

Abstract
Epidermal growth factors and their receptors (EGFRs) promote breast cancer cell proliferation and can drive tumorigenesis. However, the molecular mechanisms that mediate these effects are incompletely understood. We previously showed that mammary tumor development in the mouse model of breast cancer MMTV-neu, a model characterized by amplification of the EGFR ErbB2 in mammary tissue, correlates with a marked up-regulation of fatty acid-binding protein 5 (FABP5). FABP5 functions to deliver ligands to and enhance the transcriptional activity of the nuclear receptor peroxisome proliferator-activated receptor β/δ (PPARβ/δ), a receptor whose target genes include genes involved in cell growth and survival. We show here that in MCF-7 mammary carcinoma cells, EGFR signaling directly up-regulates the expression of FABP5. The data demonstrate that treatment of these cells with the EGFR ligand heregulin-β1 signals through the ERK and the phophatidylinositol-3-kinase cascades, resulting in activation of the transcription factor NF-κB. In turn, NF-κB induces the expression of FABP5 through two cognate response elements in the promoter of this gene. The observations further demonstrate that FABP5 and PPARβ/δ are critical mediators of the ability of EGFR to enhance cell proliferation, indicating that this transcriptional pathway plays a key role in EGFR-induced tumorigenesis. Additional observations indicate that the expression of FABP5 is down-regulated by the Krüppel-like factor KLF2, suggesting a tumor suppressor activity for this factor.
Keywords: Breast Cancer, Gene Transcription, Growth Factors, Lipid-binding Protein, NF-κB Transcription Factor, Nuclear Receptors, PPAR, Fatty Acid-binding Protein, Heregulin
Introduction
ErbB2 (HER2/neu) is a member of the epidermal growth factor receptor (EGFR)3 family that also includes EGFR/HER1, HER3/ErbB3, and HER4/ErbB4 (1, 2). ErbB2 is a tyrosine kinase oncogene whose amplification is evident in a large percentage of primary human breast cancers (3), and its occurrence is inversely correlated with long-term survival in human patients (4–8). ErbB2 lacks a ligand binding domain, and it functions as a heterodimer with other members of the EGFR family that are activated by their cognate ligands heregulins (1, 2). Overexpression of ErbB2 or of ErbB2 in conjunction with other EGFRs or treatment of cells with heregulins result in uncontrolled proliferation, resistance to apoptosis, and increased motility and invasion and can lead to oncogenic transformation (9–19). Indeed, one of the best characterized animal models of breast cancer is the TgN(MMTVneu)202Mul transgenic mouse, which displays mammary-specific amplification of ErbB2 (20, 21). In the MMTV-neu mouse model, 100% of female mice develop mammary adenocarcinomas that involve the entire epithelium in each gland with a median time for tumor progression at ∼205 days (22).
Ligand-induced heterodimerization of ErbB2 with an EGFR partner activates a complex signaling network that includes kinases such as extracellular signal-regulated kinases (ERK), phophatidylinositol-3-kinase (PI3K), mitogen-activated protein kinase, and protein kinase C (2). However, down-stream events through which heregulins, their receptors, and the associated signaling cascades trigger proliferation and cell transformation remain incompletely understood.
We previously showed that tumor development in the MMTV-neu mouse model of breast cancer is accompanied by a marked up-regulation of an intracellular lipid-binding protein (iLBP) termed fatty acid-binding protein 5 (FABP5) (23, 24). The iLBPs are small (∼15 kDa) proteins that bind a variety of retinoids and fatty acids (25–27). The functions of many of the 14 members of the iLBP family remain unknown, but it has been demonstrated that three of these cooperate with specific members of the nuclear receptor family of ligand-activated transcription factors to mediate the transcriptional activities of shared ligands. It was, thus, shown that cellular retinoic acid-binding protein II (CRABP-II), FABP4, and FABP5 function in conjunction with RAR, PPARγ, and PPARβ/δ, respectively. These proteins, which reside in the cytosol in the absence of ligands, undergo nuclear translocation upon binding of specific compounds, which activate their cognate receptors. In the nucleus these iLBPs directly associate with their cognate receptors to form a complex through which the ligand is “channeled” from the binding protein to the receptor. These iLBPs, thus, facilitate the ligation of cognate receptors and markedly enhance their transcriptional activities (23, 24, 28–33).
The increased expression level of FABP5 observed in mammary tumors that arise in MMTV-neu mice (24), and the reports that activation of the FABP5-associated receptor PPARβ/δ protects cells against apoptosis and facilitates cell growth and migration (23, 24, 34–36) suggest that ErbB2-driven tumorigenesis may involve enhanced transcriptional activity of the FABP5/PPARβ/δ pathway. This study was undertaken to delineate the mechanisms through which the expression of FABP5 is up-regulated in ErbB2-driven tumors and to obtain insights into the involvement of the FABP5/PPARβ/δ pathway in EGFR-stimulated cell proliferation.
EXPERIMENTAL PROCEDURES
Reagents
Heregulin-β1 (HRG-β1) was purchased from R&D Systems. Antibodies against FABP5 and PPARβ/δ were obtained from R&D Systems. β-Tubulin antibodies were from Sigma. Antibodies against phosphorylated Akt, ERK, ErbB2, ErbB3, and ErbB4 were from Cell Signaling. Antibody against p50 and p65 was obtained from Santa Cruz. Anti-mouse and anti-rabbit immunoglobulin horseradish peroxidase-conjugated antibodies were from Bio-Rad, and anti-goat immunoglobulin was from Santa Cruz.
Vectors
Expression vectors for dominant negative ERK (ERK-K52R) (37), dominant negative IκBα (SRIkBα) (38), and ErbB2/Neu were provided by Dr. Melanie Cobb, Dr. George Stark, and Dr. Ruth Keri, respectively.
Cells
MCF-7, MDA-MB453 and HEK 293T cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and antibiotics.
Western Blotting
Cells were lysed in buffer containing 150 mm NaCl, 10 mm Tris, pH 7.2, 0.1% SDS, 1% Triton X-100, 1% deoxycholate, 5 mm EDTA, 1 mm phenylmethylsulfonyl fluoride, 2 μg/ml leupeptin, 2 μg/ml aprotinin, and 2 μg/ml pepstatin A. Protein concentrations were determined by the Bradford assay, and 50–75 μg/lane cell lysate was resolved by SDS-PAGE and probed by Western blots using appropriate antibodies.
Quantitative Real-time PCR (Q-PCR)
Total RNA was extracted using Trizol. 2 μg of mRNA was reverse-transcribed into cDNA using the high capacity RNA to cDNA kit from Applied Biosystems (Gaithersburg, MD). Q-PCR analyses were performed in triplicate using the Taqman Gene Expression Master Mix (Applied Biosystems). TaqMan chemistry and Assays on Demand probes for FABP5 (Hs00154260-m1) and PDK1 (Hs00198887-m1) were purchased from Applied Biosystems. As an internal control, 18 S rRNA (4319413E-0710034) was used. Detection and data analysis were carried out on an ABI StepOne Plus Real-Time PCR system.
