

Abstract
After a general introduction, the main pathways of ethanol metabolism (alcohol dehydrogenase, catalase, coupling of catalase with NADPH oxidase and microsomal ethanol-oxidizing system) are shortly reviewed. The cytochrome P450 isoform (CYP2E1) specifically involved in ethanol oxidation is discussed. The acetaldehyde metabolism and the shift of the NAD/NADH ratio in the cellular environment (reductive stress) are stressed. The toxic effects of acetaldehyde are mentioned. The ethanol-induced oxidative stress: the increased MDA formation by incubated liver preparations, the absorption of conjugated dienes in mitochondrial and microsomal lipids and the decrease in the most unsaturated fatty acids in liver cell membranes are discussed. The formation of carbon-centered (1-hydroxyethyl) and oxygen-centered (hydroxyl) radicals during the metabolism of ethanol is considered: the generation of hydroxyethyl radicals, which occurs likely during the process of univalent reduction of dioxygen, is highlighted and is carried out by ferric cytochrome P450 oxy-complex (P450–Fe3+O·−2) formed during the reduction of heme-oxygen. The ethanol-induced lipid peroxidation has been evaluated, and it has been shown that plasma F2-isoprostanes are increased in ethanol toxicity.
Keywords: CYP2E1 isoform, Ethanol metabolism, Hydroxyethyl radicals, Liver-free non-protein bound iron, Oxidative stress, Plasma isoprostanes
Introduction
Ethanol unlike many other hepatotoxic chemicals is not a foreign substance for living organisms; it occurs, in fact, in small amounts in mammalian tissues [82] and is conceivably formed by the alcohol dehydrogenase-catalyzed reduction of acetaldehyde derived from the decarboxylation of the intermediary metabolite pyruvate [83]. A similar reaction also occurs in biological fermentation (such as that yielding ethyl alcohol in wine and other alcoholic beverages) in which glucose is fermented by yeast. Moreover, significant amounts of ethanol are normally formed in the gastrointestinal tract, absorbed by the portal vein and metabolized in the liver [64]; most alcohol is of microbial origin, the other portion probably arising from the acetaldehyde formed by the normal pathways of degradation of threonine, deoxyribose phosphate and β-alanine. Furthermore, ethanol is rapidly and most entirely converted to the key intermediate, acetate, which can enter a wide-spread variety of metabolic pathways. This “more physiological” aspect of ethanol when compared with other hepatotoxic drugs complicates to a large extent the study of the mechanisms involved in the pathogenetic effects of alcohol. The great interest on ethylism from both a clinical and a sociological point of views resulted in an impressive number of studies on the effects of ethyl alcohol. However, the results have not been even univocal because the models used (alcohol dosage, acute or chronic administration, animals studied, different susceptibility to ethanol consumption, sex, nutritional status, etc.) have been extremely variable. Even the choice of the model itself for the study is questionable: in fact, if the acute (single large dose of alcohol) intoxication is a model more suitable for the study of the direct effects of ethanol at the cellular, subcellular and molecular level, it is obviously away from the conditions of the alcoholic; the chronic intoxication, on the other hand, is more adherent to such conditions but is subjected to a number of variables (of nutritional order in particular) that can contribute to a great extent to the development of the lesions, so that it is more difficult to discern the net role played by alcohol. Since the aim of the present review is to focus on the mechanisms of ethanol-induced liver injury and on the role of oxidative stress, particular attention will be paid to the results obtained with acute ethanol intoxication.
Ethanol metabolism and consequent alterations of the overall metabolism of the hepatic cell
The pathophysiological bases of the hepatic alterations produced by ethanol start obviously from its metabolism. Ethanol is quickly absorbed in the gastrointestinal tract and is almost completely oxidized to carbon dioxide and water. Minor amounts only are excreted, unmetabolized, in urine, breath and sweat. The rate of alcohol metabolism varies between 50 and 180 mg/h/kg body wt [118] in adult men, while in rats and mice, notwithstanding the natural aversion to alcohol, a two to threefold higher rate has been reported [67].
Alcohol is mainly metabolized in the liver, and the metabolic pathways are summarized in Fig. 1. The most important pathway is represented by alcohol dehydrogenase, a zinc containing metalloenzyme [121] widely distributed in nature, localized in the soluble cytoplasm and NAD+-dependent. It catalyzes the oxidation of ethanol to acetaldehyde (Fig. 1A). It is generally accepted that ethanol is also metabolized by catalase, a heme-containing enzyme widely distributed in nature and particularly expressed in the peroxisomes. Catalase, which normally catalyzes the decomposition of H2O2 to H2O and O2 (catalatic reaction), can also catalyze the reduction of H2O2 to H2O if electron donors are present (peroxidatic reaction); generally such reaction is catalyzed by peroxidase, but catalase can also catalyze peroxidative type reactions, when hydrogen peroxide is produced and a source of electron donors is available. Ethanol, which acts as an electron donor, is so oxidized to acetaldehyde (Fig. 1B). Hydrogen peroxide necessary for the reaction can be produced by oxidases, some of which also occur in the peroxisomes [14].
Fig. 1.

Metabolic pathways of ethanol
Ethanol oxidation can also result from the coupling of the reactions catalyzed by NADPH oxidase (microsomal) and xanthine oxidase with catalase (Fig. 1C, D, respectively). Which is the real importance of catalase in ethanol metabolism is not clear; it seems to be the most important pathway for the metabolism of methanol [1], while it would be of minor importance for the oxidation of ethanol, at least under normal conditions.
Finally, Lieber and De Carli [69] and Lieber et al. [78] have described a third metabolic pathway for ethanol oxidation occurring in the endoplasmic reticulum of the hepatocyte and named “microsomal ethanol-oxidizing system” (MEOS) (Fig. 1E). It utilizes the terms of the enzymatic system of “drug metabolism” (mixed function oxidase system, driven by the microsomal electron transport chain). MEOS with NADPH as a cofactor, oxidizes, it too, ethanol to acetaldehyde (Fig. 1E). A long debate followed the report of MEOS as a separate and individual ethanol-oxidizing system: some authors [19, 115] claimed that microsomal ethanol oxidation could result from the coupling of the activities of NADPH oxidase, the real microsomal enzyme, with catalase or with catalase and alcohol dehydrogenase (present as contaminant in the microsomal fraction) [57]. However, the group of Lieber [117] and others [87] showed that (a) MEOS is active even in acatalasemic animals; (b) a reconstituted system consisting of the essential terms of drug metabolism, i.e., cytochrome P450, NADPH-cytochrome c reductase and synthetic phospholipids, is able to oxidize, besides benzphetamine (characteristic substrate of drug metabolism), ethanol and other alcohols too and that MEOS is adaptively increased after chronic ethanol consumption, like other drug metabolizing activities. The induction of drug metabolizing enzymes due to alcohol [56, 73, 108, 109] and the fact that ethanol in vitro inhibits in a competitive way the same enzymes, could explain, at least in part, the increased tolerance of alcoholics to sedatives when sober and the enhanced sensitivity to sedatives when inebriated. The quantitative contribution of microsomal ethanol oxidation to the overall ethanol metabolism is another much debated question. According to some estimations [115] this route does not play a significant role in the metabolism of alcohol in vivo; however, according to the calculation of [71] about ¼ of the ethanol ingested could be metabolized by MEOS in normal conditions, while, after prolonged ethanol consumption, the role of MEOS could be considerably greater. Today, it is well known that the isoform of cytochrome P450 involved in ethanol oxidation, CYP2E1 (see in the following paragraphs), has a much higher Km for ethanol (8–10 mmol/l) compared to that of alcohol dehydrogenase (0.2–2 mmol/l); however, chronic alcohol consumption increases the activity of CYP2E1 by several times [3].
