

Abstract
The recently identified small molecule, 3-amino-5-chloro-6-methoxy-4-methylthieno[2,3-b]pyridine-2-carboxylic acid cyclopropylamide (LY2033298), is the first selective allosteric modulator of the muscarinic acetylcholine receptors (mAChRs) that mediates both receptor activation and positive modulation of the endogenous agonist, acetylcholine (ACh), via the same allosteric site on the M4 mAChR. We thus utilized this novel chemical tool, as well as ACh, the bitopic (orthosteric/allosteric) agonist, McN-A-343, and the clinically efficacious M1/M4 mAChR-preferring agonist, xanomeline, in conjunction with site-directed mutagenesis of four different regions of the M4 mAChR (extracellular loops 1, 2, and 3, and transmembrane domain 7), to identify regions that govern ligand-specific modes of binding, signaling, and allosteric modulation. In the first extracellular loop (E1), we identified Ile93 and Lys95 as key residues that specifically govern the signaling efficacy of LY2033298 and its binding cooperativity with ACh, whereas Phe186 in the E2 loop was identified as a key contributor to the binding affinity of the modulator for the allosteric site, and Asp432 in the E3 loop appears to be involved in the functional (activation) cooperativity between the modulator and the endogenous agonist. In contrast, the highly conserved transmembrane domain 7 residues, Tyr439 and Tyr443, were identified as contributing to a key activation switch utilized by all classes of agonists. These results provide new insights into the existence of multiple activation switches in G protein-coupled receptors (GPCRs), some of which can be selectively exploited by allosteric agonists, whereas others represent global activation mechanisms for all classes of ligand.
Keywords: Allosteric Regulation, Drug Action, G Protein-coupled Receptors (GPCR), Receptor Structure-Function, Receptors, Acetylcholine, Muscarinic Receptor
Introduction
The GPCR3 superfamily comprises nearly 1000 7-transmembrane (TM)-spanning proteins that represent the largest target class for all current drugs (1). The mAChRs are prototypical family A GPCRs that mediate, along with the ionotropic nicotinic receptors, the actions of the neurotransmitter, ACh. As with all biogenic amines, the orthosteric binding pocket for the endogenous agonist is located within the core of the helical TM bundle of these receptors. For the mAChRs, major contact points conserved across all five receptor subtypes include Asp3.32, Tyr3.33, Tyr6.51, Asn6.52, Tyr7.39, Cys7.42, and Tyr7.43 (Ballesteros-Weinstein numbering) (2), whereas Leu3.29, Asn3.37, Thr5.39, Thr5.42, and Ala5.46 constitute “second shell” residues that stabilize the primary binding pocket (3). Understanding the activation mechanism of GPCRs is an area of intensive current research. The binding event for ACh is intimately linked to the subsequent conformational changes that result in transmission of a stimulus to the cell. Key initial events in this process include the formation of an ionic bond between the positively charged ammonium head group of ACh and the negatively charged Asp3.32 and an enclosure of the headgroup in an aromatic cage composed of Tyr3.33, Tyr6.51, Tyr7.39, and Tyr7.43 (3–5). Tightening of this aromatic cage around ACh, as well as the formation of new intramolecular contacts, triggers both a rotational and vertical movement (widening of the intracellular face of the receptor) of TM6 and TM7 relative to TM3 to ultimately transduce ACh binding into receptor activation (3, 5).
Because the mechanism of orthosteric agonist binding and activation is highly conserved across all mAChR subtypes, the development of subtype-selective mAChR activators that target this orthosteric region remains a significant challenge. However, there has been exciting recent progress toward the development of allosteric ligands that bind to a site distinct from the orthosteric site on mAChRs (6, 7). A number of prototypical allosteric ligands of the mAChRs have now been identified that bind to the receptor simultaneously with orthosteric ligands and modulate the binding and/or signaling of the latter agent (8). More recently, allosteric GPCR ligands have been reported that also appear to activate receptors in their own right, in the absence of orthosteric ligand (9). One such ligand is the novel small molecule, LY2033298 (Fig. 1E), which is a selective allosteric potentiator of ACh binding and function at the M4 mAChR subtype but is also capable of activating multiple signaling pathways linked to this receptor (10–12). The discovery of this novel allosteric modulator/agonist represents a significant advance in mAChR biology for two reasons. First, selective activation of the M4 mAChR may be a novel approach for improving cognitive symptoms associated with disorders such as schizophrenia (13). Second, LY2033298 is the first mAChR agonist whose activity has been unambiguously shown to arise purely from an allosteric binding site (12).
FIGURE 1.

Mutations and ligands investigated in the current study. A, snake diagram of the M4 mAChR highlighting mutated residues. B–E, structures of the endogenous agonist, ACh (B), the M1/M4-preferring agonist, xanomeline (C), the functionally selective bitopic agonist, McN-A-343 (D), and the allosteric ligand, LY2033298 (E).
In contrast to the relatively well defined binding and activation mechanism of orthosteric agonists such as ACh, the molecular mechanisms underlying action of allosteric agonists are largely unknown. This is in part due to the fact that most structure-function studies of putative allosteric agonists do not differentiate between effects of GPCR mutations on the affinity of the modulator for the allosteric site on the unoccupied receptor, the cooperativity between the modulator and the orthosteric ligand on the occupied receptor, and any direct signaling effects of the modulator; all of these can contribute to changes in observed allosteric agonist potency. Previous studies have found that the binding pocket for prototypical allosteric modulators of the mAChRs is located more extracellular relative to the orthosteric pocket and likely involves regions in the extracellular loops and the top of the TM helices (14–16). Preliminary mutagenesis studies suggest that such regions may also be important for the actions of LY2033298. For instance, the triple substitution of Thr-Val-Ile84–86(2.65–2.67)4 at the junction of TM2 and the E1 loop from the M2 mAChR in place of the equivalent residues (Ile-Ile-Lys93–95) of the M4 mAChR increased the ability of LY2033298 to potentiate the function of ACh in a calcium mobilization assay, whereas substitution of Ser428 and Asp432(7.32) in the E3 loop with the equivalent residues found in the M2 mAChR decreased the ability of LY2092298 to potentiate ACh function (10). However, this initial study did not identify what molecular components of the actions of LY2033298 were affected by the mutations.