Transactivation Assays
MCF-7 cells (2 × 105) were plated in 6-well plates in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. Cells were transfected with Superfect with vectors harboring a luciferase reporter driven by the FABP5 promoter (or mutant) or by a luciferase reporter controlled by three copies of a PPAR response element (PPRE-luc) in conjunction with an expression vector for PPARβ/δ. Cell were co-transfected with an expression vector for β-galactosidase, serving as an internal control. 40 h post-transfection, cells were serum-starved and then treated with 30 ng/ml HRG-β1 for 16 h. Cells were lysed, and luciferase activity was assayed using the luciferase assay buffer (Promega) and corrected for transfection efficiency by the activity of β-galactosidase.
Ectopic Overexpression
MCF-7 cells (200,000) were plated in 6-well plates in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. Cells were transiently transfected with 3 μg of the appropriate vectors using Superfect as per the manufacturer's protocol (Qiagen). For expression of KLF2, MCF-7 cells were infected with an adenoviral vectors encoding GFP or GFP-KLF2 (Welgen Inc.).
Decreasing Expression Levels
Knockdown of FABP5 and PPARβ/δ was accomplished by infecting MCF-7 cells with lentiviruses encoding respective shRNA (Open Biosystems). Viruses were packaged in HEK293T cells with 7 μg of pCMV, 8 μg of pMD26, and 6 μg of the appropriate shRNA in 100-mm plates using Superfect. Lentiviruses were harvested, and Polybrene was added to the virus-containing media. Virus-containing media were used to infect MCF-7 cells. Infection was repeated, and cells were harvested after 3 days. Expression levels of FABP5 and PPARβ/δ were examined by Western blot analyses. For knockdown of ErbB3 or ErbB4, MCF-7 cells were transiently transfected with lentiviral vectors harboring ErbB3 or ErbB4 shRNA (Open Biosystems) using Superfect according to the manufacturer's protocol.
Mutations in the FABP5 Promoter
800 base pairs upstream of the start site of the FABP5 gene were cloned upstream of a firefly luciferase gene in the plasmid pGL3-Basic. Mutations in the NF-κB binding sites in that sequence were generated using the Stratagene QuikChange kit (Stratagene, La Jolla, CA). In each case, the NF-κB sequences of the FABP5 promoter was mutated to BglII site. The mutations were confirmed by sequencing at the Case Western Reserve University Genomics Core Facility.
Bromodeoxyuridine Incorporation
MCF-7 cells were plated at 1 × 106 cells in a 100-mm plate. Cells were serum-starved for 24 h and then treated with HRG-β1 (30 ng/ml) for 24 h. Bromodeoxyuridine incorporation was assayed using the protocol of the manufacturer (BD Biosciences). Samples were analyzed at Case Western Reserve University Cell Sorting Facility using a BD Biosciences LSR II cell analyzer.
MTT Assay
Cell proliferation assay was performed using MTT cell proliferation assay kit (Cayman Chemical Co., Ann Arbor, MI). Briefly, MCF-7 cells were infected with control adenovirus (Ad-GFP) or adenovirus overexpressing KLF2 (Ad-KLF2). Cells were detached from culture plates using cell dissociation buffer (Invitrogen). 10,000 cells were plated on each 96-well tissue culture. Cells were either left untreated or treated with 30 ng/ml HRG-β1 for 24 h. Cells were incubated with MTT reagent, and formazan crystals were dissolved using crystal dissolving solution. Absorbance of each sample was recorded at 570 nm using a microplate reader.
Cell Counting
MCF-7 or MDA-MB453 cells were plated in 6-well plates, serum-starved overnight, and treated with HRG-β1 for 24 h. Cells were washed with phosphate-buffered saline, treated with trypsin, stained with trypan blue, and counted.
Chromatin Immunoprecipitation Assays
MCF-7 cells were grown to 70–80% confluency on 100-mm tissue culture dishes, serum-starved for 24 h, and incubated with 30 ng/ml HRG-β1 for 4 h at 37 °C. Cells were fixed with 1% formaldehyde (10 min) and quenched by the addition of 0.125 m glycine (5 min.). Cells were washed 3 times with phosphate-buffered saline (PBS), scraped in PBS, and harvested by centrifugation at 3000 × g (5 min, 4 °C). Cells were suspended in lysis buffer (5 mm PIPES, pH 8.0, 85 mm KCl, 0.5% Nonidet P-40, 10 μg/ml leupeptin, 1 μg/ml aprotinin, 5 μg/ml pepstatin A), incubated at 4 °C, and pelleted by centrifugation at 3000 × g (5 min, 4 °C). Pelleted cells were resuspended in nucleic lysis buffer (10 mm Tris-HCl, pH 8.1, 10 mm EDTA, 1% SDS, and 1 μg/ml leupeptin, 1 μg/ml aprotinin, 5 μg/ml pepstatin A) and sonicated 10 times for 10 s per pulse, yielding DNA fragments of 300–3000 bp in size. Sonicated samples were precleared by diluting 10 times in dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mm EDTA, 16.7 mm Tris-HCl pH 8.1, 167 mm NaCl)) and incubated for 1 h at 4 °C with 50 μl/ml 50% Protein A-Sepharose slurry. Supernatant was transferred to a tube containing 10 μg of the p50 or p65 antibody and incubated overnight. Complexes were precipitated with 50 μl of 50% Protein A-Sepharose. Beads were washed sequentially with 1 ml of dilution buffer containing 0.1% SDS and 150 mm NaCl, 1 ml of dilution buffer containing 0.1% SDS and 500 mm NaCl, and 1 ml of buffer III (10 mm Tris, pH 8.1, 0.25 m LiCl, 1% Nonidet P-40, 1% sodium deoxycholate, and 1 mm EDTA) and two washes with 1 ml of 10 mm Tris, pH 8.0, 1 mm EDTA. 200 μl of elution buffer (0.1 m NaHCO3 and 1% SDS) were added to the beads, and samples were incubated at room temperature for 15 min with agitation. Cross-link was reversed overnight at 65 °C. Supernatant was incubated (1 h, 4 °C) with 1 m Tris, pH 6.5, and 10 mg/ml proteinase K. DNA was extracted using phenol-chloroform. PCRs was carried out using Pfu turbo (Invitrogen) according to the manufacturer's directions. PCR products was separated on a 1% agarose gel, stained in ethidium bromide, and visualized with a Bio-Rad Gel Doc 2000 gel documentation system.
RESULTS
HRG-β1 Up-regulates the Expression of FABP5 in MCF-7 and MDA-MB453 Mammary Carcinoma Cells
The hallmark of the MMTV-neu mouse model of breast cancer is mammary overexpression of the growth factor receptor ErbB2. The observations that tumor development in this model is accompanied by a marked up-regulation of FABP5 (23, 24) raise the possibility that the expression of this protein is directly controlled by EGFR signaling. To examine this possibility, the ErbB2-positive MCF-7 and MDA-MB453 mammary carcinoma cells were treated with HRG-β1, and the expression level of FABP5 was assessed. In MCF-7 cells, both the mRNA and protein level of FABP5 was induced by HRG-β1 in a time-dependent manner (Fig. 1, a–c). HRG-β1 also induced the expression of FABP5 in MDA-MB 453 cells (supplemental Fig. S5a). The induction of FABP5 by HRG-β1 in MCF-7 cells was abolished in the presence of actinomycin D (Fig. 1c), indicating that the regulation was exerted at the level of transcription. To examine whether the effect reflected a direct response, MCF-7 cells were pretreated with the protein synthesis inhibitor cycloheximide before the addition of HRG-β1. Cycloheximide had no effect on the ability of HRG-β1 to induce FABP5 mRNA (Fig. 1d), demonstrating that the induction did not require de novo protein synthesis and suggesting direct transcriptional control of EGFR signaling on FABP5 expression. In support of this conclusion, transcriptional activation assays conducted using a luciferase reporter driven by an 800-bp sequence of the proximal promoter of FABP5 revealed that HRG-β1 activates the promoter and does so in a dose-dependent manner (supplemental Fig. S1).
