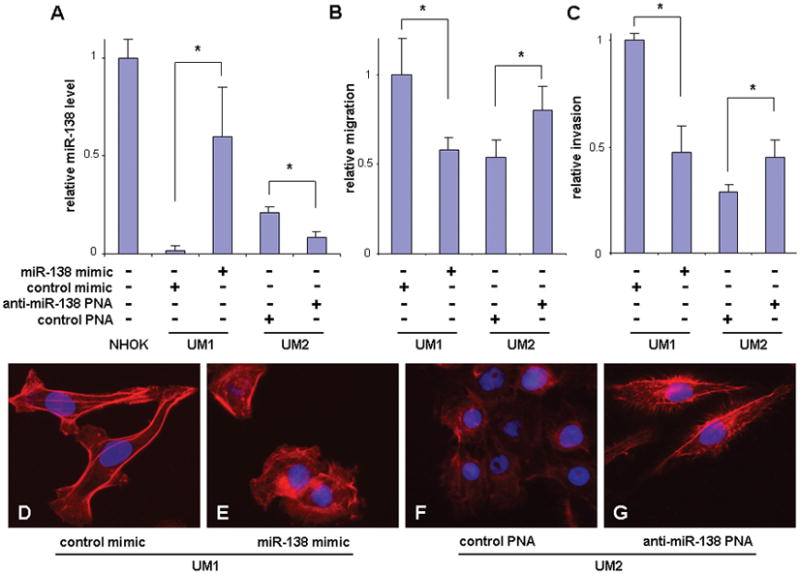
The UM1 cells were transfected with hsa-miR-138 mimic or negative control mimic. The UM2 cells were treated with anti-miR-138 PNA and negative control PNA. The relative miR-138 levels in these cells and a normal human oral keratinocyte primary culture (NHOK) were determined with quantitative RT-PCR assays (A). The miR-138 induced changes in the cell migration (B), invasion (C), and stress-fiber formation (F-actin) (D–G) were measured in these cells as described in the Material and Methods section. Additional images capturing miR-138 induced stress-fiber changes are presented in Supplementary Figure 2. Data represents at least 3 independent experiments with similar results. * indicates p < 0.05.
In order to further explore the functional roles of miR-138 in TSCC cell metastasis, a bioinformatics-based prediction was carried out to identify the potential targets for miR-138 based on a conservative two-way intersection of TargetScanHuman 5.119 and PicTar.20 A total of 86 potential targets for miR-138 were identified (see Supplementary Table 1). Among those predicted targets, 3 of them are major players in the Rho GTPase signaling cascade. These targets are: RhoC, one of the three Rho GTPases; ROCK2, a Rho-associated kinase; and ARHGEF3, one of the guanine nucleotide exchange factors (GEFs). The Rho GTPase is a subfamily of the Ras superfamily. The members of the Rho GTPase family have been described as “molecular switches” that regulate cell shape, polarity and locomotion through their effects on many aspects of intracellular actin dynamics.21 There are 3 Rho GTPases in human, RhoA, RhoB, and RhoC, which share 85% amino acid sequence identity, and exhibit distinct cellular functions.22 RhoA plays key roles in the regulation of actomyosin contractility, as well as cell proliferation and survival. RhoB, which is localized primarily on endosomes, has been shown to regulate cytokine trafficking and cell survival. RhoC plays a major role in the regulation of actin cytoskeleton, cell shape, attachment, and motility, which is highly relevant to cancer metastasis. Rho GTPases carry out these distinct functions by activating various downstream effectors, including Rho-associated kinases (such as ROCK1 and ROCK2). The activity of Rho GTPases is tightly controled by several families of regulators, including guanine nucleotide dissociation inhibitors (GDIs), GEFs, and GTPase activating proteins (GAPs). These constitute the major players in the Rho GTPase signaling pathway.22 Based on our bioinformatic analysis, as well as the observed cellular changes associated with miR-138 described above, it is logical to hypothesize that miR-138 regulates the cancer metastasis by targeting the Rho GTPase signaling cascade.