The current study thus aimed to resolve key questions surrounding the structural basis of binding, cooperativity, and efficacy of LY2033298 as a modulator and agonist of the M4 mAChR. Specifically, we focused on four key regions of the receptor, namely, the E1–E3 loops, as well as a key activation switch in TM7 that has been implicated in the efficacy of orthosteric agonists (4, 17, 18). As comparator ligands (Fig. 1, B–D), we have included the endogenous orthosteric agonist, ACh, an M1/M4-prefering agonist (xanomeline) that has shown clinical efficacy in the treatment of cognitive episodes associated with schizophrenia, and the functionally selective agonist, McN-A-343, which was recently shown to be a bitopic orthosteric/allosteric ligand at the M2 mAChR (19). We present new evidence for differential effects on allosteric agonist affinity, cooperativity, and signaling efficacy depending on the region of the receptor involved. We also identify activation mechanisms that are likely to be common to orthosteric and allosteric agonists, as well as residues involved in ligand-specific receptor activation.
EXPERIMENTAL PROCEDURES
Materials
Chinese hamster ovary-FlpIn cells were from Invitrogen, hygromycin B was purchased from Roche Applied Science (Indianapolis, IN). Dulbecco's modified Eagle's medium and fetal bovine serum (FBS) were from Invitrogen and JRH Biosciences (Lenexa, KS), respectively. Primers used for the generation of mutant receptors were purchased from Sigma and Geneworks (see supplemental Table 1). The AlphaScreen SureFire phospho-ERK1/2 reagents were kindly donated by Drs. Michael Crouch and Ron Osmond (TGR Biosciences, South Australia), whereas the AlphaScreen streptavidin donor beads and anti-IgG (protein A) acceptor beads used for phosphorylated ERK1/2 (pERK1/2) detection, [3H]quinuclidinyl benzilate ([3H]QNB; specific activity, 52 Ci/mmol) and [3H]N-methylscopolamine ([3H]NMS; specific activity, 72 Ci/mmol) were purchased from PerkinElmer Life Sciences. Xanomeline and LY2033298 were synthesized in-house at Eli Lilly (Indianapolis, IN). All other chemicals were from Sigma.
Receptor Mutagenesis and Generation of Cell Lines
The desired M4 mAChR mutations were introduced into a triple hemagglutinin-tagged human M4 mAChR in pEf5/frt/v5-dest (Invitrogen) using the Stratagene QuikChange site-directed mutagenesis kit. Purified DNA was stably transfected into a Chinese hamster ovary-FlpIn cell line, selected using 0.4 mg/ml hygromycin and maintained at 0.2 mg/ml hygromycin in media (Dulbecco's modified Eagle's medium supplemented with 10% FBS and 16 mm HEPES). Cells were harvested using 2 mm EDTA in phosphate-buffered saline (137 mm NaCl, 2.7 mm KCl, 4.3 mm Na2HPO4, 1.5 mm KH2PO4) for all experiments.
[3H]QNB Binding Assays
To determine the affinity of [3H]QNB at each of the M4 mAChR receptor constructs, saturation binding assays were performed by incubating varying concentrations of [3H]QNB with 50 μg of membrane (see Ref. 11 for membrane preparations) at 37 °C for 1 h, in a final volume of 1 ml of binding buffer (20 mm HEPES, 100 mm NaCl, and 10 mm MgCl2 at pH 7.4). Subsequently, radioligand inhibition binding assays were performed by co-incubating 50 μg of membrane with a KA concentration of [3H]QNB (see Table 1) and varying concentrations of the nonradiolabeled test compound in 1 ml of binding buffer in the presence of the guanine nucleotide, GppNHp (100 μm), which was used to promote receptor-G protein uncoupling. Interaction studies were also performed between [3H]QNB, ACh, and LY2033298 by co-incubating 50 μg of membrane, a fixed concentration of ACh (see “Results”), and ∼100 pm [3H]QNB with increasing concentrations of LY2033298. GppNHp (100 μm) was included in the 0.5-ml final volume of binding buffer, and the reaction was left to reach equilibrium for 3 h at 37 °C. For all experiments, nonspecific binding was defined in the presence of 100 μm atropine, total binding was determined in the absence of the test ligand, and vehicle effects were determined with 1% DMSO. The assays were terminated by vacuum filtration through GF-B glass fiber filters and washing three times with ice-cold 0.9% NaCl. The [3H]QNB radioactivity was measured using a Packard 1600 TR liquid scintillation beta counter.
TABLE 1.
Equilibrium binding parameter estimates for ligands at M4 mAChR constructs
Values represent the mean ± S.E. from at least three experiments performed in triplicate.
| Receptor | [3H]QNB |
pKBc |
Log αd | ||||
|---|---|---|---|---|---|---|---|
| Bmaxa | pKAb | ACh | Xanomeline | McN-A-343 | LY2033298 | ||
| pmol/mg | |||||||
| M4 WT | 0.23 ± 0.05 | 10.33 ± 0.06 | 4.74 ± 0.05 | 6.67 ± 0.08 | 4.86 ± 0.05 | 5.21 ± 0.15 | 1.72 ± 0.09 (53) |
| M4 I93T/I94V/K95I | 0.28 ± 0.05 | 10.34 ± 0.03 | 4.69 ± 0.11 | 6.64 ± 0.07 | 4.97 ± 0.02 | 4.82 ± 0.36 | 2.14 ± 0.17e (138) |
| M4 I93T | 0.28 ± 0.05 | 10.76 ± 0.03e | 4.97 ± 0.04 | 7.22 ± 0.11e | 5.23 ± 0.01e | 5.36 ± 0.09 | 2.42 ± 0.16e (263) |
| M4 I94V | 0.22 ± 0.04 | 10.32 ± 0.08 | 4.71 ± 0.06 | 6.47 ± 0.06 | 4.85 ± 0.02 | 5.17 ± 0.08 | 1.74 ± 0.07 (55) |
| M4 K95I | 0.25 ± 0.03 | 10.21 ± 0.10 | 4.86 ± 0.05 | 6.38 ± 0.05 | 5.01 ± 0.02 | 5.20 ± 0.14 | 1.24 ± 0.04e (17) |
| M4 F186A | 0.26 ± 0.04 | 10.29 ± 0.09 | 4.85 ± 0.06 | 6.50 ± 0.08 | 5.30 ± 0.04e | NDf | ND |
| M4 S428P | 0.11 ± 0.02 | 10.77 ± 0.13e | 5.14 ± 0.03e | 6.73 ± 0.07 | 5.31 ± 0.03e | 5.17 ± 0.15 | 1.81 ± 0.11 (65) |
| M4 D432N | 0.13 ± 0.03 | 10.60 ± 0.07 | 5.19 ± 0.04e | 6.72 ± 0.08 | 5.15 ± 0.01e | 5.21 ± 0.20 | 1.37 ± 0.04 (23) |
| M4 Y439A | 0.22 ± 0.01 | 10.24 ± 0.08 | 3.33 ± 0.10e | 6.30 ± 0.07e | 4.54 ± 0.02e | 5.84 ± 0.12 | 0.49 ± 0.03e (3) |
| M4 C442A | 0.26 ± 0.05 | 10.01 ± 0.06 | 4.04 ± 0.07e | 6.33 ± 0.10e | 4.87 ± 0.03 | 5.35 ± 0.06 | 1.81 ± 0.03 (65) |
| M4 Y443A | 0.17 ± 0.05 | 10.22 ± 0.09 | 3.36 ± 0.01e | 6.13 ± 0.03e | 4.44 ± 0.10e | 6.22 ± 0.05e | 1.16 ± 0.01e (15) |
a Maximum density of binding sites.