Acetaldehyde metabolism
As reported in the previous paragraphs, all the ethanol-metabolizing pathways lead to the formation of acetaldehyde. The latter can be oxidized to acetate mainly by enzymes, aldehydes dehydrogenases, which are mitochondrial, NAD+-dependent enzymes. Thus, the two main steps of ethanol metabolism, namely, the NAD+-linked oxidation of ethanol to acetaldehyde and the NAD+-linked oxidation of acetaldehyde to acetate, cause an increased concentration of the reduced pyridine coenzyme, NADH. Therefore, a decrease in the NAD+/NADH ratio occurs in the liver cell and such an imbalance would represent, according to a classic view of the problem [68, 74, 75], the central event in the explanation of the various metabolic alterations produced directly in the hepatic cell by ethanol oxidation, including triglyceride accumulation (the importance of the reductive stress environment created by ethanol oxidation will be outlined in the following paragraphs).
As is known, the reoxidation of NADH formed in the soluble cytoplasm occurs through shuttle mechanisms that transfer reducing equivalents from cytoplasmic NADH to mitochondrial electron transport chain. The latter becomes quickly saturated by reducing equivalents originating from ethanol and thus can slow down the citric acid cycle that, under normal conditions, supplies itself reducing equivalents to the respiratory chain. The slackening of the citric acid cycle leads to various metabolic consequences, the major part of which derive from decreased oxidation of acetate, which can originate, as mentioned in the previous paragraphs, directly by ethanol, even if a consistent part of the latter would be metabolized in peripheral tissues [80]. The metabolic unbalance derived from the decreased acetate oxidation is primarily reflected on fatty acid oxidation, fatty acid synthesis, ketone bodies formation and cholesterol metabolism. Decreased fatty acid oxidation and increased fatty acid synthesis have been demonstrated since a long time ago [40, 74, 95, 99, 126]. It was therefore suggested that ethanol, acting itself as a fuel for mitochondria, could replace in a “competitive way” fatty acids that, under normal conditions, represent the major source of energy for the liver cell.
Today, it is known that the ethanol-induced stimulation of hepatic triglyceride synthesis depends, to a great extent, upon the increased expression of the enzymes involved in fatty acid synthesis that are regulated by the transcription factor sterol regulatory element-binding protein (SREBP)-1 [123], located in the endoplasmic reticulum. Upon ethanol feeding, SREBP-1 is proteolytically cleaved to the active form that translocates to the nucleus, inducing the expression of the genes coding for lipogenic enzymes.
Pathogenetic mechanisms of the hepatic lesions produced by ethanol
As it is known, the long lasted debate whether nutritional deficiencies associated to alcoholism are responsible of liver injury [15, 49] or whether ethanol per se (or some proximal metabolite) exerts toxic effects on hepatic cell has been largely resolved by the introduction of a novel model for experimental studies based on liquid diets in which ethanol (36% of total calories) replaces in a isocaloric way part of carbohydrates. With these diets, considered nutritionally adequate, the alcohol assumption in the rat was greatly increased and hepatic lesions (hepatic steatosis) were obtained in both rats and humans [76, 77]. These lesions therefore apparently were independent of nutritional deficiencies. Subsequently, with the use of primates as experimental animals, a sequential production of fatty liver, inflammatory reactions, necrosis, fibrosis and sclerosis has been obtained [78] upon prolonged ethanol feeding.
The ethanol-induced ultrastructural changes of the liver cell involve both the mitochondria and the endoplasmic reticulum. The mitochondrial changes consist of enlargement, swelling, shortening and disorganization of the cristae, decreased number or absence of matrix granules and intramitochondrial crystalline inclusions. The presence of bizarre shapes and giant mitochondria [61, 63] with an increased matrix density was also reported [56, 66, 72, 97]. The changes involving the endoplasmic reticulum [109] consist of a marked proliferation of the smooth membranes with abundant vesicular structures. The alterations of the endoplasmic reticulum are, according to [56], the first morphological changes detectable in the liver cell after ethanol feeding to rats; they were observed, in fact, after few days of alcohol administration, while mitochondrial changes appeared some days later. Autophagic vacuoles, containing altered mitochondria, and residual dense bodies were also described. The presence of hyaline Mallory bodies was repeatedly documented. An increased number of peroxisomes were also reported [12], which is consistent with the increased catalase activity.
The morphological changes of mitochondria have a functional counterpart, consisting in a decrease in cytochrome a, a3 and b [21, 62, 110], of the enzymatic activities of the Krebs cycle [21, 110] and in general of the respiratory control [45, 59]. Many of these alterations are stable and can be in turn responsible for the changes in lipid metabolism produced by ethanol oxidation itself. Ethanol-induced mitochondrial changes also involve the autochthonous synthesis of some mitochondrial proteins, thus producing an unbalance between proteins synthesized in mitochondria and those synthesized in the endoplasmic reticulum. Such unbalance could influence the mitochondria biosynthesis.
The toxic effects of acetaldehyde
That ethanol-induced liver changes are in part mediated by the proximal metabolite acetaldehyde that has been proposed since a long time ago and reconsidered when it has been observed [21] that many of the earlier mentioned ethanol-induced mitochondrial changes are reproduced by acetaldehyde. In particular, the addition of acetaldehyde to isolated mitochondria at concentrations of the same order as those occurring in the liver actively oxidizing ethanol (1–3 mM), inhibits mitochondrial respiration at the level of complex I (NADH-ubiquinone oxido-reductase) and the coupling site I of oxidative phosphorylation [21, 22]. Moreover, acetaldehyde inhibits, at the same concentrations, fatty acid oxidation, and such inhibition seems to be due to the inhibition of β-oxidation, citric acid cycle and oxidative phosphorylation [24]. The inhibition of the oxidation of NAD+-dependent substrates [21] and the inhibition of fatty acid oxidation [24] do not seem to depend from a competition of acetaldehyde with such substrates for NAD+. It has been suggested [23, 25], on the other hand, that acetaldehyde could interact with sulphydryl groups involved in oxidative phosphorylation particularly with those essential for the complex I (NAD+ ubiquinone-oxydoreductase) which is the site of the respiratory chain mainly affected by both acetaldehyde and ethanol. Thus, the block of –SH groups would be responsible for the alterations. The reaction of acetaldehyde and other alkanals with cysteine has been actually known since many decades (formation of thiazolidine-carboxylic acids) and other low molecular weight thiols (reduced glutathione, GSH, in particular) can also be involved in this reaction. In effect, ethanol toxicity is always accompanied by a decrease in hepatic GSH content. If, as we will see later, lipid peroxidation is going to develop in ethanol hepatotoxicity, much more reactive aldehydes (alkenals and 4-hydroxyalkenals) will be formed and a much higher reactivity toward –SH groups and other nucleophiles has to be expected.
Finally, the acetaldehyde-produced alterations of the mitochondrial functions impair the mitochondrial acetaldehyde metabolism [51], thus producing worsening of the damage by a vicious cycle.