Based on the bioinformatics-based prediction, a highly conserved targeting site for miR-138 was identified in the 3′-UTR of the RhoC mRNA (Figure 2A). To confirm that miR-138 directly targets this sequence located in RhoC mRNA, dual luciferase reporter assay was performed using a construct in which this targeting site was cloned into the 3′-UTR of the reporter gene (pGL3-RhoC). As illustrated in Figure 2B, when cells were transfected with miR-138, the luciferase activity was significantly diminished as compared to the cells transfected with negative control. When the seed region of the targeting site was mutated (pGL3-RhoCm), the miR-138 effect on luciferase was abolished. Furthermore, as shown in Figure 2C, ectopic transfection of miR-138 reduced the protein level of RhoC in UM1 cells, and knockdown of miR-138 using anti-miR-138 PNA increased the protein level of RhoC in UM2 cells. Similar results were observed in additional OSCC cell line 1386Ln and 686Ln (Supplementary Figure 3). These miR-138-induced changes appear to be specific to RhoC as no apparent difference was observed in RhoA expression upon transfection (or knockdown) of miR-138. The other member of Rho family, RhoB, is not expressed to any significant extent and was not detectable in our cell lines (data not shown). As shown in Figure 2D with quantitative RT-PCR analysis, a significant decrease of RhoC mRNA level was detected in UM1 cells that were transfected with the miR-138 mimic. An apparent Increase in RhoC mRNA was observed when UM2 cells were treated with anti-miR-138 PNA. However, this increase is not statistically significant. These results suggested that miR-138 regulates RhoC gene expression, at least in part, by regulating the stability of RhoC mRNA. To confirm that the reduction of RhoC has a functional relevance to cell migration and invasion in our TSCC cell lines, we knocked-down RhoC using specific siRNA (Figure 2E), and demonstrated that the reduced RhoC level is associated with reduced cell migration (Figure 2F) and invasion (Figure 2G). Furthermore, the siRNA induced knockdown of RhoC resulted in a switch from the elongated cell morphology to the rounded cell morphology (Figure 2H & I). This morphology switch was accompanied by significant reduction in cellular stress fiber (filamentous-actin formation). Thus, these results suggest that miR-138 regulates TSCC migration, invasion, cell shape, and stress fiber formation, in part, by targeting RhoC mRNA. This finding is in agreement with previous studies demonstrating that the expression of RhoC is progressively increased as tumors become more aggressively metastatic, and that RhoC expression promotes metastasis.23–25
Figure 2. MiR-138 regulates cell migration and invasion by targeting RhoC.
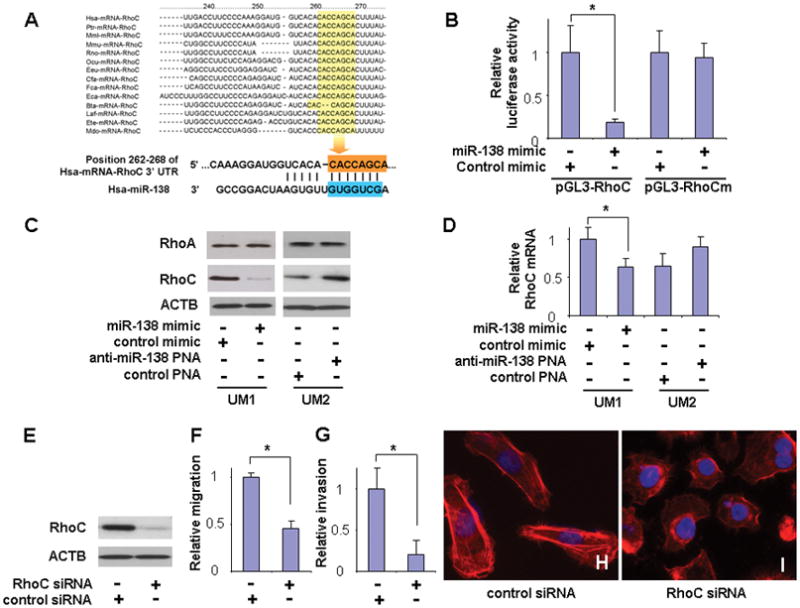
(A) The predicted highly conserved miR-138 targeting sequence located in the 3′-untranslated region (3′-UTR) of RhoC mRNA. (B) Dual luciferase reporter assays were performed to testing the interaction of miR-138 and its targeting sequence in the RhoC 3′-UTR using constructs containing the predicted targeting sequence (pGL3-RhoC) and mutated targeting sequence (pGL3-RhoCm) cloned into the 3′-UTR of the reporter gene. (C) Western blot analyses were performed to examine the effects of miR-138 on RhoC and RhoA gene expression at the protein level. (D) Quantitative RT-PCR assays were performed to examine the effects of miR-138 on RhoC gene expression at mRNA level. (E) Western blot analysis confirming the effective knockdown of RhoC expression by specific siRNA in UM1 cells. Cell migration (F) and invasion (G) were significant reduced when cells were treated with RhoC specific siRNA. (H–I) UM1 cells exhibit significant changes in cell morphology and stress-fiber formation upon treatment with RhoC specific siRNA. Data represents at least 3 independent experiments with similar results. * indicates p < 0.05.