FIGURE 1.

HRG-β1 regulates the transcription of FABP5 in MCF-7 cells. a, MCF-7 cells were serum-starved for 24 h before treatment with HRG-β1 (30 ng/ml) for the indicated times. FABP5 mRNA levels were assessed by Q-PCR. Data are the means ± S.D. (n = 3). b, serum-starved MCF-7 cells were treated with HRG-β1 (30 ng/ml) for the denoted times before lysis. Lysates were analyzed by immunoblots. c, serum-starved MCF-7 cells were treated with actinomycin D (actD, 5 μg/ml) before the addition of HRG-β1 (30 ng/ml) for the denoted times. FABP5 mRNA levels were measured by Q-PCR. Data are the mean ± S.D. (n = 3). d, serum-starved MCF-7 cells were treated with cycloheximide (CHX, 20 μg/ml) for 10 min before the addition of HRG-β1 (30 ng/ml, 4 h). Levels of FABP5 mRNA were measured by Q-PCR. Data are the mean ± S.D. (n = 3).
Induction of FABP5 by HRG-β1 Is Mediated by PI3K and ERK
EGFRs can modulate transcriptional rates through activation of a complex network including the signaling transduction pathways mediated by PI3K and ERK (2). Indeed, a 15-min treatment of MCF-7 cells with HRG-β1 triggered phosphorylation of both ERK (Fig. 2a) and the downstream effector of PI3K Akt1 (Fig. 2b). These activities could be respectively blocked by treatment with the MEK inhibitor PD098059 and the PI3K inhibitor wortmannin (Fig. 2, a and b). To identify the pathways by which HRG-β1 up-regulates FABP5, MCF-7 cells were pretreated with PD098059 or wortmannin before treatment with HRG-β1. Inhibition of either ERK or PI3K or treatment with both inhibitors markedly decreased the induction of FABP5 by HRG-β1 (Fig. 2, c and d, and data not shown). In accordance, treatment of MCF-7 cells with either wortmannin or PD98059 blocked the ability of HRG-β1 to elicit the luciferase reporter driven by the FABP5 promoter (supplemental Fig. S2). Ectopic expression of the dominant negative ERK construct ERK-K52R (37), which effectively blocked HRG-β1-induced phosphorylation of the downstream ERK substrate P90S6K (Fig. 2e), markedly decreased the ability of HRG-β1 to induce the expression of FABP5 (Fig. 2f). Ectopic expression of a constitutively active Akt1 up-regulated the basal expression of FABP5 and significantly enhanced the HRG-β1 response (Fig. 2g). Taken together, these data establish that induction of FABP5 by HRG-β1 is mediated by both the ERK and the PI3K/Akt1 pathways.
FIGURE 2.

Up-regulation of FABP5 by HRG-β1 is mediated by the ERK and PI3K pathways. a, serum-starved MCF-7 cells were treated with the MEK inhibitor PDO98059 (20 μm) for 1 h before the addition of HRG-β1 (30 ng/ml, 15 min). Cell lysates were analyzed by immunoblots using antibodies against phospho-ERK (p-ERK) and total ERK. b, serum-starved MCF-7 cells were treated with the PI3K inhibitor wortmannin (100 nm) for 1 h before the addition of HRG-β1 (30 ng/ml, 15 min). Cell lysates were analyzed by immunoblots using antibodies against phospho-Akt (p-Akt) and total Akt. c, serum-starved MCF-7 cells were treated with PDO98059 (PD, 20 μm) or wortmannin (100 nm) for 1 h before the addition of HRG-β1 (30 ng/ml, 4 h). FABP5 mRNA levels were measured by Q-PCR. Data are the mean ± S.D. (n = 3). p = 0.006 (*) and p = 0.014 (**) versus HRG-β1 alone. d, serum-starved MCF-7 cells were treated with PDO98059 (20 μm) or wortmannin (100 nm) for 1 h before the addition of HRG-β1 (30 ng/ml, 24 h). Cell lysates were analyzed by immunoblots. e, MCF-7 cells were transfected with an empty vector (ev) or an expression vector for the dominant negative ERK-K52R (DN-ERK), serum-starved overnight, and then treated with HRG-β1 (15 min.). Cell lysates were analyzed by immunoblots using antibodies recognizing ERK or its substrate p-p90S6K. β-Tubulin was used as a loading control. f, MCF-7 cells were transfected with denoted plasmids, serum-starved overnight, and then treated with HRG-β1 (4 h). Expression of FABP5 mRNA was measured by Q-PCR. Data are the mean ± S.D. (n = 3). *, p = 0.018 versus HRG-β1-treated cells transfected with an empty vector. g, MCF-7 cells were transfected with a control vector or a vector harboring Myc-tagged constitutively active (CA)-myristoylated Akt1. 36 h later cells were serum-starved overnight and treated with HRG-β1 (30 ng/ml, 4 h). FABP5 mRNA levels were measured by Q-PCR. Data are the mean ± S.D. (n = 3). *, p < 0.03 versus respective empty vector-transfected controls. Inset, shown are immunoblots demonstrating expression of myc-CA-Akt1.
Induction of FABP5 by HRG-β1 Is Mediated by NF-κB
The ERK or PI3K pathways often regulate transcription by activating NF-κB, a transcription factor that plays key roles in mediating EGFR-induced cell proliferation (39–41). Inspection of the proximal promoter of FABP5 revealed two putative NF-κB binding sites located at −784 and −131 upstream from the gene start site (Fig. 3a). The functionality of these sites was examined by transcriptional activation assays using a luciferase reporter in which these sites were mutated individually or in combination (Fig. 3b). The data indicated that both of the NF-κB sites play a role in the induction of FABP5 by HRG-β1. The functionality of the NF-κB sites and their responsiveness to HRG-β1 was further examined by chromatin immunoprecipitation assays (Fig. 3c). The observations demonstrated that both of the NF-κB subunits, p50 and p65, are recruited to both of the NF-κB sites of the FABP5 promoter in response to HRG-β1 in MCF-7 cells (Fig. 3c).
FIGURE 3.