b Negative logarithm of the radioligand equilibrium dissociation constant.
c Negative logarithm of the unlabeled ligand equilibrium dissociation constant.
d Logarithm of the cooperativity factor for the interaction between LY2033298 and ACh; antilogarithms are shown in parentheses.
e Significantly different (p < 0.05) from WT value as determined by one-way analysis of variance with Dunnett's post-hoc test.
f ND, not determined.
[3H]NMS Dissociation Kinetic Assays
[3H]NMS dissociation time-course experiments were performed by preincubating 50 μg of membrane with [3H]NMS (final assay concentration of 500 pm) for 1 h. At various time intervals (0, 2, 5, 8, 10, 15, and 30 min) atropine (100 μm) alone or in the presence of LY2033298 (30 μm) was added to the reaction containing GppNHp (100 μm) to make up a 0.5-ml final reaction volume. Total binding was defined at the 0 time interval. Nonspecific binding was defined in the presence of atropine over the entire time course. Following full dissociation time-course experiments, two time points were chosen (10 and 40 min) to define dissociation in the presence of multiple concentrations of LY2033298 (10 μm, 30 μm, and 100 μm). Nonspecific and total binding were defined as before, and assays were terminated as described above.
pERK1/2 Assays
Cells were seeded and washed as described previously (11) and serum-starved at 37 °C overnight. Concentration-response assays were performed by incubating increasing concentrations of each ligand (5 min for ACh or LY2033298 and 8 min for xanomeline or McN-A-343 as determined in time-course experiments) at 37 °C with Dulbecco's modified Eagle's medium in a final volume of 200 μl per well. For all experiments, 10% FBS was used as an internal control, and media or 0.1% DMSO were used as vehicle controls. The reactions were terminated and measured as described previously (11).
Data Analysis
Data were analyzed using Prism 5.03 (GraphPad, San Diego, CA). For radioligand saturation binding, nonspecific and total binding data were analyzed as described previously (20) to derive radioligand dissociation constant (KA) and receptor density (Bmax) estimates. For radioligand orthosteric inhibition binding experiments, specific binding of each ligand was analyzed according to a one-site binding equation (20). The negative logarithm of the equilibrium dissociation constant (pKB) of each ligand was determined using the Cheng and Prusoff equation (21). Interaction experiments between [3H]QNB, ACh, and LY2033298 were fitted to the following allosteric ternary complex model,
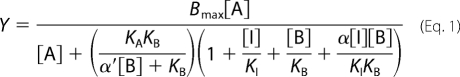 |
where KA, KB, and KI represent the equilibrium dissociation constants of the radioligand, allosteric ligand, and the orthosteric inhibitor, respectively, [A], [B], and [I] denote their respective concentrations, and α′ and α are the cooperativity factors for the interaction between the allosteric ligand and radioligand or unlabeled ligand, respectively. In all instances, the value of α′ was not significantly different from 1 and was fixed as such for all analyses.
Dissociation kinetic experiments were fitted to a monoexponential decay equation while concentration-response data generated from ERK1/2 phosphorylation assays were normalized to the 10% FBS response and fitted to a sigmoid concentration-response equation (20), see supplemental Table 2, as well as to the following operational model of agonism (22),
 |
where Em is the maximal possible response of the system (not the agonist), Basal is the basal level of response in the absence of agonist, KA denotes the functional equilibrium dissociation constant of the agonist (A), τ is an index of the coupling efficiency (or efficacy) of the agonist. To define the Em and τ for each mutant and assay, the KA for each agonist was constrained to equal the KB value derived from radioligand binding assays (see “Results”) in the nonlinear regression procedure.
All parametric measures of potency, affinity, operational efficacy, and cooperativity were estimated as logarithms (23). A one-way analysis of variance with a Dunnett's post test was used to determine statistical differences between the wild-type M4 mAChR and the M4 mutants, where p < 0.05 was considered significant.
RESULTS
Effects of Mutations on Orthosteric and Allosteric Ligand Affinity
Fig. 1A shows a snake diagram highlighting the TM helices of the M4 mAChR and identifies the residues mutated in the current study. Ile-Ile-Lys93–95(2.65–2.67) (E1), and Ser428 and Asp432(7.32) (E3/TM7 junction), were mutated to the equivalent residues in the M2 mAChR based on our recent finding that the combined mutation of these residues can affect the potency of LY2033298 in modulating ACh activity (10); in addition to the triple substitution of Ile-Ile-Lys93–95(2.65–2.67) we also made the individual point substitutions. In the E2 loop, we mutated Phe186 to Ala because all mAChRs (except the M5 mAChR) possess an aromatic amino acid residue in this region, and it has previously been shown that Tyr177 in this position of M2 mAChR is important for the activity of prototypical mAChR modulators, as well as the bitopic agonist, McN-A-343 (19, 24). Finally, we created alanine substitutions of the following conserved TM7 residues, Tyr439(7.39), Cys442(7.42), and Tyr443(7.43), because these have previously been identified to be critical for the binding and signaling of a variety of mAChR agonists, suggesting that they may contribute to a global activation switch (4, 17).