RhoC functions through direct interaction with its downstream signaling molecule, Rho-associated kinases (e.g., ROCK1 and ROCK2), which in turn phosphorylate both a range of cytoskeletal proteins—allowing for the generation of contractile forces—and the ezrin-family proteins that link the actin cytoskeleton to the plasma membrane. Interestingly, we also identified ROCK2 as another direct miR-138 target gene in the Rho GTPase signaling pathway, and a highly conserved targeting site for miR-138 was identified in the 3′-UTR of the ROCK2 mRNA (Figure 3A). The direct interaction between miR-138 and the targeting sequence in ROCK2 mRNA was tested using a luciferase reporter gene construct containing this targeting sequence in the 3′-UTR (pGL3-ROCK2). As illustrated in Figure 3B, when cells were transfected with miR-138 mimic, the luciferase activity was significantly reduced when compared to the cells transfected with negative control. However, when the seed region of the targeting site was mutated (pGL3-ROCK2m), the miR-138 effect on luciferase was abolished. Furthermore, as shown in Figure 3C, ectopic transfection of miR-138 reduced the protein level of ROCK2 in UM1 cells, and knockdown of miR-138 using anti-miR-138 PNA increased the protein level of ROCK2 in UM2 cells. Similar results were observed in additional OSCC cell line 1386Ln and 686Ln (Supplementary Figure 3). Since no apparent difference was observed in the expression of ROCK1 upon transfection (or knockdown) of miR-138, the miR-138-induced changes in ROCK family proteins appears to be specific to ROCK2. As shown in Figure 3D, no significant change in ROCK2 mRNA level was detected when cells were transfected with either miR-138 mimic or anti-miR-138 PNA. These results, together with the observed changes at ROCK2 protein levels, suggested that miR-138 regulates ROCK2 gene expression primarily by inhibiting translation. To confirm that the reduction of ROCK2 has a functional effect on cell migration and invasion, we knocked-down ROCK2 using specific siRNA (Figure 3E), and demonstrated that the reduced ROCK2 level is associated with reduced cell migration (Figure 3F), invasion (Figure 3G), and the reduction in stress fiber formation (Figure 3H & I). Thus, these results suggest that miR-138 regulates TSCC migration, invasion, cell shape, and stress fiber formation by targeting ROCK2 mRNA. It is worth knowing that RhoC is believed to be a better activator of ROCKs in epithelial cells than RhoA and RhoB,22, 26 which might account for its specific role in cell locomotion and metastasis.27 By targeting RhoC and ROCK2 concurrently, miR-138 thus selectively inhibits the RhoC specific cell migration and invasion.
Figure 3. MiR-138 regulates cell migration and invasion by targeting ROCK2.