Up-regulation of FABP5 by HRG-β1 is mediated by NF-κB. a, putative NF-κB response elements in the FABP5 promoter are shown. b, MCF-7 cells were transiently transfected with a luciferase reporter driven by 800 bp of the proximal FABP5 promoter (wild type (WT)) or FABP5 promoter constructs mutated at the NFκB sites, individually (mut131 or mut784) or in combination (mut131/mut784). Cells were treated with HRG-β1 (30 ng/ml) for 16 h, lysed, and analyzed for luciferase activity. Luciferase activity was normalized to β-galactosidase. The mutations of the NFκB sites at 784 and 131 are in bold: AGATCTCCGC and AGATCTCCCG. c, serum-starved MCF-7 cells were treated with HRG-β1 (30 ng/ml), and chromatin immunoprecipitation assays were carried out (see “Experimental Procedures”) using antibodies against the NF-κB subunit p50 and p65 or nonspecific IgG. Immunoprecipitated DNA was amplified using primers specific for the NF-κB elements of the FABP5 gene. d, MCF-7 cells were treated with the NF-κB inhibitor PDTC for 1 h before the addition of HRG-β1 (30 ng/ml, 24 h). Cell lysates were analyzed by immunoblots. e, serum-starved MCF-7 cells were treated with PDTC for 1 h before the addition of HRG-β1 (30 ng/ml, 4 h). FABP5 mRNA was assessed by Q-PCR. Data are the mean ± S.D. (n = 3). *, p = 0.0176 versus HRG-β1 alone. Inset, shown are immunoblots demonstrating decreased expression of IκB-α in the presence of PDTC. f, MCF-7 cells were transfected with a control vector or a vector harboring dominant negative IκB-α (SRIκB). 36 h later cells were serum-starved overnight and treated with HRG-β1 (30 ng/ml, 4 h). FABP5 mRNA levels were measured by Q-PCR. Data are the mean ± S.D. (n = 3). *, p = 0.006 versus HRG-β1-treated cells transfected with an empty vector (e.v.). Inset, shown is immunoblots demonstrating expression of SRIκB.
The involvement of NF-κB in mediating the induction of FABP5 by HRG-β1 was further demonstrated by the observations that treatment with the inhibitor of NF-κB activation pyrrolidine dithiobicarbamate (PDTC) or ectopic expression of the dominant negative construct for the NF-κB regulator IκBα, IkBα-S32A/S36A (SRIkBα (38)), markedly inhibited the ability of HRG-β1 to up-regulate FABP5 expression (Fig. 3, d–f). PDTC also inhibited the ability of HRG-β1 to activate the FABP5 promoter (supplemental Fig. S2).
HRG-β1-induced Up-regulation of FABP5 Is Mediated by ErbB2 in Conjunction with ErbB3 and ErbB4
ErbB2 is not itself activated by ligands, but it functions as a heterodimer with ligand-responsive EGFRs, most often ErbB3 and ErbB4 (42–44). The involvement of these EGFRs in the induction of FABP5 by HRG-β1 was, thus, examined. Treatment of MCF-7 cells with the ErbB2 inhibitor AG825, which blocked the ability of HRG-β1 to induce the phosphorylation of Akt1 (Fig. 4a), markedly decreased the expression of FABP5 (Fig. 4, a and b). To further assess the role of ErbB2 in controlling FABP5 expression, the expression level of the receptor was varied. Decreasing ErbB2 expression using shRNA methodologies suppressed HRG-β1-induced FABP5 expression (Fig. 4c), and ectopic overexpression of the receptor enhanced the ability of HRG-β1 to up-regulate FABP5 (Fig. 4d). Hence, ErbB2 is closely involved in HRG-β1-induced expression of FABP5.
FIGURE 4.

ErbB2, ErbB3, and ErbB4 are involved in regulating FABP5 expression. a, serum-starved MCF-7 cells were treated with the ErbB2 inhibitor AG825 for 1 h before the addition of HRG (30 ng/ml, 15 min.). Cell lysates were analyzed by immunoblots with antibodies recognizing the denoted proteins. b, serum-starved MCF-7 cells were treated AG825 for 1 h before the addition of HRG (30 ng/ml). FABP5 mRNA was measured by Q-PCR. Data are the means ± S.D. (n = 3). *, p = 0.058 versus HRG-β1 alone. c, MCF-7 cells were transiently transfected with a control vector or with expression vectors harboring ErbB2shRNA. 36 h after transfection, cells were serum-starved overnight and treated with HRG-β1 for 4 h. FABP5 mRNA levels were measured by Q-PCR. Data are the mean ± S.D. (n = 3). *, p = 0.0395 versus HRG-β1 treated control. Inset, immunoblots demonstrating decreased expression of ErbB2 are shown. d, MCF-7 cells were transfected with a control vector or expression vectors for wild-type ErbB2. 36 h later, cells were serum-starved overnight and treated with HRG-β1 for 4 h. FABP5 mRNA was measured by Q-PCR. *, p = 0.0207 versus HRG-β1 treated control. Inset, shown are immunoblots demonstrating expression of ErbB2. ev, empty vector. e, MCF-7 cells were transfected with a control vector or with expression vectors harboring shRNA for ErbB3 or ErbB4. 36 h after transfection, cells were serum-starved overnight and treated with HRG-β1 for 4 h. FABP5 mRNA levels were measured by Q-PCR. Data are the mean ± S.D. (n = 3). p = 0.029 (*) and p = 0.039 (**) versus HRG-β1-treated control. Inset, shown are immunoblots demonstrating decreased expression of ErbB3 and ErbB4.
To determine the dimerization partner of ErbB2 that is involved in the induction of FABP5 by HRG-β1, the expression of either ErbB3 or ErbB4 was decreased using corresponding shRNAs (Fig. 4e). Reducing the expression of either ErbB3 or ErbB4 inhibited HRG-β1-induced FABP5 expression. Taken together, the observations indicate that ErbB2, ErbB3, and ErbB4 are all involved in regulating FABP5 expression in response to HRG-β1 in MCF-7 cells.
HRG-β1 and NF-κB Orchestrate Cross-talk between FABP5 and KLF2
Several Krüppel-like factors (KLFs) have been implicated in playing important roles in cancer by either enhancing or suppressing cell proliferation (45–50). Of these, it has been reported that KLF2 inhibits cancer cell growth (49, 50). To examine possible cross-talk between KLF2 and FABP5, the effect of ectopic overexpression of KLF2 on expression of FABP5 in MCF-7 cells was examined. Overexpression of KLF2 (Fig. 5a) reduced FABP5 mRNA levels (Fig. 5b) and markedly decreased the level of FABP5 protein (Fig. 5c). Conversely, knock-down of KLF2 induced FABP5 expression and enhanced the ability of HRG-β1 to up-regulate FABP5 expression (Fig. 5d). In accordance with the conclusion that KLF2 suppresses FABP5 expression, overexpression of this factor suppressed the activity of the FABP5 promoter as well as its response to HTG-β1 (supplemental Fig. S3). KLF2 has been shown to suppress cell proliferation in T-cell leukemia cells and in ovarian cancer cells (49, 50). In accordance with these reported antiproliferative activities, overexpression of KLF2 in MCF-7 cells, suppressed HRG-β1-induced cell proliferation (Fig. 5e). Interestingly, although FABP5 expression was up-regulated by HRG-β1 (Fig. 1a), the growth factor decreased the expression of KLF2 (Fig. 5, f and g). In addition, although activation of NF-κB up-regulated FABP5, this transcription factor suppressed the expression of KLF2, as demonstrated by increased KLF2 levels upon blocking NF-κB activity by using either a chemical inhibitor or a dominant negative construct (Fig. 5, f and g). Moreover, HRG-β1 markedly increased KLF2 expression in cells in which NF-κB was inhibited (Fig. 5, f and g). These observations, thus, demonstrate that FABP5 expression is negatively regulated by KLF2 and that HRG-β1 and NF-κB inversely regulate the expression of FABP5 and KLF2. The data further suggest that EGFRs regulate the expression of KLF2 in a complex fashion, displaying both negative and positive regulation depending on the state of activation of NF-κB.