Radioligand saturation binding with the orthosteric antagonist, [3H]QNB, demonstrated that each receptor was expressed at approximately equivalent levels compared with the wild type (Table 1). The affinity of [3H]QNB was also similar between receptors, with the exception of Ile93(2.65) → Thr and Ser428 → Pro, where modest increases were noted (Table 1). Substitution of Ile-Ile-Lys93–95(2.65–2.67) for Thr-Val-Ile had no significant effect on the binding affinity of any of the agonists tested (Table 1 and Fig. 2), although the individual mutation of Ile93(2.65) → Thr caused an increase in the affinity of xanomeline and McN-A-343 for the M4 mAChR.
FIGURE 2.

Agonist affinity estimates are differentially modified by M4 mAChR mutations. Bars represent the difference in pKB of each agonist, derived using either a competitive or allosteric ternary complex model (see “Experimental Procedures”), relative to the wild-type receptor value for that agonist. Data represent the mean ± S.E. of at least three experiments performed in triplicates. n.d. indicates that there was no detectable binding; *, significantly different to wild-type, p < 0.05, one-way analysis of variance, Dunnett's post-test.
In contrast, the mutation of Phe186 → Ala had a striking inhibitory effect on the affinity of LY2033298, as estimated by application of a ternary complex model to the interaction between the modulator, [3H]QNB, and ACh, where a pKB value for the modulator could not be determined (Fig. 3 and Table 1). This latter finding could reflect an inability of LY2033298 to bind to the mutant receptor, or it could be due to neutral cooperativity (α′ = α = 1) between the modulator and both the orthosteric radioligand and orthosteric agonist. To differentiate between the two mechanisms, we examined the effects of LY2033298 on the dissociation kinetics of another orthosteric antagonist, [3H]NMS (which has substantially faster dissociation kinetics than [3H]QNB); the interaction between [3H]NMS and LY2033298 is characterized by weakly negative to almost neutral cooperativity (10, 12), and thus the potency of the modulator to inhibit radioligand dissociation from the [3H]NMS-occupied receptor will be close to (albeit slightly below) its affinity of the modulator for the allosteric site (25). As shown in Fig. 4, mutation of Phe186 → Ala had a profound inhibitory effect on the ability of LY2033298 to reduce [3H]NMS dissociation, with a resulting pEC50 estimate of 4.0 ± 0.1 compared with 5.0 ± 0.2 (n = 3) for the wild type. In contrast, this mutation had no effect on any of the other agonists with the exception of McN-A-343, where a slight enhancement of its affinity was noted (Table 1 and Fig. 2). Collectively, these results indicate that Phe186 is possibly a direct contact residue for the binding of LY2033298; the effect on McN-A-343 binding may be indirect as this compound is bitopic and thus interacts appreciably with the orthosteric site as well as an allosteric site (19).
FIGURE 3.

Identification of residues that govern LY2033298 affinity and cooperativity with ACh at the M4 mAChR. The competition between 100 pm [3H]QNB and a fixed concentration of ACh was determined in the absence (bars) or presence (circles) of increasing concentrations of LY2033298 at the indicated mAChR constructs. The fixed concentration of ACh was 10 μm for all experiments except for F186A (30 μm) and Y439A (1 mm). The curves drawn through the points represent the best global fit of an allosteric ternary complex model (Equation 2) to each pair of datasets, with the cooperativity between LY2033298 and [3H]QNB (α′) fixed to a value of 1. Data points represent the mean ± S.E. of at least three experiments performed in triplicates.
FIGURE 4.
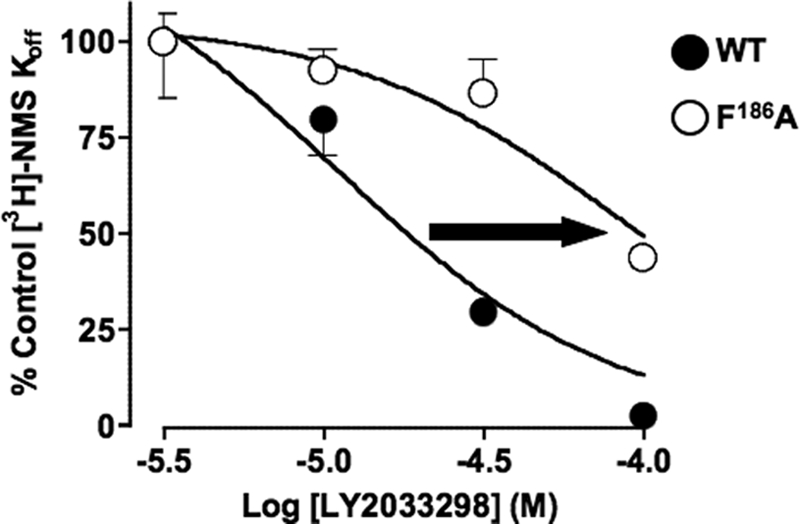
[3H]NMS dissociation kinetic studies confirm that Phe186 likely contributes to the allosteric binding site for LY2033298 at the M4 mAChR. Concentration-effect relationships for LY2033298 on the dissociation rate of [3H]NMS at the wild type or F186A M4 mAChR stably expressed in membranes from Chinese hamster ovary-FlpIn cells. Data represent mean ± S.E. of three experiments performed in triplicates.
Although our previous study suggested a possible role for the combination of the E3/TM7 mutants, Asp432(7.32) → Asn and Ser428 → Pro, in the functional potency of LY2033298, our binding interaction study has now revealed no significant effect of either (individual) mutation on the affinity of the modulator for the M4 mAChR allosteric site. This suggests that any functional effect of these residues is likely to reflect a change in the cooperativity between LY2033298 and ACh, or the signaling efficacy of either ligand (see below); it is of note that these mutations did have a slight but significant enhancing effect on the affinity of ACh and McN-A-343 (Table 1 and Fig. 2).