Ethanol-induced oxidative stress
It has been known since a long time ago that prior administration for a variety of substances known as antioxidants affords a marked protection against the liver damage induced by CCl4 and other hepatotoxins [44, 54]. It was subsequently demonstrated [29, 31, 36] that CCl4 both administered in vivo or added in vitro to liver preparations markedly increases the peroxidation of liver lipids, as measured by malonil dialdehyde (MDA) formation. Such a pro-oxidant effect of CCl4 was immediately confirmed by Recknagel and Ghoshal [100, 101]. The theoretical background for these studies was suggested by [18] and [125], who proposed that in the hepatic cell, CCl4 could undergo a homolytic cleavage yielding free radicals (CCl·3, CCl3OO·, etc.). The latter could rapidly interact with neighboring molecules, such as proteins, nucleic acid, thiols and membrane unsaturated fatty acids. The latter interaction would set into motion lipid peroxidation that seriously affects membrane structure and function. Since antioxidant pretreatment [37] was also found to be effective in ethanol-induced liver damage, it was suggested [38] that the liver injury produced by CCl4 or ethanol could have a common pathogenetic mechanism, namely, oxidative stress1 and peroxidation of liver lipids. It was subsequently found [30, 32] that, as in the case of CCl4, the MDA production by incubated liver homogenates is greatly enhanced during 1, 2, 4, 6 and 12 h after acute ethanol administration. The increase in liver lipid peroxidation precedes the accumulation of triglycerides in the liver, thus excluding that the enhanced MDA formation could result from the increased hepatic fat content; the latter possibility is also ruled out by the fact that no increased MDA production occurs 24 h after ethanol dosing when the hepatic triglyceride level is maximum. Furthermore, it was shown [32] that ethanol added in vitro to liver homogenates has a pro-oxidant effect although to a lesser extent than carbon tetrachloride. The effect is specific for the liver tissue, since ethanol, either in vitro or in vivo, has no effect on the peroxidation of brain [48] or other tissue homogenates [114].
It was proposed [32] that ethanol or its metabolites stress the “peroxidative balance” of the liver cell [16] toward autoxidation, either acting as pro-oxidant or lowering the cellular antioxidant level. Direct evidence for increased hepatic lipoperoxidation in vivo after acute ethanol intoxication was forwarded by Kalish and Di Luzio [58], who showed that the peroxide content was increased in the liver in ethanol-treated rats. Hashimoto and Recknagel [50], on the contrary, found no evidence of conjugated diene absorption characteristic of peroxidized lipids [17] in the lipids of any subcellular fraction at any time after acute ethanol intoxication. On the basis of these results, it was concluded that in the case of ethanol-induced liver injury, there is no direct evidence for the in vivo occurrence of hepatic lipoperoxidation. Di Luzio [39] questioned the above results and showed that the absorption of conjugated dienes can be detected in the mitochondrial but not in the microsomal lipids of ethanol-treated rats. On the other hand, Corongiu et al. [35] demonstrated the absorption of conjugated dienes in microsomal lipids of ethanol-treated animals by the second-derivative spectroscopy.
An approach to the problem with different technical procedures was then devised. Since the end result of lipoperoxidation is a decrease in the most highly unsaturated fatty acids, which are the major peroxidizable substrates, a decrease in their content in the lipids of isolated subcellular fractions could indicate, among other possibilities, that a peroxidative breakdown of these moieties had actually occurred in vivo. As a matter of fact, a progressive decrease in the arachidonic acid content of liver microsomal phospholipids was observed [33] shortly after carbon tetrachloride intoxication; it was also observed, in contrast with liver phospholipids, that hepatic triglycerides do not show any change in arachidonic acid content after poisoning, again suggesting that lipid peroxidation involves structural lipids rather than the lipids accumulating in the liver as a result of the intoxication. A clear decrease in the arachidonic and docosahexaenoic acid content of liver mitochondrial lipids from acutely ethanol-treated rats was actually found [34]. In contrast with the mitochondrial changes, ethanol did not induce a decrease in the most unsaturated components of the fatty acid pattern of liver microsomal phospholipids [34].
A decrease in arachidonic as well as an increase in linoleic acid content of liver mitochondrial lipids was also observed by French et al. [43] after chronic ethanol administration, but these changes were mainly attributed to alterations in the activity of the chain elongation desaturation system.
Implication of oxidative stress in ethanol toxicity would imply that either ethanol is converted, during its metabolism, to a free radical intermediate or that ethanol or its metabolites react with some nucleophile in an antioxidant molecule, thus blocking the molecule and decreasing the antioxidant potential. The latter possibility has been shown above (reaction of acetaldehyde with –SH groups of cysteine or GSH), but the loss of GSH is by far smaller than that occurring with many other GSH depletors (bromobenzene, allyl alcohol, etc. [28]) and cannot account by itself for the induction of lipid peroxidation. The former possibility—the formation of a free radical during ethanol metabolism—was postulated by Slater [113] since many years ago. Ethanol may enter free radical reaction relatively easily [111], through the interaction with some endogenous radical; the latter could give rise to a homolytic cleavage of ethanol yielding a reducing ethoxy radical (CH3–CH2O·), which in the presence of some oxidant would be converted to acetaldehyde:
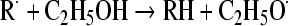 |
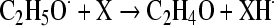 |
(from [113]).
Several endogenous radicals are known to be involved in the NADPH-cytochrome P450 chain; ethanol may interact at this site during its metabolism in MEOS. Also, in the scheme proposed for the action of catalase-free radical intermediates of the hydrogen donor are formed; if ethanol is the donor, free radical intermediates from ethanol can result.
More recent studies have conclusively shown that ethoxy radical is really generated during ethanol oxidation and that an oxidative stress is imposed on the liver cell as a result of ethanol metabolism [94]. Several sources of such an oxidative stress have been described. Ethanol oxidation results in the production of free radicals, which can derive from both oxygen and ethanol itself. Oxygen radicals can originate as follows: microsomal NADPH-cytochrome c reductase and cytochrome P450 (components of MEOS) can generate O·−2 and H2O2 [52, 53, 65, 93, 124]; the same oxygen species can be produced by aldehydes oxidase and xanthine oxidase [85], both involved in the metabolism of ethanol-derived acetaldehyde; O·−2 and H2O2 can also be generated by microsomal NADPH oxidase, which has been shown to be increased after acute [120] or chronic [70, 104, 119, 122] ethanol administration; during NADPH oxidation liver microsomes produce significant amount of OH· (being H2O2 the precursor), which in turn appears to be required for ethanol oxidation [20, 26, 55].
