

Abstract
Virus infection elicits a robust innate antiviral response dominated by the production of type 1 IFN. In non-professional innate immune cells such as fibroblasts, type 1 IFN is rapidly produced following the recognition of viral dsRNA and the subsequent activation of the constitutively expressed transcription factor IFN regulatory factor 3 (IRF3). While origin, localization and length are factors in mediating dsRNA recognition and binding by cellular dsRNA binding proteins, the biological significance of differential dsRNA binding is unclear, since the subsequent signaling pathways converge on IRF3. Here, we show a dsRNA length dependent activation of IRFs, IFNs and IFN stimulated genes in mouse fibroblasts. The length dependence was exacerbated in fibroblasts deficient in the mitochondria-associated adaptor IPS-1 and IRF3, suggesting that antiviral gene induction mediated by short and long dsRNA molecules is predominantly IPS-1 and IRF3-dependent and –independent, respectively. Furthermore, we provide evidence of an innate antiviral response in fibroblasts in the absence of both IRF3 and type 1 IFN induction. Even with these key modulators missing, a 60% to 90% inhibition of virus replication was observed following 24-hour treatment with short or long dsRNA molecules, respectively. These data provide evidence of a novel antiviral pathway that is dependent on dsRNA length, but independent of the type 1 IFN system.
Introduction
Viral infection of susceptible cells leads to the induction of a cellular antiviral response aimed at limiting virus replication and spread. DsRNA, a byproduct in the replication cycle of virtually all viruses (1) is a potent inducer of innate and adaptive immune responses. Three different families of pattern recognition receptors, the toll-like receptors (TLRs), the retinoic acid-inducible gene-I (RIG-I)3-like receptors (RLRs) and the nucleotide oligomerization domain (NOD)-like receptors (NLRs) bind dsRNA and initiate cellular signaling pathways (2). The TLRs and RLRs elicit type 1 interferon (IFN) and cytokine production while NLRs promote interleukin 1β maturation through caspase-1 activation. In non-professional innate immune cells such as fibroblasts, TLR3 and the RLRs RIG-I and MDA-5 (melanoma differentiation-associated gene 5) bind dsRNA and mediate antiviral signaling through the adaptors TRIF (Toll/Interleukin-1 receptor domain-containing adaptor inducing IFNβ) and IPS-1 (IFNβ promoter stimulator 1), respectively (2). Both adaptors subsequently activate the cellular transcription factors NF-κB and IFN regulatory factor 3 (IRF3), leading to IFNβ and cytokine production. Similar to the cytoplasmic RLRs, protein kinase regulated by RNA (PKR) also binds dsRNA and activates IRF3 through IPS-1 (3). IFNβ signaling through the JAK-STAT pathway, which includes IRF9 as an essential component, leads to the induction of IRF7 and amplification of the cellular antiviral response through the induction of IFNα species and IFN stimulated genes (ISGs) (4).
Recent studies have begun to characterize the dsRNA binding properties of TLR3, PKR, MDA-5 and RIG-I. Binding of dsRNA to TLR3 depends strongly on pH and dsRNA length, suggesting that TLR3 signaling initiates within an endosome through a pH-dependent binding mechanism and that sufficient dsRNA length (approximately 45 bp) is required to bind to both N-terminal and C-terminal regions of the TLR3 ectodomain (5-8). A similar minimal length dependence was observed for dsRNA to bind two PKR monomers and elicit PKR autophosphorylation and activation (9, 10). With the cytosolic RLRs RIG-I and MDA-5, both dsRNA origin and length influence recognition and binding. RIG-I preferentially recognizes RNA from a variety of RNA viruses while MDA-5 binds the dsRNA mimic polyinosinic polycytidylic acid (poly IC) and picornavirus RNA (11). Furthermore, RIG-I and MDA-5 were shown to preferentially bind to short and long dsRNA molecules, respectively (12). Regardless of which cellular protein senses the dsRNA, current models suggest that activation of all sensors leads to IRF3 activation and subsequence ISG and type 1 IFN induction.
The importance of IRF3 in the early and late phases of type 1 IFN expression and antiviral immunity was demonstrated through the generation of IRF3 and IRF3&9 null mice (4). Mice deficient for IRF3 were more vulnerable to virus infection and IFN production was reduced 20 to 50 fold. In the absence of both IRF3 and IRF9, type 1 IFN production was completely ablated (4). Since IRF7 gene expression is dependent on type 1 IFN signaling, levels of this transcription factor were significantly diminished in cells lacking IRF3 and absent in cells lacking IRF3 and IRF9 following virus infection. These studies led to a model in which constitutive expression of low levels of type 1 IFN in uninfected cells supports basal expression of equally low levels of IRF7. Upon viral infection, activation of IRF3 initiates a positive feedback loop resulting in robust IFNβ, IRF7, IFNα and ISG induction (13). Subsequent studies in IRF7 and IRF3&7 null mice confirmed the requirement for IRF3 and IRF7 in virus-mediated IFNβ and IFNα production, respectively (14).