FIGURE 5.

KLF2 negatively regulates FABP5, and the two proteins are inversely regulated by HRG-β1 and NF-κB. a–c, MCF-7 cells were infected with adenoviruses harboring GFP or GFP-KLF2 for 48 h. Expression level of mRNA for KLF2 (a) or FABP5 (b) were measured by Q-PCR. Data are the mean ± S.D. (n = 3). *, p < 0.025. c, cells were lysed 24 or 48 h after transfection, and lysates were analyzed by immunoblots using denoted antibodies. d, MCF-7 cells were transfected with si-control or vector encoding Si-KLF2 (Santa Cruz Biotechnology, Inc.), and cells were treated with HRG-β1 (30 ng/ml, 6 h). Expression levels of FABP5 and the internal control cyclophilin were measured by quantitative PCR (FABP5, forward (gcagacccctctctgcac) and reverse (tcgcaaagctattcccactc); cyclophilin, forward (cctaaagcatacgggtcctg) and reverse (tttcactttgccaaacacca)). *, p = 0.0004 versus non-treated cells transfected with si-control; **, p = 0.0003 versus non-treated cells transfected with si-control; ***, p = 0.01 versus HRG-β-treated cells transfected with si-control. e, MCF-7 cells were infected with control adenovirus (Ad-GFP) or adenovirus overexpressing KLF2 (Ad-KLF2) and treated with HRG-β1 for 24 h. Cell proliferation was assessed by MTT assays. *, p = 0.001 versus non-treated cells infected with Ad-GFP; **, p < 0.001 versus si-control-infected untreated cells; ***, p < 0.001 versus Ad-GFP-infected, HRG-β1-treated cells. f, serum-starved MCF-7 cells were treated with the NF-κB inhibitor PDTC (100 μm) for 1 h before the addition of HRG-β1 (30 ng/ml, 4 h). KLF2 mRNA levels were measured by Q-PCR. Data are the mean ± S.D. (n = 3). p = 0.009 (*) and p = 0.05 (**) versus non-treated control. g, MCF-7 cells were transfected with a control vector or a vector harboring dominant negative IκB-α (SRIκB) for 36 h. ev, empty vector. Cells were serum-starved overnight and treated with HRG-β1 for 4 h. FABP5 mRNA levels were measured by Q-PCR. Data are the mean ± S.D. (n = 3). p = 0.015 (*) and p = 0.02 (**) versus empty vector-transfected non-treated empty vector control.
The FABP5/PPARβ/δ Pathway Is Critical for HRG-β1-induced Cell Proliferation
FABP5 functions by delivering ligands to PPARβ/δ, thereby enhancing the transcriptional activity of the receptor (23, 24, 33). The observations that HRG-β1 induces the expression of FABP5, thus, suggest that EGFR signaling may activate PPARβ/δ-mediated transcriptional regulation. As direct target genes for PPARβ/δ include genes involved in survival pathways, such as PDK1 (51, 52), and proliferation and angiogenesis, such as vascular endothelial growth factor (53), the FABP5/PPARβ/δ path may play a role in EGFR-induced carcinoma cell proliferation. Treatment of MCF-7 cells with HRG-β1 induced the expression of the direct PPARβ/δ target gene PDK1, and decreasing the expression of either FABP5 or PPARβ/δ inhibited the effect (Fig. 6, a and d). Hence, EGFR signaling activates the FABP5/PPARβ/δ pathway in these cells. To further examine the notion that HRG-β1 enhances the transcriptional activity of FABP5/PPARβ/δ, transcriptional activation assays using a luciferase reported driven by PPAR response elements were carried out. In these assays, to ensure that observed transcriptional activities emanated specifically from PPARβ/δ, cells were also transfected with an expression vector for this PPAR isotype. The data (supplemental Fig. S4) demonstrated that, indeed, HRG-β1 elicited the transcriptional activity of PPARβ/δ (supplemental Fig. S4). Attesting to the involvement of FABP5 in the response, decreasing the expression level of the binding protein hampered the ability of HRG-β1 to activate the reporter (supplemental Fig. S4).
FIGURE 6.

Induction of MCF-7 cell proliferation by HRG-β1 is mediated by FABP5 and PPARβ/δ. a and d, MCF-7 cells were infected with lentiviruses harboring shRNA for FABP5 (a) or PPARβ/δ (d). Insets, immunoblots demonstrate down-regulation of denoted proteins 3 days after infection. Three days after infection, cells were serum-starved before the addition of HRG-β1 for 4 h. Levels of PDK1 mRNA were measured by Q-PCR. Data are the mean ± S.D. (n = 3). *, p < 0.05 versus HRG-β1 control. ctr, control. b, c, and e, MCF-7 cells were infected with an empty lentivirus or lentivirus harboring shRNA for FABP5 (b and c) or PPARβ/δ (e). 3 days after infection, cells were treated with HRG-β1 for 24 h. Cell proliferation was assessed by bromodeoxyuridine (BRDU) incorporation (b and e) and by cell counting (c). Data are the mean ± S.D. p = 0.01 (*), p = 0.05 (**), and p = 0.02 (***) versus HRG-β1-treated controls.
As expected, treatment of MCF-7 cells with HRG-β1 markedly enhanced cell growth (Fig. 6, b, c, and e). Strikingly, the ability of HRG-β1 to induce cell proliferation was all but completely abolished upon decreasing the expression of either FABP5 or PPARβ/δ (Fig. 6, b, c, and e). Similar to the response of MCF-7 cells, reducing the expression level of FABP5 hampered the ability of HRG-b1 to enhance the proliferation of MDA-MB453, another ErbB2-positive mammary carcinoma cell (supplemental Fig. S5b). Hence, transcriptional signaling by the FABP5/PPARβ/δ pathway is a key mediator of EGFR-induced cell growth.
DISCUSSION
Amplification of ErbB2 and the resulting enhancement of EGFR signaling in mammary epithelium result in cellular transformation and tumorigenesis (3–8, 20, 21). EGFRs function through a complex array of cascades, including the PI3K/Akt and the ERK1/2 pathways. In many cells these pathways converge to activate the transcription factor NF-κB that, in turn, enhances cellular motility and invasion, survival, and proliferation (54–59). However, the nature of NF-κB target genes involved in these oncogenic activities remains incompletely understood.