With regard to ethanol-derived radicals, it has been shown [4, 5] that ethanol is activated to a free radical intermediate by the ethanol inducible form of cytochrome P450, i.e., the specific isoenzymatic form involved in MEOS, CYP2E1. With the use of electron spin resonance (ESR) spectroscopy in combination with the spin trapping agent 4-pyridyl-1-oxo-t-butyl nitrone (4-POBN), it has been demonstrated [4, 5] that rat liver microsomes incubated with ethanol and NADPH can produce a free radical intermediate, identified as 1-hydroxyethyl radical. Free radical intermediates are also produced by liver microsomes during the metabolism of various aliphatic alcohols (1-propanol, 2-propanol, 1-butanol, 2-butanol and 1-pentanol), indicating the existence of a common activating pathway for these compounds [5, 7]. The formation of radical intermediates has been confirmed in the whole animal in vivo with the use of 4-POBN [8, 60, 102, 103]. The generation of ethanol radicals would occur during the process of univalent reduction of dioxygen and possibly would be carried out by ferric cytochrome P450 oxy-complex (P450–Fe3+O·−2) [10, 11] formed during the reduction of heme-oxygen. In such a state, cytochrome would be sufficiently reactive to abstract a proton from the 1-carbon of ethanol, yielding a carbon-centered radical and H2O2 [116]. Alternatively, hydroxyethyl radicals could be produced by addition to ethanol of OH· radicals generated by liver microsomes [81]. However, generated hydroxyethyl radicals bind to microsomal protein [9], particularly CYP2E1, and probably play an important role in the induction of lipid peroxidation [6, 42]. The binding of alcohol radical to protein represents another mechanism of hepatic protein alkylation in addition to that operated by acetaldehyde [41] and known to contribute to overall liver cell damage. Furthermore, the hydroxyethyl radical-derived protein adducts are immunogenic and give rise to antibodies different from those generated by acetaldehyde-derived protein adducts [88]. Chronic alcohol feeding of rats leads to the production of antibodies that recognize hydroxyethyl rat serum albumin but do not recognize rat serum albumin [88]. Moreover, sera of alcoholic cirrhotic patients contain IgG and IgA antibodies that recognize proteins modified by hydroxyethyl radicals [27]. Such antibodies may play an important role in the immunologic reactions triggered by ethanol and due to antibodies against liver cells found in the serum of patients with alcoholic liver injury [46].
Ethanol-induced oxidative stress as measured by F2-isoprostane determination
A great advance in the study of oxidative stress has been represented by the demonstration [89, 90, 106] of the formation of a series of prostaglandin F2-like compounds, named F2-isoprostanes, which originate in vitro and in vivo from the peroxidation of phospholipids bound arachidonic acid. Since F2-isoprostanes, which are initially formed in situ on phospholipids [91], are released into the blood compartment and since these prostanoids are much less reactive than other lipid peroxidation products such as lipoperoxides and aldehydes, they can be found more easily in plasma and urine. Elevated levels of F2-isoprostanes have been found in various human pathologies [105]. Therefore, we reconsidered the whole problem of ethanol-induced oxidative stress with this methodological approach, which is nowadays considered as the most sophisticated and reliable technique to evaluate oxidative stress, at least when the determinations are carried out by gas-chromatography mass spectrometry. Plasma F2-isoprostanes are increased in ethanol toxicity ([92, 86, 13, 96], Comporti et al. unpublished work).
Final considerations
Owing to the fact that this review is concerned with the mechanisms inducing oxidative stress upon ethanol exposure (particularly acute exposure), no attention has been paid to the development of alcoholic liver disease and its progression to liver fibrosis. Thus, no mention has been done of the impairment of endogenous antioxidant defences, accumulation of unfolded proteins and endoplasmic reticulum stress [84, 112], AMP-dependent protein Kinase (AMPK) and adiponectin-regulated hepatic lipid metabolism [107], translocation of gut derived endotoxins to portal circulation [98], activation of Kupffer cells and release of proinflammatory cytokines (TNFα in particular), role of immune reactions in alcoholic-induced inflammation and progression to liver fibrosis through activation of hepatic stellate cells.
Extensive information about the above topics can be found in several reviews [2, 3, 79].
Footnotes
Oxidative stress is generally considered as a disturbance in the pro-oxidant/antioxidant balance in favor of the former, leading to potential damage [47].
References
- 1.Aebi H, Koblet H, Wartburg JP. Ueber den mechanismus der biologischen methanoloxydation. Elv Physiol Pharmacol Acta. 1957;15:384–399. [PubMed] [Google Scholar]
- 2.Albano E. Alcohol, oxidative stress and free radical damage. Proc Nutr Soc. 2006;65:278–290. doi: 10.1079/PNS2006496. [DOI] [PubMed] [Google Scholar]
- 3.Albano E. New concepts in the pathogenesis of alcoholic liver disease. Expert Rev Gastroenterol Hepatol. 2008;2:749–759. doi: 10.1586/17474124.2.6.749. [DOI] [PubMed] [Google Scholar]
- 4.Albano E, Tomasi A, Gloria-Gatti L, Poli G, Vannini V, Dianzani MU. Free radical metabolism of alcohols by rat liver microsomes. Free Radic Res Commun. 1987;3:243–249. doi: 10.3109/10715768709069789. [DOI] [PubMed] [Google Scholar]
- 5.Albano E, Tomasi A, Gloria-Gatti L, Dianzani MU. Spin trapping of free radical species produced during the microsomal metabolism of ethanol. Chem Biol Interact. 1988;65:223–234. doi: 10.1016/0009-2797(88)90108-1. [DOI] [PubMed] [Google Scholar]
- 6.Albano E, Poli G, Tomasi A, Gloria-Gatti L, Dianzani MU. Study of the mechanisms responsible for ethanol-induced oxidative damage using isolated rat hepatocytes. In: Hayaishi O, Niki E, Kondo M, Yoshikawa T, editors. Medical biochemical and chemical aspects of free radicals. Amsterdam: Elsevier Biomedical; 1988. pp. 1389–1392. [Google Scholar]
- 7.Albano E, Tomasi A, Persson J, Terelius Y, Gloria-Gatti L, Ingelman-Sundberg M, Dianzani MU. Role of ethanol-inducible cytochrome P450 (P450IIE1) in catalyzing the free radical activation of aliphatic alcohols. Biochem Pharmacol. 1991;41:1895–1902. doi: 10.1016/0006-2952(91)90129-S. [DOI] [PubMed] [Google Scholar]