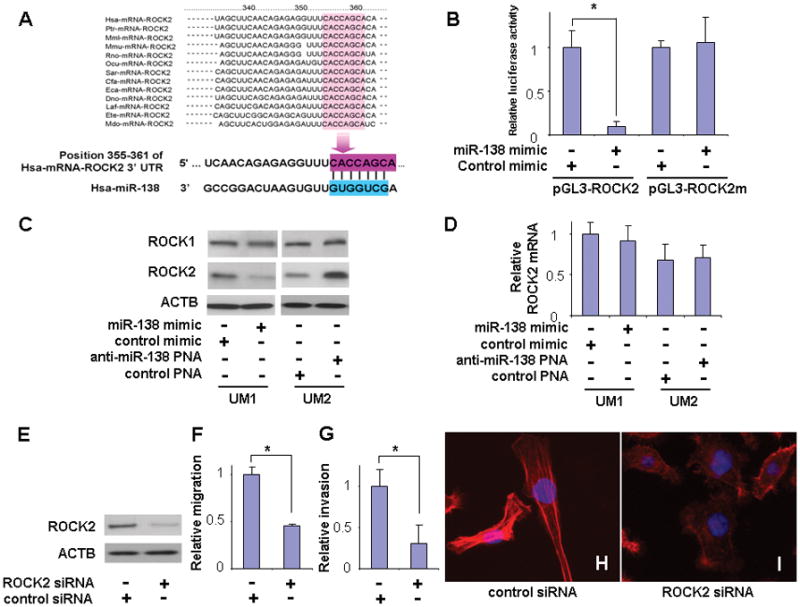
(A) The predicted highly conserved miR-138 targeting sequence in the ROCK2 3′-untranslated region (3′-UTR). (B) Dual luciferase reporter assays were performed to testing the interaction of miR-138 and its targeting sequence in the ROCK2 3′-UTR using constructs containing the predicted targeting sequence (pGL3-ROCK2) and mutated targeting sequence (pGL3-ROCK2m) cloned into the 3′-UTR of the reporter gene. (C) Western blot analyses were performed to examine the effects of miR-138 on ROCK1 and ROCK2 gene expression at the protein level. (D) Quantitative RT-PCR assays were performed to examine the effects of miR-138 on ROCK2 gene expression at mRNA level. (E) Western blot analysis confirming the effective knockdown of ROCK2 expression by specific siRNA in UM1 cells. Cell migration (F) and invasion (G) were significant reduced when cells were treated with ROCK2 specific siRNA. (H–I) UM1 cells exhibit significant changes in cell morphology and stress-fiber formation upon treatment with ROCK2 specific siRNA. Data represents at least 3 independent experiments with similar results. * indicates p < 0.05.
The third predicted target for miR-138 in Rho GTPase signaling pathway is ARHGEF3. Guanine nucleotide exchange factors, such as ARHGEF3, accelerate the GTPase activity of Rho GTPases by catalyzing their release of bound GDP. A previous study demonstrated that ARHGEF3 interacts with RhoA and RhoB, but not with RhoC.28 A highly conserved binding site for miR-138 was predicted at position 1279 to 1285 in the 3′-UTR. While our luciferase reporter gene assay suggested that the conserved sequence in the 3′-UTR of the ARHGEF3 is a functional binding site for miR-138, ectopic transfection or knockdown of miR-138 did not change the expression level of ARHGEF3 to any significant extent (data not shown). Therefore, it appears that ARHGEF3 is not functionally targeted by miR-138 in our system. Nevertheless, it remains possible that miR-138 may functionally target ARHGEF3 in other cell types or different biological systems. It is worth knowing that ARHGEF3 does have a tissue-specific expression pattern 28 although its function and intracellular localization have yet to be fully documented.
Metastasis requires passage across tissue boundaries, which is facilitated by increased cancer cell motility due to cytoskeletal remodeling. The Rho GTPases signaling cascade plays a central role in regulating cell adhesion, migration and the cytoskeleton.21 While Rho family gene mutations are relatively rare in tumors, overexpressions of these genes are common events in cancer cells.29 The overexpression of key genes in Rho GTPases signaling cascade, including RhoC and ROCK2, have been frequently linked to enhanced metastatic potential in various cancer types.30–33 As illustrated in Figure 4, our results suggest a novel paradigm in which miR-138 regulates RhoC-specific GTPase signaling cascade by targeting both RhoC and ROCK2 mRNAs concurrently, and suppress their expression at post-transcriptional levels. The reduction of miR-138 in highly metastatic OTSCC thus leads to the enhanced expression of RhoC and ROCK2 and consequently enhances the activity of the Rho GTPase signaling cascade, which in turn leads to increased metastatic phenotypes (increase in migration and invasion).