The expression of the intracellular lipid-binding protein FABP5 is highly up-regulated in mammary tumors that arise from amplification of ErbB2 in the mouse model of breast cancer MMTV-neu (23, 24) as well as in human pancreas, breast, bladder, and prostate cancers (60, 61). FABP5 functions in conjunction with the nuclear receptor PPARβ/δ, a receptor whose activation protects cells against apoptosis and facilitates cell growth and migration (23, 24, 34–36). These considerations suggest that the FABP5/PPARβ/δ pathway may be closely involved in tumor development.
The data described here show that, in MCF-7 mammary carcinoma cells, activation of NF-κB by EGFRs, functioning through both the PI3K and the ERK pathways, up-regulates the expression of FABP5 through two NF-κB sites within the FABP5 promoter (Fig. 3). The observations show further that the increased expression of FABP5 brought about in response to EGFR signaling enhances the transcriptional activity of PPARβ/δ, as reflected by induction of the PPARβ/δ target gene PDK1 (Fig. 6). Strikingly, down-regulation of either FABP5 or its cognate receptor PPARβ/δ completely abolished the ability of HRG-β1 to enhance MCF-7 cell growth (Fig. 6, c and f). These observations establish a direct connection between EGFR signaling and FABP5 expression and demonstrate that the FABP/PPARβ/δ pathway plays a key role in EGFR-induced carcinoma cell proliferation. The reports that the PPARβ/δ target gene vascular endothelial growth factor is highly overexpressed in ErbB2-positive cells (62) as well as in FABP5-overexpressing prostate cancer cells (63, 64) support the notion that the FABP/PPARβ/δ pathway plays an important role in the development of ErbB2-driven as well as other tumors.
Additional data show that the expression of FABP5 is also regulated by KLF2, a member of the family of Krüppel-like factors (65, 66). Several KLFs have been implicated in roles in cancer biology. For example, in different contexts, KLF4 functions either as a tumor suppressor or as an oncogene, a function it exerts through suppression of the transcriptional repressor p53 (46, 67). The role of KLF2 in cancer is poorly understood, but it has been reported that this factor enhances cell quiescence (68) and inhibits Jurkat leukemia cell growth (49). The observations that KLF2 suppresses the expression of FABP5 (Fig. 5, a and b) suggest the possibility that KLF2 exerts anti-carcinogenic activities and that it does so through inhibition of the transcriptional activities of FABP5/PPARβ/δ. Interestingly, the data indicate that although EGFR signaling induces the expression of FABP5, it suppresses KLF2, and that both activities are mediated by NF-κB (Figs. 3 and 5, c and d). These observations are in accordance with the report that NF-κB suppresses the expression of KLF2 in endothelial cells, where the factor plays anti-inflammatory roles (69, 70). The data show further that although HRG-β1 suppresses KLF2 in the presence of active NF-κB, it up-regulates the expression of the factor when NF-κB is inhibited. A model for the mechanisms by which EGFRs orchestrate cross-talk between FABP5 and KLF2 to regulate proliferation is depicted in Fig. 7. The involvement of KLF2 in carcinogenesis, the mechanism by which KLF2 suppresses the expression of FABP5, and the molecular events that underlie the complex response of KLF2 to EGFR signaling remain to be clarified.
FIGURE 7.
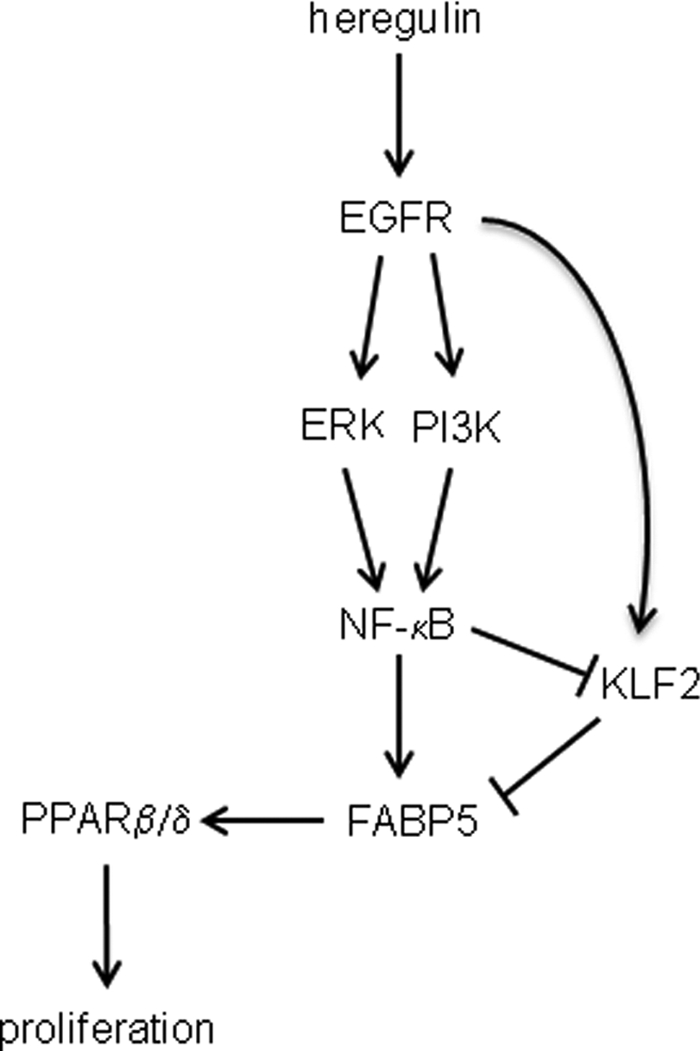
Model for the involvement of FABP5 in EGFR-induced cell proliferation. EGFR signaling through ERK and PI3K leads to activation of NF-κB, which in turn directly up-regulates the expression of FABP5. FABP5 augments the transcriptional activity of PPARβ/δ, resulting in enhanced cell survival and proliferation. Induction of FABP5 by EGFRs may be further enhanced by EGFR-induced suppression of KLF2, a negative regulator of FABP5. EGFR signaling regulates the expression of KLF2 in a complex manner. It suppresses KLF2 upon activation of NFκB but up-regulates KLF2 expression when NF-κB is inhibited.
Taken together, the observations point at FABP5 as a potential therapeutic target for treatment of ErbB2/HER2-positive breast cancer. Notably, although therapy using the HER2 antibodies trastuzumab/Herceptin is generally successful, not all HER2-positive cancers respond to trastuzumab, and some tumors develop resistance (71). Targeting FABP5 may bypass such a resistance. Although no antagonists for FABP5 currently exist, an inhibitor for the homologous protein FABP4 was recently reported (72). The development of compounds that target FABP5 could be assisted by the recent delineation of the structural features that underlie the ability of specific ligands to activate intracellular lipid-binding proteins (31, 32, 73).
Supplementary Material
Acknowledgments
We thank George Stark, Melanie Cobb, and Ruth Keri for providing plasmids and Youwei Zhang for antibodies.
This work was supported, in whole or in part, by National Institutes of Health Grants DK060684 and CA068150 (to N. N.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S5.