- 8.Albano E, Tomasi A, Parola M, Comoglio A, Ingelman-Sundberg M, Dianzani MU. Mechanisms responsible for free radical formation during ethanol metabolism and their role in causing oxidative injury by alcohol. In: Corongiu F, Banni S, Dessi MA, Rice Evans C, editors. Free radicals and antioxidants nutrition. London: Richelieu Press; 1993. pp. 77–96. [Google Scholar]
- 9.Albano E, Parola M, Comoglio A, Dianzani MU. Evidence for the covalent binding of hydroxyethyl radicals to rat microsomal proteins. Alcohol Alcohol. 1993;28:453–459. [PubMed] [Google Scholar]
- 10.Albano E, Tomasi A, Ingelman-Sundberg M. Spin trapping of alcohol-derived radicals in microsomes and reconstituted systems by electron spin resonance. Methods Enzymol. 1994;233:117–127. doi: 10.1016/S0076-6879(94)33014-X. [DOI] [PubMed] [Google Scholar]
- 11.Albano E, French SW, Ingelman-Sundberg M. Hydroxyethyl radicals in ethanol hepatotoxicity. Front Biosci. 1999;4:533–540. doi: 10.2741/Albano. [DOI] [PubMed] [Google Scholar]
- 12.Albot G, Parturier-Albot M, Nicolasen P. Histology, ultrastructure and histobiology of subacute alcoholic hepatitis and cirrhogenic alcoholic hepatitis. Rev Intern Hepatol. 1968;18:117–154. [PubMed] [Google Scholar]
- 13.Aleynik SI, Leo MA, Aleynik MK, Lieber CS. Increased circulating products of lipid peroxidation in patients with alcoholic liver disease. Alcohol Clin Exp Res. 1998;22:192–196. [PubMed] [Google Scholar]
- 14.Baudhuin P. Liver peroxisomes, cytology and function. Ann NY Acad Sci. 1969;168:214–228. doi: 10.1111/j.1749-6632.1969.tb43111.x. [DOI] [PubMed] [Google Scholar]
- 15.Best CH, Hartroft WS, Lucas CC, Ridout JH. Liver damage produced by feeding alcohol or sugar and its prevention by choline. Br Med J. 1949;11:1001–1006. doi: 10.1136/bmj.2.4635.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bieri JG, Anderson AA. Peroxidation of lipids in tissue homogenates as related to vitamin E. Arch Biochem Biophys. 1960;90:105–110. doi: 10.1016/0003-9861(60)90619-6. [DOI] [Google Scholar]
- 17.Bolland JL, Koch HP (1945) The course of autoxidation reactions in polyisoprenes and allied compounds. Part IX. The primary thermal oxidation product of ethyl linoleate. J Chem Soc 445–447
- 18.Butler TC. Reduction of carbon tetrachloride in vivo and reduction of carbon tetrachloride and chloroform in vitro by tissues and tissue constituents. J Pharmacol Exp Ther. 1961;134:311–319. [PubMed] [Google Scholar]
- 19.Carter EA, Isselbacher KJ. The role of microsomes in the hepatic metabolism of ethanol. Ann NY Acad Sci. 1971;179:282–294. doi: 10.1111/j.1749-6632.1971.tb46907.x. [DOI] [PubMed] [Google Scholar]
- 20.Cederbaum AI, Dicker E. Inhibition of microsomal oxidation of alcohols and of hydroxyl-radical-scavenging by the iron-chelating agent desferrioxamine. Biochem J. 1983;210:107–113. doi: 10.1042/bj2100107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cederbaum AI, Rubin E. Molecular injury to mitochondria produced by ethanol and acetaldehyde. Fed Proc. 1975;34:2045–2051. [PubMed] [Google Scholar]
- 22.Cederbaum AI, Lieber CS, Rubin E. Effects of chronic ethanol treatment on mitochondrial functions. Arch Biochem Biophys. 1974;165:560–569. doi: 10.1016/0003-9861(74)90283-5. [DOI] [PubMed] [Google Scholar]
- 23.Cederbaum AI, Lieber CS, Rubin E. The effects of acetaldehyde on mitochondrial function. Arch Biochem Biophys. 1974;161:26–39. doi: 10.1016/0003-9861(74)90231-8. [DOI] [PubMed] [Google Scholar]
- 24.Cederbaum AI, Lieber CS, Rubin E. Effect of acetaldehyde on fatty acid oxidation and ketogenesis by hepatic mitochondria. Arch Biochem Biophys. 1975;169:29–41. doi: 10.1016/0003-9861(75)90313-6. [DOI] [PubMed] [Google Scholar]
- 25.Cederbaum AI, Lieber CS, Rubin E. Effect of chronic ethanol consumption and acetaldehyde on partial reactions of oxidative phosphorylation and CO2 production from citric acid cycle intermediates. Arch Biochem Biophys. 1976;176:525–538. doi: 10.1016/0003-9861(76)90196-X. [DOI] [PubMed] [Google Scholar]
- 26.Cederbaum AI, Dicker E, Cohen G. Effect of hydroxyl radical scavengers on microsomal oxidation of alcohols and associated microsomal reactions. Biochemistry. 1978;17:3058–3064. doi: 10.1021/bi00608a018. [DOI] [PubMed] [Google Scholar]
- 27.Clot P, Bellomo G, Tebone M, Aricòt S, Albano E. Detection of antibodies against proteins modified by hydroxyethyl free radicals in patients with alcoholic cirrhosis. Gastroenterology. 1995;108:201–207. doi: 10.1016/0016-5085(95)90025-X. [DOI] [PubMed] [Google Scholar]
- 28.Comporti M. Glutathione depleting agents and lipid peroxidation. Chem Phys Lipids. 1987;45:143–169. doi: 10.1016/0009-3084(87)90064-8. [DOI] [PubMed] [Google Scholar]
- 29.Comporti M, Saccocci C. Stimulation of the lipid peroxidation of rat liver homogenates by low concentration of CC1-4 in vitro. Boll Soc Ital Biol Sper. 1965;41:1066–1069. [PubMed] [Google Scholar]
- 30.Comporti M, Di Luzio NR. Effect of ethanol administration on the peroxidation of liver lipids. Physiologist. 1966;9:157. [Google Scholar]
- 31.Comporti M, Saccocci C, Dianzani MU. Effect on CCl4 in vitro and in vivo on lipid peroxidation of rat liver homogenates and subcellular fractions. Enzymologia. 1965;29:185–204. [PubMed] [Google Scholar]
- 32.Comporti M, Hartman A, Di Luzio NR. Effect of in vivo and in vitro ethanol administration on liver lipid peroxidation. Lab Invest. 1967;16:616–624. [PubMed] [Google Scholar]
- 33.Comporti M, Burdino E, Ugazio G. Changes in fatty acid pattern of liver microsomal phospholipids in rats treated with carbon tetrachloride. Ital J Biochem. 1971;20:156–165. [PubMed] [Google Scholar]
- 34.Comporti M, Burdino E, Raja F. Fatty acid composition of mitochondrial and microsomal lipids of rat liver after acute ethanol intoxication. Life Sci. 1971;10:855–866. doi: 10.1016/0024-3205(71)90198-6. [DOI] [PubMed] [Google Scholar]
- 35.Corongiu FP, Lai M, Milia A. Carbon tetrachloride, bromotrichloromethane and ethanol acute intoxication. Biochem J. 1983;212:625–631. doi: 10.1042/bj2120625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dianzani MU, Baccino FM, Comporti M. The direct effect of carbon tetrachloride on subcellular particles. Lab Invest. 1966;15:149–156. [PubMed] [Google Scholar]
- 37.Di Luzio NR. Prevention of the acute ethanol-induced fatty liver by the simultaneous administration of antioxidants. Life Sci. 1964;3:113–118. doi: 10.1016/0024-3205(64)90189-4. [DOI] [PubMed] [Google Scholar]