Figure 4.
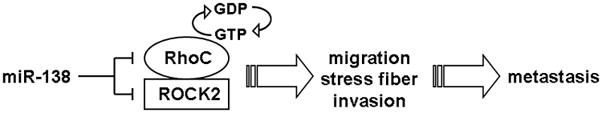
Potential roles of miR-138 on Rho GTPase signaling cascade and cancer cell metastasis.
While our study provided evidence suggesting the roles of miR-138 in cancer cell migration and invasion, several other functional roles for miR-138 have been previously suggested. Our previous study suggested that miR-138 also promotes apoptosis in head and neck SCC.6 It has been reported previously that hTERT is targeted by miR-138 in thyroid cancer.9 We did not observe any apparent change in hTERT expression after the transfection of miR-138 mimic in our cell lines (data not shown). This apparent difference in the miR-138 effect on hTERT may be due to the differences in cancer types. Alternatively, this may be cell line specific. It is possible that the cell lines we tested here (or the human anaplastic thyroid carcinoma cell line used in the previous study) may have specific mutation(s) that dictates the miR-138 effects on hTERT. More in-depth functional analysis will be needed to fully evaluate the effect of miR-138 on hTERT. In addition, miR-138 appears to play important roles in cardiac morphogenesis during embryonic development,34 and in dendritic spine morphogenesis,35 a phenomenon associated with long-lasting memory.
In summary, miR-138 is a multi-functional molecule regulator that regulates a variety of biological processes. One of its major roles in cancer progression is regulating cancer cell migration and invasion. The results from the present study demonstrated the molecular mechanisms of miR-138 in OTSCC cell migration and invasion, and its potential as a novel therapeutic target for OTSCC patients at risk of metastasis.
Supplementary Material
Acknowledgments
This work was supported in part by NIH PHS grants K22DE014847, RO1CA139596, RO3CA135992, and a grant from Prevent Cancer Foundation (to X.Z.). We thank Ms. Katherine Long for her editorial assistance.
Abbreviation
- GAPs
GTPase activating proteins
- GDIs
guanine nucleotide dissociation inhibitors
- KGM
keratinocyte growth medium
- LOH
loss of heterozygosity
- miRNAs
MicroRNAs
- NHOK
normal human oral keratinocyte
- OSCC
oral squamous cell carcinomas
- TSCC
tongue squamous cell carcinoma
References
- 1.Franceschi D, Gupta R, Spiro RH, Shah JP. Improved survival in the treatment of squamous carcinoma of the oral tongue. Am J Surg. 1993;166:360–5. doi: 10.1016/s0002-9610(05)80333-2. [DOI] [PubMed] [Google Scholar]
- 2.Fakih AR, Rao RS, Borges AM, Patel AR. Elective versus therapeutic neck dissection in early carcinoma of the oral tongue. Am J Surg. 1989;158:309–13. doi: 10.1016/0002-9610(89)90122-0. [DOI] [PubMed] [Google Scholar]
- 3.Salvi A, Sabelli C, Moncini S, Venturin M, Arici B, Riva P, Portolani N, Giulini SM, De Petro G, Barlati S. MicroRNA-23b mediates urokinase and c-met downmodulation and a decreased migration of human hepatocellular carcinoma cells. Febs J. 2009;276:2966–82. doi: 10.1111/j.1742-4658.2009.07014.x. [DOI] [PubMed] [Google Scholar]
- 4.Liu X, Yu J, Jiang L, Wang A, Shi F, Ye H, Zhou X. MicroRNA-222 Regulates Cell Invasion by Targeting Matrix Metalloproteinase 1 (MMP1) and Manganese Superoxide Dismutase 2 (SOD2) in Tongue Squamous Cell Carcinoma Cell Lines. Cancer Genomics Proteomics. 2009;6:131–9. [PMC free article] [PubMed] [Google Scholar]
- 5.Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007;449:682–8. doi: 10.1038/nature06174. [DOI] [PubMed] [Google Scholar]