While the prototypic response to virus infection and generation of dsRNA in non-professional innate immune cells is the activation of IRF3 and subsequent production of type 1 IFN, induction of ISGs or an antiviral response has been observed in the absence of either IRF3 or IFN production. Infection of IRF3-/- mouse embryonic fibroblasts (MEFs) with Newcastle disease virus induced a panel of IRF3-independent direct response genes, including IFN-inducible p200 family proteins p203 and p204 and Mx2 (15). Although siRNA mediated knockdown of IRF3 in a human hepatoma cell line dampened ISG induction in response to poly IC and Sendai virus infection, it had no effect on ISG56 and MxA induction in response to Sin Nombre virus particle treatment (16). Similarly, expression of a dominant negative form of IRF3 in 293 cells failed to impact on the ability of poly IC to block hepatitis C virus replication (17). Finally, consistent with previous observations (4), IRF3-/- mice were more vulnerable to infection with West Nile virus (WNv) than their wild type counterparts (18), despite the ability of this virus to elicit type 1 IFN production in knock out animals (18, 19). While these studies suggest that IRF3 is important, but not essential, for virus-mediated type 1 IFN production, IRF3 has been shown to play a critical role in induction of ISGs and an antiviral response in the absence of IFN production (20-22). This activity stems from the ability of IRF3 to bind directly to the promoter region of a subset of ISGs (23).
Despite recent advancements in characterizing viral dsRNA mediated antiviral immunity in non-professional innate immune cells, two issues remain outstanding. First, the biological consequence of differential dsRNA recognition remains unclear, particularly since downstream signaling pathways converge on IRF3. Second, while both IRF3 and type 1 IFN play central roles in mediating the antiviral response to dsRNA and can each function in the absence of the other, it is unclear whether dsRNA can induce antiviral activity in the absence of both IRF3 and type 1 IFN. Here we provide evidence that the innate antiviral response to viral dsRNA is length dependent in wild type mouse fibroblasts and that the length dependence is exacerbated in the absence of IRF3. Furthermore, we provide evidence of an IRF3 and type 1 IFN independent antiviral response that is more robustly activated by longer dsRNA molecules.
Materials and Methods
Cells and viruses
Murine embryonic fibroblasts (MEFs) were derived from wildtype (WT) C57Bl/6, IRF3-/- (4), IRF3&9-/- (4), IPS-1-/- and IPS-1+/+ littermate control mice and were maintained in α -minimal essential medium (MEM) supplemented with 10% FBS, 100 U mL-1 penicillin, 100 μg mL-1 streptomycin and 2mM L-glutamine. Experiments were performed with cells at passage 3-8. All cells were incubated at 37°C in a humidified 5% CO2 incubator. Vesicular stomatitis virus expressing green fluorescent protein (VSVgfp; kindly provided by B. Lichty), Herpes simplex type 1 (strain KOS) expressing green fluorescent protein (HSV-1gfp) (24) and WNv (Kunjin subtype; kindly provided by M. Diamond) were propagated on Vero cells (American Type Culture Collection).
DsRNA synthesis and treatments
DsRNA was synthesized by in vitro transcription using the Megascript RNAi kit (Ambion). 1 μg of PCR fragments amplified from portions of the cloned WNv genome was used as a template (Table I). DsRNA lengths of 200, 500 and 1000 bp (ν200, ν500, ν1000) were derived from the E protein sequence and a 3000 bp dsRNA fragment was derived from the NS3-NS4B sequence (ν3000). The average length for the poly IC (GE Healthcare, UK) used in this study was approximately 4000 bp as determined by marker size comparison using agarose gel electrophoresis and a 1kb Plus DNA ladder (Fermentas, ON).
Table I1.
| dsRNA | Source gene | Length (bp) | Primers2 |
|---|---|---|---|
| ν200 | WNv E | 200 | Sense: TCCTCCAACTGCGAGAAACGTG |
| Antisense: AAAGGAGCGCAGAGACTAGCCG | |||
| ν500 | WNv E | 500 | Sense: TCCTCCAACTGCGAGAAACGTG |
| Antisense: TGGCACGGATGGACCTTG | |||
| ν1000 | WNv E | 1000 | Sense: TCCTCCAACTGCGAGAAACGTG |
| Antisense: ACACATGCGCCAAATTTGCC | |||
| ν3000 | NS3-NS4B | 3000 | Sense: CATGACAACCAACCCCCACGCATGATG |
| Antisense: GCGGGCGTGATGGTTGAAGGTGT | |||
Primer sequences used for amplifying PCR product templates for dsRNA in vitro transcription.
All primers included a T7 sequence tag (5′ taatacgactcactataggg 3′) used by the T7 polymerase during dsRNA synthesis.