- 38.Di Luzio NR. A mechanism of the acute ethanol-induced fatty liver and modification of liver injury by antioxidants. Lab Invest. 1966;15:50–63. [PubMed] [Google Scholar]
- 39.Di Luzio NR. Role of lipid peroxidation and antioxidants in ethanol-induced lipid alterations. Exp Mol Pathol. 1968;8:394–402. doi: 10.1016/S0014-4800(68)80008-5. [DOI] [PubMed] [Google Scholar]
- 40.Di Luzio NR, Poggi M. Abnormal lipid tolerance and hyperlipemia in acute ethanol-treated rats. Life Sci. 1963;2:751–758. doi: 10.1016/0024-3205(63)90082-1. [DOI] [PubMed] [Google Scholar]
- 41.Donohur TM, Tuma DJ, Sorrell MF. Acetaldehyde adducts with proteins: bindings of 14C-acetaldehyde to serum albumin. Arch Biochem Biophys. 1983;220:239–246. doi: 10.1016/0003-9861(83)90406-X. [DOI] [PubMed] [Google Scholar]
- 42.Ekström G, Ingelman-Sundberg M. Rat liver microsomal NADPH-supported oxidase activity and lipid peroxidation dependent or ethanol-inducible cytochrome P450 (P450 II E1) Biochem Pharmacol. 1989;38:1313–1319. doi: 10.1016/0006-2952(89)90338-9. [DOI] [PubMed] [Google Scholar]
- 43.French SW, Ihrig TJ, Morin RJ. Lipid composition of RBC ghosts, liver mitochondria and microsomes of ethanol-fed rats. Q J Stud Alcohol. 1970;31:801–809. [PubMed] [Google Scholar]
- 44.Gallagher CH. The effect of antioxidants on poisoning by carbon tetrachloride. Austral J Exp Biol Med Sci. 1962;40:241–254. doi: 10.1038/icb.1962.28. [DOI] [PubMed] [Google Scholar]
- 45.Gordon ER. Mitochondrial functions in an ethanol-induced fatty liver. J Biol Chem. 1973;248:8271–8280. [PubMed] [Google Scholar]
- 46.Grossley IR, Neuberger J, Davis M, Williams R, Eddleston ALWF. Ethanol metabolism in generation of new antigenic determinations on liver cells. Gut. 1986;27:186–189. doi: 10.1136/gut.27.2.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Halliwell B, Gutteridge JMC. Free radicals in biology and medicine. 3. Oxford: Oxford Science Publication; 1999. [Google Scholar]
- 48.Hartman A, Comporti M, Di Luzio NR. Peroxidation of lipids in the pathogenesis of acute ethanol induced liver injury. Gastroenterology. 1967;52:316. [Google Scholar]
- 49.Hartroft WS, Porta EA. Alcohol, diet, and experimental hepatic injury. Can J Physiol Pharmacol. 1968;46:463–473. doi: 10.1139/y68-069. [DOI] [PubMed] [Google Scholar]
- 50.Hashimoto S, Recknagel RO. No chemical evidence of hepatic lipid peroxidation in acute ethanol toxicity. Exp Mol Pathol. 1968;8:225–242. doi: 10.1016/0014-4800(68)90018-X. [DOI] [PubMed] [Google Scholar]
- 51.Hasumura Y, Teschke R, Lieber CS. Acetaldehyde oxidation by hepatic mitochondria: decrease after chronic ethanol consumption. Science. 1975;198:727–729. doi: 10.1126/science.168641. [DOI] [PubMed] [Google Scholar]
- 52.Hildebrandt AG, Roots I. Reduced nicotinamide adenine dinucleotide phosphate (NADPH)-dependent formation and breakdown of hydrogen peroxide during mixed function oxidation reactions in liver microsomes. Arch Biochem Biophys. 1975;171:385–397. doi: 10.1016/0003-9861(75)90047-8. [DOI] [PubMed] [Google Scholar]
- 53.Hildebrandt AG, Speck M, Roots I. Possible control of hydrogen peroxide production and degradation in microsomes during mixed function oxidase reaction. Biochem Biophys Res Commun. 1973;54:968–975. doi: 10.1016/0006-291X(73)90789-4. [DOI] [PubMed] [Google Scholar]
- 54.Hove EL. Interrelation between alpha-tocopherol and protein metabolism; the protective effect of vitamin E and certain nitrogenous compounds against CCl4 poisoning in rats. Arch Biochem. 1948;17:467–473. [PubMed] [Google Scholar]
- 55.Ingelman-Sundberg M, Johanson I. Mechanisms of radical formation and ethanol oxidation by ethanol inducible and other forms of rabbit liver microsomal cytochromes P450. J Biol Chem. 1984;249:6447–6458. [PubMed] [Google Scholar]
- 56.Iseri OA, Lieber CS, Gottlieb LS. The ultrastructure of fatty liver induced by prolonged ethanol ingestion. Am J Pathol. 1966;48:535–555. [PMC free article] [PubMed] [Google Scholar]
- 57.Isselbacher KJ, Carter EA. Ethanol oxidation by liver microsomes: evidence against a separate and distinct enzyme system. Biochem Biophys Res Commun. 1970;39:530–535. doi: 10.1016/0006-291X(70)90610-8. [DOI] [PubMed] [Google Scholar]
- 58.Kalish GH, Di Luzio NR. Peroxidation of liver lipids in the pathogenesis of the ethanol-induced fatty liver. Science. 1966;152:1390–1392. doi: 10.1126/science.152.3727.1390-a. [DOI] [PubMed] [Google Scholar]
- 59.Kiessling KH, Pilström L. Effect of ethanol on rat liver. V. Morphological and functional changes after prolonged consumption of various alcoholic beverages. Q J Stud Alcohol. 1968;29:819–827. [PubMed] [Google Scholar]
- 60.Knecht KT, Bradfort BU, Mason RP, Thurman GR. In vivo formation of radical metabolite of ethanol. Mol Pharmacol. 1990;38:26–30. [PubMed] [Google Scholar]
- 61.Koch OR, Porta EA, Hartroft WS. A new experimental approach in the study of chronic alcoholism. III. Role of alcohol versus sucrose or fat derived calories in hepatic damage. Lab Invest. 1968;18:379–386. [PubMed] [Google Scholar]
- 62.Koch OR, Boveris A, Sirotzky de Favelukes S, Schwarck de Tarlowsky M, Stoppani AOM. Biochemical lesions of liver mitochondrial from rats after chronic alcohol consumption. Exp Mol Pathol. 1977;27:213–220. doi: 10.1016/0014-4800(77)90031-4. [DOI] [PubMed] [Google Scholar]
- 63.Koch OR, Roatta De Conti L, Pacheco Bolaños L, Stoppani AOM. Ultrastructural and biochemical aspects of liver mitochondria during recovery from ethanol-induced alterations. Am J Pathol. 1978;90:325–344. [PMC free article] [PubMed] [Google Scholar]
- 64.Krebs HA, Perkins JR. The physiological role of liver alcohol dehydrogenase. Biochem J. 1970;118:635–644. doi: 10.1042/bj1180635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lai CS, Grover TA, Piette LH. Hydroxyl radical production in a purified NADPH-cytochrome c (P450 reductase system) Arch Biochem Biophys. 1979;193:373–378. doi: 10.1016/0003-9861(79)90042-0. [DOI] [PubMed] [Google Scholar]
- 66.Lane BP, Lieber CS. Ultrastructural alterations in human hepatocytes following ingestion of ethanol with adequate diets. Am J Pathol. 1966;49:593–603. [PMC free article] [PubMed] [Google Scholar]
- 67.Lester D, Keokosky WZ, Felzenberg F. Effect of pyrazoles and other compounds on alcohol metabolism. Q J Stud Alcohol. 1968;29:449–454. [PubMed] [Google Scholar]
- 68.Lieber CS, Davidson CS. Some metabolic effects of ethyl alcohol. Am J Med. 1962;33:319–327. doi: 10.1016/0002-9343(62)90229-2. [DOI] [PubMed] [Google Scholar]