- 6.Liu X, Jiang L, Wang A, Yu J, Shi F, Zhou X. MicroRNA-138 suppresses invasion and promotes apoptosis in head and neck squamous cell carcinoma cell lines. Cancer Lett. 2009;286:217–22. doi: 10.1016/j.canlet.2009.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kozaki K, Imoto I, Mogi S, Omura K, Inazawa J. Exploration of tumor-suppressive microRNAs silenced by DNA hypermethylation in oral cancer. Cancer Res. 2008;68:2094–105. doi: 10.1158/0008-5472.CAN-07-5194. [DOI] [PubMed] [Google Scholar]
- 8.Wong TS, Liu XB, Wong BY, Ng RW, Yuen AP, Wei WI. Mature miR-184 as Potential Oncogenic microRNA of Squamous Cell Carcinoma of Tongue. Clin Cancer Res. 2008;14:2588–92. doi: 10.1158/1078-0432.CCR-07-0666. [DOI] [PubMed] [Google Scholar]
- 9.Mitomo S, Maesawa C, Ogasawara S, Iwaya T, Shibazaki M, Yashima-Abo A, Kotani K, Oikawa H, Sakurai E, Izutsu N, Kato K, Komatsu H, et al. Downregulation of miR-138 is associated with overexpression of human telomerase reverse transcriptase protein in human anaplastic thyroid carcinoma cell lines. Cancer Sci. 2008;99:280–6. doi: 10.1111/j.1349-7006.2007.00666.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seike M, Goto A, Okano T, Bowman ED, Schetter AJ, Horikawa I, Mathe EA, Jen J, Yang P, Sugimura H, Gemma A, Kudoh S, et al. MiR-21 is an EGFR-regulated anti-apoptotic factor in lung cancer in never-smokers. Proc Natl Acad Sci U S A. 2009 doi: 10.1073/pnas.0905234106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Obernosterer G, Leuschner PJ, Alenius M, Martinez J. Post-transcriptional regulation of microRNA expression. Rna. 2006;12:1161–7. doi: 10.1261/rna.2322506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Piccinin S, Gasparotto D, Vukosavljevic T, Barzan L, Sulfaro S, Maestro R, Boiocchi M. Microsatellite instability in squamous cell carcinomas of the head and neck related to field cancerization phenomena. Br J Cancer. 1998;78:1147–51. doi: 10.1038/bjc.1998.644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hogg RP, Honorio S, Martinez A, Agathanggelou A, Dallol A, Fullwood P, Weichselbaum R, Kuo MJ, Maher ER, Latif F. Frequent 3p allele loss and epigenetic inactivation of the RASSF1A tumour suppressor gene from region 3p21.3 in head and neck squamous cell carcinoma. Eur J Cancer. 2002;38:1585–92. doi: 10.1016/s0959-8049(01)00422-1. [DOI] [PubMed] [Google Scholar]
- 14.Wang X, Gleich L, Pavelic ZP, Li YQ, Gale N, Hunt S, Gluckman JL, Stambrook PJ. Cervical metastases of head and neck squamous cell carcinoma correlate with loss of heterozygosity on chromosome 16q. Int J Oncol. 1999;14:557–61. doi: 10.3892/ijo.14.3.557. [DOI] [PubMed] [Google Scholar]
- 15.Nakayama S, Sasaki A, Mese H, Alcalde RE, Matsumura T. Establishment of high and low metastasis cell lines derived from a human tongue squamous cell carcinoma. Invasion Metastasis. 1998;18:219–28. doi: 10.1159/000024515. [DOI] [PubMed] [Google Scholar]
- 16.Park N-H, Min B-M, Li S-L, Huan MZ, Cherrick HM, Doniger J. Immortalization of normal human oral keratinocytes with type 16 human papillomavirus. Carcinogenesis. 1991;1:1627–31. doi: 10.1093/carcin/12.9.1627. [DOI] [PubMed] [Google Scholar]