As expected, mutation of the key TM7 residues, Tyr439(7.39) → Ala, Cys442(7.42) → Ala, and Tyr443(7.43) → Ala, had significant inhibitory effects on the binding of ACh (Fig. 2 and Table 1). The Tyr mutations also reduced the binding of xanomeline and McN-A-343 but not to the extent of the effect on ACh (Fig. 2 and Table 1). The binding of xanomeline was weakly reduced at the Cys442(7.42) → Ala mutant, although McN-A-343 was unaffected. Interestingly, the affinity of LY2033298 was only sensitive to mutation of Tyr443(7.43) → Ala, but in the opposite direction to the other agonists, i.e. its affinity was increased at this mutant by a factor of ∼10. Although this surprising effect may suggest that Tyr443(7.43) represents another contact residue for LY2033298, this cannot be the case because the residue has been well established as a key contact point for ACh (4, 18), and LY2033298 prefers to bind to the ACh-occupied receptor. Thus, the alternative interpretation of this finding is that the effect of the Tyr443(7.43) → Ala mutation is an indirect one through a network of contacts onto the allosteric binding pocket.
Effects of Mutations on Allosteric Modulation
The interaction studies performed between [3H]QNB, ACh, and LY2033298 afforded not only the determination of modulator binding affinity at the various M4 mAChR constructs but also the cooperativity between the modulator and ACh. As illustrated in Fig. 3 for some of the key mutations, the experimental paradigm involved the determination of LY2033298 titration curves in the presence of a fixed concentration of both [3H]QNB and ACh; the allosteric effect of LY2033298 on ACh affinity was inferred from the changes in the ability of the orthosteric agonist to displace the radiolabeled antagonist, and the parameters describing the interaction estimated from the application of Equation 1 to this two-curve design.
These studies revealed that a key effect of the triple Ile-Ile-Lys93–95(2.65–2.67)Thr-Val-Ile substitution was to substantially increase the positive cooperativity between LY2033298 and ACh (Table 1). Analysis of the individual mutations indicated that this was largely governed by Ile93(2.65) → Thr (Fig. 3 and Table 1), because Ile94(2.66) → Val had no effect, whereas Lys95(2.67) → Ile actually reduced the cooperativity (Table 1). We propose that this cooperativity affect is a key molecular mechanism for the increase in functional LY2033298 potency noted following mutation of these three residues in our initial study of the effects of the modulator on ACh-mediated Ca2+ mobilization (10).
With regards to the role of the E2 loop, no interaction between ACh and LY2033298 was noted at the Phe186 → Ala mutant (Fig. 3 and Table 1), because, as described above, this residue is likely to constitute a direct contact point for the modulator in the allosteric binding pocket. In the E3 loop/TM7 region, the mutation of Asp432(7.32) → Asn reduced the cooperativity between ACh and LY2033298 from ∼50-fold to ∼20-fold, although this was not statistically significant; Ser428 → Pro had no effect on the binding cooperativity between the two agents.
Interestingly, both Tyr mutations in TM7 resulted in a significant blunting of the positive cooperativity between LY2033298 and ACh (Fig. 3 and Table 1). Given that these residues also have a profound effect on the binding affinity of ACh, this suggests that they form a key region in the network of interactions that govern the transmission of cooperativity between the orthosteric and allosteric sites.
Effects of Mutations on Signaling Efficacy
Finally, to investigate the effects of the selected mutations on the signaling capacity of the M4 mAChR, we determined the concentration-response profile for ERK1/2 phosphorylation in response to each of the orthosteric, allosteric, and bitopic agonists. The potency (pEC50) and maximal agonist effect (Emax) parameters for these studies are shown in supplemental Table 2. However, because potency is a composite of both affinity and signaling efficacy, we applied an operational model of agonism (Equation 2) incorporating the pKB values determined for each ligand at each receptor construct (Table 1) to estimate the effect of the mutation on the operational efficacy (τ) of each ligand, without the confounding influence of any effects on affinity. The estimated log τ values are summarized in Table 2.
TABLE 2.
Coupling efficiency of ligands at M4 mAChR constructs
Values represent the mean ± S.E. from at least three experiments performed in triplicate.
| Receptor | Log τa |
|||
|---|---|---|---|---|
| ACh | Xanomeline | McN-A-343 | LY2033298 | |
| M4 WT | 2.33 ± 0.04 | 0.73 ± 0.06 | 0.70 ± 0.08 | 1.35 ± 0.07 |
| M4 I93T/I94V/K95I | 2.62 ± 0.08 | 0.86 ± 0.12 | 0.68 ± 0.11 | 2.32 ± 0.18b |
| M4 I93T | 2.39 ± 0.06 | 0.44 ± 0.05 | 0.35 ± 0.06 | 2.43 ± 0.21b |
| M4 I94V | 2.33 ± 0.07 | 0.72 ± 0.19 | 0.52 ± 0.09 | 1.23 ± 0.06 |
| M4 K95I | 2.02 ± 0.03 | 0.66 ± 0.06 | 0.37 ± 0.03 | 0.63 ± 0.07b |
| M4 F186A | 2.02 ± 0.09 | 0.74 ± 0.13 | 0.31 ± 0.08 | NDc |
| M4 S428P | 1.87 ± 0.07 | 0.08 ± 0.01b | 0.02 ± 0.12b | 1.28 ± 0.18 |
| M4 D432N | 2.16 ± 0.10 | 0.31 ± 0.28 | 0.55 ± 0.11 | 1.15 ± 0.17 |
| M4 Y439A | 0.59 ± 0.17b | 0.11 ± 0.14 | ND | ND |
| M4 C442A | 1.52 ± 0.14b | 0.65 ± 0.10 | ND | 0.85 ± 0.30 |
| M4 Y443A | 0.44 ± 0.14b | 0.54 ± 0.76 | ND | ND |
a Logarithm of the operational efficacy parameter, τ, determined via nonlinear regression of the concentration-response data to an operational model of agonism.
b Significantly different (p < 0.05) from WT value as determined by one-way analysis of variance with Dunnett's post-hoc test.
c ND, not determined.