- EGFR
- epidermal growth factor receptor
- ERK
- extracellular signal-regulated kinase
- PPAR
- peroxisome proliferator-activated receptor
- PI3K
- phophatidylinositol 3-kinase
- iLBP
- intracellular lipid-binding protein
- FABP5
- fatty acid-binding protein 5
- HRG-β1
- heregulin-β1
- Q-PCR
- quantitative real-time PCR
- PDTC
- pyrrolidine dithiobicarbamate
- KLF
- Krüppel-like factors
- GFP
- green fluorescent protein
- shRNA
- short hairpin RNA
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- Ad
- adenovirus
- PIPES
- 1,4-piperazinediethanesulfonic acid.
REFERENCES
- 1.Hynes N. E., Lane H. A. (2005) Nat. Rev. Cancer 5, 341–354 [DOI] [PubMed] [Google Scholar]
- 2.Prenzel N., Fischer O. M., Streit S., Hart S., Ullrich A. (2001) Endocr. Relat. Cancer 8, 11–31 [DOI] [PubMed] [Google Scholar]
- 3.King C. R., Kraus M. H., Aaronson S. A. (1985) Science 229, 974–976 [DOI] [PubMed] [Google Scholar]
- 4.van de Vijver M. J., Peterse J. L., Mooi W. J., Wisman P., Lomans J., Dalesio O., Nusse R. (1988) N. Engl. J. Med. 319, 1239–1245 [DOI] [PubMed] [Google Scholar]
- 5.Slamon D. J., Clark G. M., Wong S. G., Levin W. J., Ullrich A., McGuire W. L. (1987) Science 235, 177–182 [DOI] [PubMed] [Google Scholar]
- 6.Slamon D. J., Godolphin W., Jones L. A., Holt J. A., Wong S. G., Keith D. E., Levin W. J., Stuart S. G., Udove J., Ullrich A. (1989) Science 244, 707–712 [DOI] [PubMed] [Google Scholar]
- 7.Reese D. M., Slamon D. J. (1997) Stem Cells 15, 1–8 [DOI] [PubMed] [Google Scholar]
- 8.Hung M. C., Matin A., Zhang Y., Xing X., Sorgi F., Huang L., Yu D. (1995) Gene 159, 65–71 [DOI] [PubMed] [Google Scholar]
- 9.Ménard S., Tagliabue E., Campiglio M., Pupa S. M. (2000) J. Cell Physiol. 182, 150–162 [DOI] [PubMed] [Google Scholar]
- 10.Sledge G. W., Jr., Miller K. D. (2003) Eur. J. Cancer 39, 1668–1675 [DOI] [PubMed] [Google Scholar]
- 11.Di Fiore P. P., Pierce J. H., Fleming T. P., Hazan R., Ullrich A., King C. R., Schlessinger J., Aaronson S. A. (1987) Cell 51, 1063–1070 [DOI] [PubMed] [Google Scholar]
- 12.Kokai Y., Myers J. N., Wada T., Brown V. I., LeVea C. M., Davis J. G., Dobashi K., Greene M. I. (1989) Cell 58, 287–292 [DOI] [PubMed] [Google Scholar]
- 13.Alimandi M., Romano A., Curia M. C., Muraro R., Fedi P., Aaronson S. A., Di Fiore P. P., Kraus M. H. (1995) Oncogene 10, 1813–1821 [PubMed] [Google Scholar]
- 14.Siegel P. M., Ryan E. D., Cardiff R. D., Muller W. J. (1999) EMBO J. 18, 2149–2164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsai P. W., Shiah S. G., Lin M. T., Wu C. W., Kuo M. L. (2003) J. Biol. Chem. 278, 5750–5759 [DOI] [PubMed] [Google Scholar]
- 16.Yao J., Xiong S., Klos K., Nguyen N., Grijalva R., Li P., Yu D. (2001) Oncogene 20, 8066–8074 [DOI] [PubMed] [Google Scholar]
- 17.Mazumdar A., Adam L., Boyd D., Kumar R. (2001) Cancer Res. 61, 400–405 [PubMed] [Google Scholar]
- 18.Lupu R., Cardillo M., Cho C., Harris L., Hijazi M., Perez C., Rosenberg K., Yang D., Tang C. (1996) Breast Cancer Res. Treat. 38, 57–66 [DOI] [PubMed] [Google Scholar]
- 19.Lewis G. D., Lofgren J. A., McMurtrey A. E., Nuijens A., Fendly B. M., Bauer K. D., Sliwkowski M. X. (1996) Cancer Res. 56, 1457–1465 [PubMed] [Google Scholar]
- 20.Guy C. T., Webster M. A., Schaller M., Parsons T. J., Cardiff R. D., Muller W. J. (1992) Proc. Natl. Acad. Sci. U.S.A. 89, 10578–10582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li B., Rosen J. M., McMenamin-Balano J., Muller W. J., Perkins A. S. (1997) Mol. Cell. Biol. 17, 3155–3163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muller W. J., Sinn E., Pattengale P. K., Wallace R., Leder P. (1988) Cell 54, 105–115 [DOI] [PubMed] [Google Scholar]
- 23.Schug T. T., Berry D. C., Shaw N. S., Travis S. N., Noy N. (2007) Cell 129, 723–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schug T. T., Berry D. C., Toshkov I. A., Cheng L., Nikitin A. Y., Noy N. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 7546–7551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Storch J., Corsico B. (2008) Annu. Rev. Nutr. 28, 73–95 [DOI] [PubMed] [Google Scholar]
- 26.Noy N. (2000) Biochem. J. 348, 481–495 [PMC free article] [PubMed] [Google Scholar]
- 27.Veerkamp J. H., Maatman R. G. (1995) Prog. Lipid Res. 34, 17–52 [DOI] [PubMed] [Google Scholar]
- 28.Dong D., Ruuska S. E., Levinthal D. J., Noy N. (1999) J. Biol. Chem. 274, 23695–23698 [DOI] [PubMed] [Google Scholar]
- 29.Budhu A. S., Noy N. (2002) Mol. Cell. Biol. 22, 2632–2641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Manor D., Shmidt E. N., Budhu A., Flesken-Nikitin A., Zgola M., Page R., Nikitin A. Y., Noy N. (2003) Cancer Res. 63, 4426–4433 [PubMed] [Google Scholar]
- 31.Sessler R. J., Noy N. (2005) Mol. Cell 18, 343–353 [DOI] [PubMed] [Google Scholar]
- 32.Ayers S. D., Nedrow K. L., Gillilan R. E., Noy N. (2007) Biochemistry 46, 6744–6752 [DOI] [PubMed] [Google Scholar]
- 33.Tan N. S., Shaw N. S., Vinckenbosch N., Liu P., Yasmin R., Desvergne B., Wahli W., Noy N. (2002) Mol. Cell. Biol. 22, 5114–5127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tan N. S., Michalik L., Noy N., Yasmin R., Pacot C., Heim M., Flühmann B., Desvergne B., Wahli W. (2001) Genes Dev. 15, 3263–3277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tan N. S., Icre G., Montagner A., Bordier-ten-Heggeler B., Wahli W., Michalik L. (2007) Mol. Cell. Biol. 27, 7161–7175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tan N. S., Michalik L., Desvergne B., Wahli W. (2004) Expert Opin. Ther. Targets 8, 39–48 [DOI] [PubMed] [Google Scholar]