- 17.Zhou X, Temam S, Oh M, Pungpravat N, Huang BL, Mao L, Wong DT. Global expression-based classification of lymph node metastasis and extracapsular spread of oral tongue squamous cell carcinoma. Neoplasia. 2006;8:925–32. doi: 10.1593/neo.06430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 19.Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, Rajewsky N. Combinatorial microRNA target predictions. Nat Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 21.Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR. Cell migration: integrating signals from front to back. Science. 2003;302:1704–9. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 22.Wheeler AP, Ridley AJ. Why three Rho proteins? RhoA, RhoB, RhoC, and cell motility. Exp Cell Res. 2004;301:43–9. doi: 10.1016/j.yexcr.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 23.Ridley AJ. Rho proteins and cancer. Breast Cancer Res Treat. 2004;84:13–9. doi: 10.1023/B:BREA.0000018423.47497.c6. [DOI] [PubMed] [Google Scholar]
- 24.Wu M, Wu ZF, Kumar-Sinha C, Chinnaiyan A, Merajver SD. RhoC induces differential expression of genes involved in invasion and metastasis in MCF10A breast cells. Breast Cancer Res Treat. 2004;84:3–12. doi: 10.1023/B:BREA.0000018426.76893.21. [DOI] [PubMed] [Google Scholar]
- 25.Fingleton B. Molecular targets in metastasis: lessons from genomic approaches. Cancer Genomics Proteomics. 2007;4:211–21. [PubMed] [Google Scholar]
- 26.Sahai E, Marshall CJ. ROCK and Dia have opposing effects on adherens junctions downstream of Rho. Nat Cell Biol. 2002;4:408–15. doi: 10.1038/ncb796. [DOI] [PubMed] [Google Scholar]
- 27.Riento K, Ridley AJ. Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol. 2003;4:446–56. doi: 10.1038/nrm1128. [DOI] [PubMed] [Google Scholar]
- 28.Arthur WT, Ellerbroek SM, Der CJ, Burridge K, Wennerberg K. XPLN, a guanine nucleotide exchange factor for RhoA and RhoB, but not RhoC. J Biol Chem. 2002;277:42964–72. doi: 10.1074/jbc.M207401200. [DOI] [PubMed] [Google Scholar]
- 29.Sahai E, Marshall CJ. RHO-GTPases and cancer. Nat Rev Cancer. 2002;2:133–42. doi: 10.1038/nrc725. [DOI] [PubMed] [Google Scholar]
- 30.Kleer CG, Teknos TN, Islam M, Marcus B, Lee JS, Pan Q, Merajver SD. RhoC GTPase expression as a potential marker of lymph node metastasis in squamous cell carcinomas of the head and neck. Clin Cancer Res. 2006;12:4485–90. doi: 10.1158/1078-0432.CCR-06-0376. [DOI] [PubMed] [Google Scholar]
- 31.Clark EA, Golub TR, Lander ES, Hynes RO. Genomic analysis of metastasis reveals an essential role for RhoC. Nature. 2000;406:532–5. doi: 10.1038/35020106. [DOI] [PubMed] [Google Scholar]
- 32.Wong CC, Wong CM, Tung EK, Man K, Ng IO. Rho-kinase 2 is frequently overexpressed in hepatocellular carcinoma and involved in tumor invasion. Hepatology. 2009;49:1583–94. doi: 10.1002/hep.22836. [DOI] [PubMed] [Google Scholar]
- 33.Narumiya S, Tanji M, Ishizaki T. Rho signaling, ROCK and mDia1, in transformation, metastasis and invasion. Cancer Metastasis Rev. 2009;28:65–76. doi: 10.1007/s10555-008-9170-7. [DOI] [PubMed] [Google Scholar]
- 34.Morton SU, Scherz PJ, Cordes KR, Ivey KN, Stainier DY, Srivastava D. microRNA-138 modulates cardiac patterning during embryonic development. Proc Natl Acad Sci U S A. 2008;105:17830–5. doi: 10.1073/pnas.0804673105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Siegel G, Obernosterer G, Fiore R, Oehmen M, Bicker S, Christensen M, Khudayberdiev S, Leuschner PF, Busch CJ, Kane C, Hubel K, Dekker F, et al. A functional screen implicates microRNA-138-dependent regulation of the depalmitoylation enzyme APT1 in dendritic spine morphogenesis. Nat Cell Biol. 2009;11:705–16. doi: 10.1038/ncb1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.