- 69.Lieber CS, Carli LM. Ethanol oxidation by hepatic microsomes: adaptive increase after ethanol feeding. Science. 1968;162:917–918. doi: 10.1126/science.162.3856.917. [DOI] [PubMed] [Google Scholar]
- 70.Lieber CS, Carli LM. Reduced nicotinamide-adenine dinucleotide phosphate oxidase: activity enhanced by ethanol consumption. Science. 1970;170:78–80. doi: 10.1126/science.170.3953.78. [DOI] [PubMed] [Google Scholar]
- 71.Lieber CS, Carli LM. The role of the hepatic microsomal ethanol oxidizing system (MEOS) for ethanol metabolism in vivo. J Pharmacol Exp Ther. 1972;181:278–287. [PubMed] [Google Scholar]
- 72.Lieber CS, Carli LM. An experimental model of alcohol feeding and liver injury in the baboon. J Med Primatol. 1974;3:153–163. doi: 10.1159/000459999. [DOI] [PubMed] [Google Scholar]
- 73.Lieber CS, Rubin E. Alcoholic fatty liver in man on a high protein and low fat diet. Am J Med. 1968;44:200–206. doi: 10.1016/0002-9343(68)90151-4. [DOI] [PubMed] [Google Scholar]
- 74.Lieber CS, Schmid R. The effect of ethanol on fatty acid metabolism: stimulation of hepatic fatty acid synthesis in vitro. J Clin Invest. 1961;40:394–399. doi: 10.1172/JCI104266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lieber CS, Carli LM, Schmid R. Effect of ethanol on fatty acid metabolism in liver slices. Biochem Biophys Res Commun. 1959;1:302–306. doi: 10.1016/0006-291X(59)90043-9. [DOI] [Google Scholar]
- 76.Lieber CS, Jones DP, Carli LM. Effects of prolonged ethanol intake: production of fatty liver despite adequate diets. J Clin Invest. 1965;44:1009–1021. doi: 10.1172/JCI105200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lieber CS, Lefévre A, Spritz N, Feinman L, Carli LM. Difference in hepatic metabolism of long- and medium-chain fatty acids: the role of fatty acid chain length in the production of the alcoholic fatty liver. J Clin Invest. 1967;46:1451–1460. doi: 10.1172/JCI105637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lieber CS, Teschke R, Hasumura Y, Carli LM. Difference in hepatic and metabolic changes after acute and chronic alcohol consumption. Fed Proc. 1975;34:2060–2074. [PubMed] [Google Scholar]
- 79.Lu Y, Cederbaum AL. CYP2E1 and oxidative liver injury by alcohol. Free Radic Biol Med. 2008;44:723–738. doi: 10.1016/j.freeradbiomed.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lundquist F, Tygstrup N, Winkler K, Mellemgaard K, Munck-Petersen S. Ethanol metabolism and production of free acetate in the human liver. J Clin Invest. 1962;41:955–961. doi: 10.1172/JCI104574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Malhotra JD, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress: a vicious circle or a double-edged sword? Antioxid Redox Signal. 2007;9:2277–2293. doi: 10.1089/ars.2007.1782. [DOI] [PubMed] [Google Scholar]
- 85.Massey V, Strickland S, Mayhew SG, Hawell LG, Engel PC, Matthews RG, Schuman M, Sullivan PA. The production of superoxide anion radicals in the reaction of reduced flavins and flavoproteins with molecular oxygen. Biochem Biophys Res Commun. 1969;36:891–897. doi: 10.1016/0006-291X(69)90287-3. [DOI] [PubMed] [Google Scholar]
- 81.McCay PB, Reinke LA, Rau JM. Hydroxyl radicals are generated by hepatic microsomes during NADPH oxidation relationship to ethanol metabolism. Free Radic Res Commun. 1992;15:335–346. doi: 10.3109/10715769209049149. [DOI] [PubMed] [Google Scholar]
- 82.McManus IR, Contag AO, Olson RE. Characterization of endogenous ethanol in the mammal. Science. 1960;131:102–103. doi: 10.1126/science.131.3393.102. [DOI] [PubMed] [Google Scholar]
- 83.McManus IR, Contag AO, Olson RE. Studies on the identification and origin of ethanol in mammalian tissues. J Biol Chem. 1966;241:349–356. [PubMed] [Google Scholar]
- 86.Meagher EA, Barry OP, Burke A, Lucey MR, Lawson JA, Rokach J, FitzGerald GA. Alcohol-induced generation of lipid peroxidation products in humans. J Clin Invest. 1999;104:805–813. doi: 10.1172/JCI5584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mezey E, Potter JJ, Reed WD. Ethanol oxidation by a component of liver microsomes rich in cytochrome P450. J Biol Chem. 1973;248:1183–1187. [PubMed] [Google Scholar]
- 88.Moncada C, Torres V, Varghese G, Albano E, Israel Y. Ethanol-derived immunoreactive species formed by free radical mechanisms. Mol Pharmacol. 1994;46:786–791. [PubMed] [Google Scholar]
- 89.Morrow JD, Harris TM, Roberts LJ. Noncyclooxygenase oxidative formation of a series of novel prostaglandins: analytical ramifications for measurement of eicosanoids. Anal Biochem. 1990;184:1–10. doi: 10.1016/0003-2697(90)90002-Q. [DOI] [PubMed] [Google Scholar]
- 90.Morrow JD, Hill KE, Burk RF, Nammour TM, Badr KF, Roberts LJ. A series of prostaglandin F2-like compounds are produced in vivo in humans by a non-cyclooxygenase, free radical-catalyzed mechanism. Proc Natl Acad Sci USA. 1990;87:9383–9387. doi: 10.1073/pnas.87.23.9383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Morrow JD, Awad JA, Boss HJ, Blair IA, Roberts LJ. Non-cyclooxygenase derived prostanoids (F2-isoprostanes) are formed in situ on phospholipids. Proc Natl Acad Sci USA. 1992;89:10721–10725. doi: 10.1073/pnas.89.22.10721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nanji AA, Khwaja S, Tahan SR, Sadrzadeh SM. Plasma levels of a novel noncyclooxygenase-derived prostanoid (8-isoprostane) correlate with severity of liver injury in experimental alcoholic liver disease. J Pharmacol Exp Ther. 1994;269:1280–1285. [PubMed] [Google Scholar]
- 93.Nordblom GD, Coon MJ. Hydrogen peroxide formation and stoichiometry of hydroxylation reactions catalyzed by highly purified liver microsomal cytochrome P450. Arch Biochem Biophys. 1977;180:343–347. doi: 10.1016/0003-9861(77)90047-9. [DOI] [PubMed] [Google Scholar]
- 94.Nordmann R, Ribière C, Rouach H. Implication of free radical mechanisms in ethanol-induced cellular injury. Free Radic Biol Med. 1992;12:219–240. doi: 10.1016/0891-5849(92)90030-K. [DOI] [PubMed] [Google Scholar]
- 95.Ontko JA. Effects of ethanol on the metabolism of free fatty acids in isolated liver cells. J Lipid Res. 1973;14:78–86. [PubMed] [Google Scholar]
- 96.Pemberton PW, Smith A, Warnes TW. Non-invasive monitoring of oxidant stress in alcoholic liver disease. Scand J Gastroenterol. 2005;40:1102–1108. doi: 10.1080/00365520510023495. [DOI] [PubMed] [Google Scholar]