Interestingly, the effect of the E1 loop mutations on LY2033298 efficacy trended with the effects noted on the cooperativity with ACh, i.e. both the triple substitution (Fig. 6 and Table 2) and the individual Ile93(2.65) → Thr mutation (Figs. 5 and 6 and Table 2) increased the operational efficacy of LY2033298 as an agonist, whereas Ile94(2.66) → Val had no effect and Lys95(2.67) → Ile reduced allosteric ligand efficacy (Figs. 5 and 6 and Table 2). This finding suggests that the cooperativity between ACh and LY2033298 is linked to the ability of the modulator to drive the receptor toward an active state. However, there were no appreciable effects of these mutations on any of the other agonists, indicating a degree of specificity for mediating allosteric, as opposed to orthosteric, agonism.
FIGURE 6.

Agonist signaling efficacy estimates are differentially modified by M4 mAChR mutations. Bars represent the difference in log τ of each agonist, derived from an operational model of agonism (see “Experimental Procedures”), relative to the wild-type receptor value for that agonist. Data represent the mean ± S.E. of at least three experiments performed in triplicate. n.d. indicates that there was no detectable response; *, significantly different to wild type, p < 0.05, one-way analysis of variance, Dunnett's post-test.
FIGURE 5.

Identification of residues that govern orthosteric and allosteric agonist signaling efficacy at the M4 mAChR. Peak levels of pERK1/2 were assessed as described under “Experimental Procedures” and normalized to the response elicited by 10% FBS. The curves drawn through the points at each receptor construct represent the best global fit of an operational model of agonism (Equation 1) to each family of datasets, with the affinity of each agonist fixed to the pKB value determined in separate binding assays (Table 1). Data points represent the mean ± S.E. of at least three experiments performed in triplicates.
As expected, mutation of Phe186 → Ala in the E2 loop completely abolished functional responses to LY2033298 while having no significant effect on any of the other agonists (Fig. 5 and Table 2). A slightly more complex profile of responses was noted at the E3 loop/TM7 junction mutations, with no effect on ACh, a small reduction in the efficacy of xanomeline and McN-A-343 and, surprisingly, no substantial effect on the signaling efficacy of LY2033298 (Fig. 5 and Table 2). Given the overall modest nature of the effects of mutations in this region on signaling, however, it is unlikely that these residues play a major role in the activation of the receptor by either orthosteric or allosteric agonists, at least with respect to ERK1/2 phosphorylation.
In contrast, the ability of all classes of agonists to stimulate ERK1/2 phosphorylation was hindered following Ala substitutions at Tyr439(7.39) and Tyr443(7.43). The effects on xanomeline did not reach statistical significance, however, due to the large errors associated with determination of response at this mutation. Irrespectively, it is interesting to note that this latter agonist was the most resistant to the mutations, whereas the responses to ACh were markedly diminished, and those of McN-A-343 and LY2033298 were completely abolished (Fig. 5 and Table 2). This finding suggests that these aromatic residues likely contribute to a “global” activation switch that affects the ability of both orthosteric and allosteric agonists to activate the receptor. Substitution of Cys442(7.42) → Ala had a variable effect on agonist efficacy, reducing ACh and McN-A-343 efficacy, but having no significant effect on xanomeline and LY2033298 signaling.
DISCUSSION
Recent years have witnessed the discovery of a number of novel selective agonists of the mAChRs, particularly of the M1 subtype, that have been proposed to achieve their selectivity by virtue of an allosteric mode of action (9, 26–28). However, a definitive demonstration that these agonists activate the receptor and modulate the actions of orthosteric ligands via a common allosteric site (as opposed to separate actions at both the orthosteric and an allosteric site) is currently lacking (19, 29). In contrast, we have recently shown that LY2033298 indeed mediates both receptor activation and potentiation of the actions of ACh at the M4 mAChR via the same, allosteric site on that receptor (12). The discovery of LY2033298 as the first “pure” allosteric agonist and modulator of an mAChR thus afforded the current opportunity to explore molecular determinants that govern allosteric agonism at this GPCR family and whether such mechanisms diverge from those of orthosteric agonists. Our study has now identified important residues of the M4 mAChR that govern the binding of the modulator to its allosteric site (e.g. Phe186), the cooperativity between the modulator and the endogenous agonist (e.g. Ile93(2.65), Lys95(2.67), and Asp432(7.32)), and the ability of the modulator to drive the receptor into an active state (e.g. Ile93(2.65), Tyr439(7.39), and Tyr443(7.43)). The differential effects of some of these mutations on orthosteric and a (putative) bitopic M4 mAChR agonist, relative to LY2033298, clearly indicate the potential for allosteric agonists to promote unique states of a GPCR that are not necessarily shared by other agonists. However, the finding that key residues in a TM7 activation region affect the efficacy of both LY2033298 and other agonists suggests that there are also likely to be global activation mechanisms shared by all GPCR agonists irrespective of the mode of binding.
LY2033298 selectively potentiates the binding and function of ACh at the M4 mAChR, while having minimal to no effect on this agonist at other mAChR subtypes despite being able to bind to them (10).5 This indicates that there are sufficient structural differences between the mAChRs to accommodate such a degree of selectivity via an allosteric mechanism. Our earlier study thus used M2/M4 chimeric receptors to identify potential residues involved in the subtype-selective actions of LY2033298 and identified mutations in the E1 and E3 loops that resulted in gain and loss of function, respectively (10). We have now made a number of important findings that place these previous observations in a new mechanistic light, as well as highlighting a vital consideration when performing structure-function studies of allosteric ligands. First, the E1/E3 mutations actually had little effect on the binding affinity of the allosteric ligand to the free receptor, suggesting that they do not contribute directly to the binding pocket for this modulator. Second, the gain in function mediated by mutation of the E1 loop region is due to an increase in both the positive binding cooperativity between the modulator and ACh (Log α, Table 1), and the intrinsic efficacy of the LY2033298 as an allosteric agonist (Log τ, Table 2). Moreover, this effect is attributable to the single mutation of Ile93(2.65) → Thr, because mutation of Ile94(2.66) → Thr had no effect while Lys95(2.37) → Ile actually reduced the cooperativity and efficacy of LY2033298. Given that this region did not affect the efficacy of the other agonists, it also suggests that the E1 loop region may be a selective mediator of allosteric agonist signaling. Third, the loss in functional cooperativity noted upon combined substitution of Ser428 → Pro or Asp432(7.32) → Asn, in our original study, could not be attributed to an effect of either mutation on LY2033298 efficacy nor on the binding cooperativity between LY2033298 and ACh. This lack of effect of the single point Ser428 → Pro mutation is not surprising, given our previous study that noted minimal effect of the mutation alone on LY2033298-mediated Ca2+ mobilization, but the finding with the Asp432(7.32) → Asn mutation was surprising, given the large loss of functional allosterism noted in our initial study (10). Thus, we interpret these findings to suggest that this E3 loop residue is primarily involved in the transmission of activation/functional cooperativity between LY2033298 and the endogenous agonist, rather than binding cooperativity or modulator affinity. Collectively, these observations highlight the different mechanisms that can underlie changes in the potency of allosteric modulators when determined using functional approaches, and that a change in potency does not necessarily reflect a change in affinity of the modulator for the receptor.