- 37.Robbins D. J., Zhen E., Cheng M., Xu S., Vanderbilt C. A., Ebert D., Garcia C., Dang A., Cobb M. H. (1993) J. Am. Soc. Nephrol. 4, 1104–1110 [DOI] [PubMed] [Google Scholar]
- 38.Shultz D. B., Fuller J. D., Yang Y., Sizemore N., Rani M. R., Stark G. R. (2007) J. Interferon Cytokine Res. 27, 875–884 [DOI] [PubMed] [Google Scholar]
- 39.Biswas D. K., Iglehart J. D. (2006) J. Cell Physiol. 209, 645–652 [DOI] [PubMed] [Google Scholar]
- 40.Singh S., Shi Q., Bailey S. T., Palczewski M. J., Pardee A. B., Iglehart J. D., Biswas D. K. (2007) Mol. Cancer Ther. 6, 1973–1982 [DOI] [PubMed] [Google Scholar]
- 41.Biswas D. K., Shi Q., Baily S., Strickland I., Ghosh S., Pardee A. B., Iglehart J. D. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 10137–10142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Plowman G. D., Green J. M., Culouscou J. M., Carlton G. W., Rothwell V. M., Buckley S. (1993) Nature 366, 473–475 [DOI] [PubMed] [Google Scholar]
- 43.King C. R., Borrello I., Bellot F., Comoglio P., Schlessinger J. (1988) EMBO J. 7, 1647–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sliwkowski M. X., Schaefer G., Akita R. W., Lofgren J. A., Fitzpatrick V. D., Nuijens A., Fendly B. M., Cerione R. A., Vandlen R. L., Carraway K. L., 3rd (1994) J. Biol. Chem. 269, 14661–14665 [PubMed] [Google Scholar]
- 45.Simmen R. C., Pabona J. M., Velarde M. C., Simmons C., Rahal O., Simmen F. A. (2010) J. Endocrinol. 204, 223–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rowland B. D., Peeper D. S. (2006) Nat. Rev. Cancer 6, 11–23 [DOI] [PubMed] [Google Scholar]
- 47.Ghaleb A. M., Nandan M. O., Chanchevalap S., Dalton W. B., Hisamuddin I. M., Yang V. W. (2005) Cell Res. 15, 92–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Frigo D. E., Sherk A. B., Wittmann B. M., Norris J. D., Wang Q., Joseph J. D., Toner A. P., Brown M., McDonnell D. P. (2009) Mol. Endocrinol. 23, 1385–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu J., Lingrel J. B. (2004) Oncogene 23, 8088–8096 [DOI] [PubMed] [Google Scholar]
- 50.Wang F., Zhu Y., Huang Y., McAvoy S., Johnson W. B., Cheung T. H., Chung T. K., Lo K. W., Yim S. F., Yu M. M., Ngan H. Y., Wong Y. F., Smith D. I. (2005) Oncogene 24, 3875–3885 [DOI] [PubMed] [Google Scholar]
- 51.Di-Poï N., Michalik L., Tan N. S., Desvergne B., Wahli W. (2003) J. Steroid Biochem. Mol. Biol. 85, 257–265 [DOI] [PubMed] [Google Scholar]
- 52.Di-Poï N., Tan N. S., Michalik L., Wahli W., Desvergne B. (2002) Mol. Cell 10, 721–733 [DOI] [PubMed] [Google Scholar]
- 53.Wang D., Wang H., Guo Y., Ning W., Katkuri S., Wahli W., Desvergne B., Dey S. K., DuBois R. N. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 19069–19074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Prasad S., Ravindran J., Aggarwal B. B. (2010) Mol. Cell. Biochem. 336, 25–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen F., Castranova V. (2007) Cancer Res. 67, 11093–11098 [DOI] [PubMed] [Google Scholar]
- 56.Madrid L. V., Mayo M. W., Reuther J. Y., Baldwin A. S., Jr. (2001) J. Biol. Chem. 276, 18934–18940 [DOI] [PubMed] [Google Scholar]
- 57.Lee F. S., Peters R. T., Dang L. C., Maniatis T. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 9319–9324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huang C., Jacobson K., Schaller M. D. (2004) J. Cell Sci. 117, 4619–4628 [DOI] [PubMed] [Google Scholar]
- 59.Katso R., Okkenhaug K., Ahmadi K., White S., Timms J., Waterfield M. D. (2001) Annu. Rev. Cell Dev. Biol. 17, 615–675 [DOI] [PubMed] [Google Scholar]
- 60.Adamson J., Morgan E. A., Beesley C., Mei Y., Foster C. S., Fujii H., Rudland P. S., Smith P. H., Ke Y. (2003) Oncogene 22, 2739–2749 [DOI] [PubMed] [Google Scholar]
- 61.Morgan E. A., Forootan S. S., Adamson J., Foster C. S., Fujii H., Igarashi M., Beesley C., Smith P. H., Ke Y. (2008) Int. J. Oncol. 32, 767–775 [PubMed] [Google Scholar]
- 62.Lorusso V. (2008) Biologics 2, 813–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jing C., Beesley C., Foster C. S., Rudland P. S., Fujii H., Ono T., Chen H., Smith P. H., Ke Y. (2000) Cancer Res. 60, 2390–2398 [PubMed] [Google Scholar]
- 64.Jing C., Beesley C., Foster C. S., Chen H., Rudland P. S., West D. C., Fujii H., Smith P. H., Ke Y. (2001) Cancer Res. 61, 4357–4364 [PubMed] [Google Scholar]
- 65.Pearson R., Fleetwood J., Eaton S., Crossley M., Bao S. (2008) Int. J. Biochem. Cell Biol. 40, 1996–2001 [DOI] [PubMed] [Google Scholar]
- 66.Atkins G. B., Jain M. K. (2007) Circ. Res. 100, 1686–1695 [DOI] [PubMed] [Google Scholar]
- 67.Rowland B. D., Bernards R., Peeper D. S. (2005) Nat. Cell Biol. 7, 1074–1082 [DOI] [PubMed] [Google Scholar]
- 68.Buckley A. F., Kuo C. T., Leiden J. M. (2001) Nat. Immunol. 2, 698–704 [DOI] [PubMed] [Google Scholar]
- 69.Kumar A., Lin Z., SenBanerjee S., Jain M. K. (2005) Mol. Cell. Biol. 25, 5893–5903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.SenBanerjee S., Lin Z., Atkins G. B., Greif D. M., Rao R. M., Kumar A., Feinberg M. W., Chen Z., Simon D. I., Luscinskas F. W., Michel T. M., Gimbrone M. A., Jr., García-Cardeña G., Jain M. K. (2004) J. Exp. Med. 199, 1305–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mariani G., Fasolo A., De Benedictis E., Gianni L. (2009) Nat. Clin. Pract. Oncol. 6, 93–104 [DOI] [PubMed] [Google Scholar]
- 72.Furuhashi M., Tuncman G., Görgün C. Z., Makowski L., Atsumi G., Vaillancourt E., Kono K., Babaev V. R., Fazio S., Linton M. F., Sulsky R., Robl J. A., Parker R. A., Hotamisligil G. S. (2007) Nature 447, 959–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gillilan R. E., Ayers S. D., Noy N. (2007) J. Mol. Biol. 372, 1246–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.