- 97.Porta EA, Bergman BJ, Stein AA. Acute alcoholic hepatitis. Am J Pathol. 1965;46:657–689. [PMC free article] [PubMed] [Google Scholar]
- 98.Rao RK, Serth A, Sheth P. Recent advances in alcoholic liver disease I. Role of intestinal permeability and endotoxemia in alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol. 2004;286:G881–G884. doi: 10.1152/ajpgi.00006.2004. [DOI] [PubMed] [Google Scholar]
- 99.Reboucas G, Isselbacher KJ. Studies on the pathogenesis of the ethanol-induced fatty liver. I. Synthesis and oxidation of fatty acids by the liver. J Clin Invest. 1961;40:1355–1362. doi: 10.1172/JCI104366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Recknagel RO, Ghoshal AK. Quantitative estimation of peroxidative degeneration of rat liver microsomal and mitochondrial lipids after carbon tetrachloride poisoning. Exp Mol Pathol. 1966;5:413–426. doi: 10.1016/0014-4800(66)90023-2. [DOI] [PubMed] [Google Scholar]
- 101.Recknagel RO, Ghoshal AK. Lipoperoxidation as a vector in carbon tetrachloride hepatotoxicity. Lab Invest. 1966;15:132–148. [PubMed] [Google Scholar]
- 102.Reinke LA, Lai EK, Du Bose CM, Mc Cay PB. Reactive free radical generation in vivo in heart and liver of ethanol-fed rats: correlation with radical formation in vitro. Proc Natl Acad Sci USA. 1987;84:9223–9227. doi: 10.1073/pnas.84.24.9223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Reinke LA, Kotake Y, McCay PB, Janzen EG. Spin-trapping studies of hepatic free radicals formed following the acute administration of ethanol to rats: in vivo detection of 1-hydroxyethyl radicals with PBN. Free Radic Biol Med. 1991;11:31–39. doi: 10.1016/0891-5849(91)90185-6. [DOI] [PubMed] [Google Scholar]
- 104.Reitz RC. A possible mechanism for the peroxidation of lipids due to chronic ethanol ingestion. Biochem Biophys Acta. 1975;380:145–154. doi: 10.1016/0005-2760(75)90001-6. [DOI] [PubMed] [Google Scholar]
- 105.Roberts LJ, II, Morrow JD. Measurement of isoprostanes to assess oxidant stress status in vivo. In: Yoshikawa T, Toyokuni S, Yamamoto Y, editors. Free radicals in chemistry, biology and medicine. TN: Nashville; 2000. pp. 329–340. [Google Scholar]
- 106.Roberts LJ, II, Morrow JD. Products of the isoprostane pathway: unique bioactive compounds and markers of lipid peroxidation. Cell Mol Life Sci. 2002;59:808–820. doi: 10.1007/s00018-002-8469-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rogers CQ, Ajmo JM, You M. Adiponectin and alcoholic fatty liver disease. IUBMB Life. 2008;60:790–797. doi: 10.1002/iub.124. [DOI] [PubMed] [Google Scholar]
- 108.Rubin E, Lieber CS. Hepatic microsomal enzymes in man and rat: induction and inhibition by ethanol. Science. 1968;162:690–691. doi: 10.1126/science.162.3854.690. [DOI] [PubMed] [Google Scholar]
- 109.Rubin E, Hutterer F, Lieber CS. Ethanol increases hepatic smooth endoplasmic reticulum and drug-metabolizing enzymes. Science. 1968;159:1469–1470. doi: 10.1126/science.159.3822.1469. [DOI] [PubMed] [Google Scholar]
- 110.Rubin E, Beattie DS, Lieber CS. Effects of ethanol on the biogenesis of mitochondrial membranes and associated mitochondrial functions. Lab Invest. 1970;23:620–627. [PubMed] [Google Scholar]
- 111.Schönberg A. Preparative organic photochemistry. Berlin: Springer; 1968. [Google Scholar]
- 112.Schröder M. Endoplasmic reticulum stress responses. Cell Mol Life Sci. 2008;65:862–894. doi: 10.1007/s00018-007-7383-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Slater TF. Free radical mechanisms in tissue injury. London: Pion Limited; 1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Takada A, Ikegami F, Okamura Y, Asumura Y, Kanayama R, Takeuchi J. Effect of alcohol on the liver of rats. III. The role of lipid peroxidation and sulphydryl compounds in ethanol-induced liver injury. Lab Invest. 1970;23:421–428. [PubMed] [Google Scholar]
- 115.Tephly TR, Tinelli F, Watkins WD. Alcohol metabolism: role of microsomal oxidation in vivo. Science. 1969;166:627–628. doi: 10.1126/science.166.3905.627. [DOI] [PubMed] [Google Scholar]
- 116.Terelius Y, Lindros KO, Albano E, Ingelman-Sundberg M. Isozyme-specificity of cytochrome P450-mediated hepatotoxicity. In: Ruckpaul K, Rein H, editors. Frontiers in biotransformation. Berlin: Akademie Verlag; 1993. pp. 186–232. [Google Scholar]
- 117.Teschke R, Hasumura Y, Joly J-G, Ishii H, Lieber CS. Microsomal ethanol-oxidizing system (MEOS): purification and properties of a rat liver system free of catalase and alcohol dehydrogenase. Biochem Biophys Res Commun. 1972;49:1187–1193. doi: 10.1016/0006-291X(72)90594-3. [DOI] [PubMed] [Google Scholar]
- 118.Thompson GN. Alcoholism. Springfield: Thomas C.C. Ed; 1956. [Google Scholar]
- 119.Thurman RG. Induction of hepatic microsomal reduced nicotinamide adenine dinucleotide phosphate-dependent production of hydrogen peroxide by chronic prior treatment with ethanol. Mol Farmacol. 1973;9:670–675. [PubMed] [Google Scholar]
- 120.Valenzuela A, Fernandez N, Fernandez V, Ugarte G, Videla LA, Guerra R, Villanueva A. Effect of acute ethanol ingestion on lipoperoxidation and on the activity of the enzymes related to peroxide metabolism in rat liver. FASEB Lett. 1980;111:11–13. doi: 10.1016/0014-5793(80)80750-2. [DOI] [PubMed] [Google Scholar]
- 121.Vallee BL. Metal and enzyme interactions: correlation of composition, function and structure. In: Boyer PD, Lardy H, Myrbäck K, editors. The enzymes. New York: Academic Press; 1960. pp. 225–276. [Google Scholar]
- 122.Videla L, Bernstein J, Isdrael I. Metabolic alterations produced in the liver by chronic ethanol administration: increased oxidative capacity. Biochem J. 1973;134:507–514. doi: 10.1042/bj1340507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.White RE, Coon MJ. Oxygen activation by cytochrome P450. Annu Rev Biochem. 1980;49:325–356. doi: 10.1146/annurev.bi.49.070180.001531. [DOI] [PubMed] [Google Scholar]
- 125.Wirtschafter ZT, Cronyn MW. Free radical mechanism for solvent toxicity. Arch Environ Health. 1964;9:186–191. doi: 10.1080/00039896.1964.10663818. [DOI] [PubMed] [Google Scholar]
- 126.Wooles WR. Depressed hepatic fatty acid oxidation as a factor in the etiology of acute ethanol-induced fatty liver. Life Sci. 1966;5:267–276. doi: 10.1016/0024-3205(66)90147-0. [DOI] [Google Scholar]
- 123.You M, Crabb DW. Recent advances in alcoholic liver disease. II. Minireview: molecular mechanisms of alcoholic fatty liver. Am J Physiol Gastrointest Liver Physiol. 2005;287:G4–G63. doi: 10.1152/ajpgi.00056.2004. [DOI] [PubMed] [Google Scholar]