In contrast to the E1 and E3 loop mutations, we did identify a key residue in the E2 loop that was essential for the binding of LY2033298 to the M4 mAChR. Alanine substitution of Phe186 profoundly reduced the binding of LY2033298 to the receptor, suggesting that this residue may act as a direct contact residue in the binding site of LY2033298. In the M2 mAChR, the equivalent Tyr177 in the E2 loop is important for the binding of prototypical allosteric modulators, such as C7/3-phth, and for the functional selectivity that is part of the mechanism of action of the bitopic agonist, McN-A-343 (19, 24). Similar to observations made for Tyr177 → Ala at the M2 mAChR, we found only a moderate (3-fold) change in the binding affinity of McN-A-343 upon alanine substitution of Phe186 in the M4 mAChR. In contrast to the M2 mAChR, however, the efficacy of McN-A-343 was not significantly altered, suggesting that McN-A-343 interacts with the M4 mAChR in a different manner to the M2 mAChR. This may also explain, at least partly, why McN-A-343 is more efficacious at the M4 mAChR than at the M2 mAChR. Not surprisingly, there was no effect on the binding and function of the other agonists, indicating that Phe186 in the E2 loop is primarily essential for the binding of LY2033298. This adds to the growing body of evidence pointing to a key role of the E2 loop in the action of mAChR modulators. In this regard, it is of note that a recent study of a novel allosteric potentiator at the M1 mAChR, benzyl quinolone carboxylic acid, also found that the equivalent aromatic residue in the E2 loop of that receptor (Tyr179) was vital for modulator potency (30).
In addition to regions of the receptor that were primarily important for the actions of LY2033298, we also identified residues in TM7 that were critical for the activity of all classes of agonist. Perhaps unsurprisingly, these residues are completely conserved across all five mAChR subtypes. As expected, alanine substitution of two of these, Tyr439(7.39) and Tyr443(7.43), caused a significant reduction in the binding affinity of ACh, highlighting their likely roles as contact points for the endogenous agonist. The Cys442(7.42) → Ala mutation resulted in a smaller reduction in ACh binding, suggesting that this residue does not directly contribute to the binding pocket for ACh. Inhibitory effects of the Tyr439(7.39), Cys442(7.42), and Try443(7.43) substitutions on xanomeline and McN-A-343 binding were smaller than those observed for ACh, consistent with the hypothesis that these compounds likely adopt different poses within the orthosteric pocket of the receptor compared with the endogenous agonist. In contrast, these mutations either had no effect (Tyr439(7.39) → Ala and Cys442(7.42) → Ala) or caused a surprising increase (Tyr443(7.43) → Ala) in the affinity of LY2033298 for the receptor; given the location of these residues, we did not anticipate any direct effects on the binding affinity of the modulator. The magnitude of the effect of the Tyr443(7.43) → Ala suggests that this residue either promotes some sort of steric hindrance to the binding of the modulator in the more extracellular regions of the receptor, which is unlikely given that the residue is a direct contact point for ACh, or more likely contributes to modulator binding via a network of, currently unknown, interactions.
Despite the differential effects of the TM7 residues on ligand binding affinity, the common observation for all agonists was that this region was important for their signaling efficacy. The pronounced effect on ACh signaling, especially upon mutation of the Tyr residues, was not surprising given similar findings at other mAChRs (4). However, the substantial impairment in the signaling of LY2033298 and McN-A-343, together with the more modest effects on the signaling of xanomeline, is a novel finding consistent with the hypothesis that GPCRs possess global activation switches that can be engaged irrespective of the binding locus of the agonist (31).
In summary, we have identified key regions in the M4 mAChR that are involved in the selective binding and signaling of LY2033298, and in the transmission of binding and efficacy cooperativity between the orthosteric and an allosteric binding site. We have also identified Tyr7.39 and Tyr7.43 to be key activation switches that trigger global receptor activation irrespective of whether the agonist is orthosteric, allosteric, or bitopic.
Supplementary Material
This work was supported by Program Grant 519461 of the National Health and Medical Research Council (NHMRC) of Australia.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables 1 and 2.
Numbers in parentheses indicate Ballesteros and Weinstein (2) numbering.
V. Nawaratne, K. Leach, C. C. Felder, P. M. Sexton, and A. Christopoulos, unpublished observations.
- GPCR
- G protein-coupled receptor
- TM
- transmembrane
- mAChR
- muscarinic acetylcholine receptor
- ACh
- acetylcholine
- FBS
- fetal bovine serum
- ERK
- extracellular signal-regulated kinase
- pERK
- phosphorylated ERK
- QNB
- quinuclidinyl benzilate
- NMS
- N-methylscopolamine
- GppNHp
- guanosine 5′-(β,γ-imido)triphosphate.
REFERENCES
- 1.Harmar A. J., Hills R. A., Rosser E. M., Jones M., Buneman O. P., Dunbar D. R., Greenhill S. D., Hale V. A., Sharman J. L., Bonner T. I., Catterall W. A., Davenport A. P., Delagrange P., Dollery C. T., Foord S. M., Gutman G. A., Laudet V., Neubig R. R., Ohlstein E. H., Olsen R. W., Peters J., Pin J. P., Ruffolo R. R., Searls D. B., Wright M. W., Spedding M. (2009) Nucleic Acids Res. 37, D680–D685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ballesteros J. A., Weinstein H. (1995) Methods Neurosci. 25, 366–428 [Google Scholar]
- 3.Hulme E. C., Lu Z. L., Saldanha J. W., Bee M. S. (2003) Biochem. Soc. Trans. 31, 29–34 [DOI] [PubMed] [Google Scholar]
- 4.Lu Z. L., Saldanha J. W., Hulme E. C. (2001) J. Biol. Chem. 276, 34098–34104 [DOI] [PubMed] [Google Scholar]
- 5.Wess J., Han S. J., Kim S. K., Jacobson K. A., Li J. H. (2008) Trends Pharmacol. Sci. 29, 616–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neubig R. R., Spedding M., Kenakin T., Christopoulos A. (2003) Pharmacol. Rev. 55, 597–606 [DOI] [PubMed] [Google Scholar]
- 7.Christopoulos A. (2002) Nat. Rev. Drug Discov. 1, 198–210 [DOI] [PubMed] [Google Scholar]
- 8.Gregory K. J., Sexton P. M., Christopoulos A. (2007) Curr. Neuropharmacol. 5, 157–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Langmead C. J., Christopoulos A. (2006) Trends Pharmacol. Sci. 27, 475–481 [DOI] [PubMed] [Google Scholar]
- 10.Chan W. Y., McKinzie D. L., Bose S., Mitchell S. N., Witkin J. M., Thompson R. C., Christopoulos A., Lazareno S., Birdsall N. J., Bymaster F. P., Felder C. C. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 10978–10983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nawaratne V., Leach K., Suratman N., Loiacono R. E., Felder C. C., Armbruster B. N., Roth B. L., Sexton P. M., Christopoulos A. (2008) Mol. Pharmacol. 74, 1119–1131 [DOI] [PubMed] [Google Scholar]
- 12.Leach K., Loiacono R. E., Felder C. C., McKinzie D. L., Mogg A., Shaw D. B., Sexton P. M., Christopoulos A. (2010) Neuropsychopharmacology 35, 855–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shekhar A., Potter W. Z., Lightfoot J., Lienemann J., Dubé S., Mallinckrodt C., Bymaster F. P., McKinzie D. L., Felder C. C. (2008) Am. J. Psychiatry 165, 1033–1039 [DOI] [PubMed] [Google Scholar]
- 14.Gnagey A. L., Seidenberg M., Ellis J. (1999) Mol. Pharmacol. 56, 1245–1253 [DOI] [PubMed] [Google Scholar]
- 15.Krejcí A., Tucek S. (2001) Mol. Pharmacol. 60, 761–767 [PubMed] [Google Scholar]
- 16.Huang X. P., Prilla S., Mohr K., Ellis J. (2005) Mol. Pharmacol. 68, 769–778 [DOI] [PubMed] [Google Scholar]
- 17.Urizar E., Claeysen S., Deupí X., Govaerts C., Costagliola S., Vassart G., Pardo L. (2005) J. Biol. Chem. 280, 17135–17141 [DOI] [PubMed] [Google Scholar]
- 18.Wess J., Gdula D., Brann M. R. (1991) EMBO J. 10, 3729–3734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Valant C., Gregory K. J., Hall N. E., Scammells P. J., Lew M. J., Sexton P. M., Christopoulos A. (2008) J. Biol. Chem. 283, 29312–29321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Christopoulos A., Motulsky H. (2004) Fitting Models to Biological Data Using Linear and Nonlinear Regression. A Practical Guide to Curve Fitting, Oxford University Press, New York [Google Scholar]
- 21.Cheng Y. C., Prusoff W. H. (1973) Biochem. Pharmacol. 22, 3099–3108 [DOI] [PubMed] [Google Scholar]
- 22.Black J. W., Leff P. (1983) Proc. R. Soc. Lond. B. Biol. Sci. 220, 141–162 [DOI] [PubMed] [Google Scholar]
- 23.Christopoulos A. (1998) Trends Pharmacol. Sci. 19, 351–357 [DOI] [PubMed] [Google Scholar]
- 24.May L. T., Avlani V. A., Langmead C. J., Herdon H. J., Wood M. D., Sexton P. M., Christopoulos A. (2007) Mol. Pharmacol. 72, 463–476 [DOI] [PubMed] [Google Scholar]
- 25.Lazareno S., Birdsall N. J. (1995) Mol. Pharmacol. 48, 362–378 [PubMed] [Google Scholar]
- 26.Spalding T. A., Trotter C., Skjaerbaek N., Messier T. L., Currier E. A., Burstein E. S., Li D., Hacksell U., Brann M. R. (2002) Mol. Pharmacol. 61, 1297–1302 [DOI] [PubMed] [Google Scholar]
- 27.Sur C., Mallorga P. J., Wittmann M., Jacobson M. A., Pascarella D., Williams J. B., Brandish P. E., Pettibone D. J., Scolnick E. M., Conn P. J. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 13674–13679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones C. K., Brady A. E., Davis A. A., Xiang Z., Bubser M., Tantawy M. N., Kane A. S., Bridges T. M., Kennedy J. P., Bradley S. R., Peterson T. E., Ansari M. S., Baldwin R. M., Kessler R. M., Deutch A. Y., Lah J. J., Levey A. I., Lindsley C. W., Conn P. J. (2008) J. Neurosci. 28, 10422–10433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gregory K. J., Hall N. E., Tobin A. B., Sexton P. M., Christopoulos A. (2010) J. Biol. Chem. 285, 7459–7474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma L., Seager M. A., Wittmann M., Jacobson M., Bickel D., Burno M., Jones K., Graufelds V. K., Xu G., Pearson M., McCampbell A., Gaspar R., Shughrue P., Danziger A., Regan C., Flick R., Pascarella D., Garson S., Doran S., Kreatsoulas C., Veng L., Lindsley C. W., Shipe W., Kuduk S., Sur C., Kinney G., Seabrook G. R., Ray W. J. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 15950–15955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwartz T. W., Frimurer T. M., Holst B., Rosenkilde M. M., Elling C. E. (2006) Annu. Rev. Pharmacol. Toxicol. 46, 481–519 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.