Cells were treated with dsRNA in equal molar amounts to ensure an equal number of molecules per dsRNA length, unless otherwise noted. DsRNA treatments were performed in serum free OptiMEM media (Gibco) in the presence of 50 μg/mL DEAE-dextran (Pharmacia) for 1h followed by additional indicated amounts of time in full growth media, unless otherwise noted. DEAE-dextran is a cationic polymer that binds negatively charged nucleic acids and enables a closer association between the negatively charged cell membrane and the nucleic acid of interest (25). In all experiments, DEAE-dextran was utilized in dsRNA-untreated controls to ensure that the polymer alone was not influencing subsequent cellular responses.
DsRNA immunofluorescence microscopy
WT MEFs were seeded on glass coverslips to ∼60% confluency and allowed to attach over night. Cells were then infected with Kunjin virus at an MOI of 10 for 1.5h, after which the virus was removed and fresh media added to the wells. As a positive control, cells were treated with 100 μg mL-1 poly IC for 8h. At specific time points cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and blocked in blocking buffer [3% BSA, 3% goat serum, 0.02% Tween-20 in PBS]. Coverslips were incubated with the mouse anti-dsRNA antibody, J2, (English and Scientific Consulting, Hungary) at a 1:200 dilution, followed by an Alexafluor 488 labeled anti-mouse secondary antibody, also at 1:200 (Molecular Probes, OR). All antibody dilutions were performed in blocking buffer. Nuclei were stained with Hoechst stain. Images were captured using a Leica DM-IRE2 inverted microscope.
DsRNA immunoblot
RNA was harvested from cells infected with Kunjin virus or treated with dsRNA as indicated using Trizol reagent (Invitrogen). 20μg RNA was electrophoresed in a 10% non-denaturing polyacrylamide gel and transferred onto a nylon membrane (Hybond N+, GE Healthcare) using a semi dry transfer apparatus. The membranes were blocked with 5% milk in TBS, blotted with the J2 antibody followed by a goat anti-mouse IgG secondary antibody. DsRNA was visualized by enhanced chemiluminescence system. In vitro transcribed RNA was electrophoresed on the same gel as a size comparison for samples.
PCR arrays
RNA was isolated from dsRNA treated cells using Trizol reagent, and 2.5 μg of RNA was DNase treated using the DNA-free™ kit (Ambion, TX). RNA integrity and quantity was measured using the Agilent 2100 Bio-Analyzer (Agilent, Santa Clara, CA). 300 ng of high quality, total RNA was reverse transcribed using the RT2 First Strand kit (SABiosciences, MD). The cDNA was applied to the Mouse Interferons and Receptors RT2Profiler™ PCR Array (SABiosciences) as per the manufacturer's instructions. The arrays were run on the ABI PRISM 7900HT Sequence Detection System using the Sequence Detector Software version 2.2 (Applied Biosystems). Data were analyzed using the online SABiosience RT2 Profiler™ PCR array data analysis tool, based on the ΔΔCt method. Specifically, gene expression was normalized to five housekeeping genes (Gusb, Hprt, Hspcb, GAPDH, β actin) and expressed as fold change over the control group (cells treated with DEAE-dextran alone).
Quantitative RT-PCR
300 ng of total RNA was reverse transcribed with 0.2 ng of random 6mer primer and 50 U of Superscript II (Invitrogen) in a total reaction volume of 20 μL. Real-time quantitative PCR was performed in triplicate, in a total volume of 25 μL, using Universal PCR Master Mix and gene specific Taqman primers (Applied Biosystems). Data were analyzed using the ΔΔCt method. Specifically, gene expression was normalized to the housekeeping gene (GAPDH) and expressed as fold change over the control group (cells treated with DEAE-dextran alone).
Antiviral assays
Cells were seeded in 12-well dishes and treated with serial dilutions of nM quantities of dsRNA for specified lengths of time. Cells were subsequently infected with 0.1 pfu/cell VSVgfp for 1h in serum-free media. This amount of virus was determined to be the maximal dose for which signal saturation in untreated cells did not occur. The viral inoculum was then removed and replaced with DMEM containing 1% methylcellulose. GFP fluorescence intensity was measured 24 hours later on a Typhoon Trio (GE Healthcare) and quantified using the ImageQuant™ TL software. A dose response curve was generated for each dsRNA molecule and an EC50 value was calculated using GraphPad Prism software. No differences in VSVgfp infection rates in untreated cells were observed (Supplementary Figure 1).
Statistical analysis
Data were expressed as means ± standard error of the mean (unless otherwise indicated). Statistical analysis was performed using a one-way analysis of variance (ANOVA) with a tukey's post hoc test for pair-wise comparisons, a Dunnett's post test for comparisons with control treatments, and an unpaired t-test in the case of a comparison between two values. All statistical analyses were performed using GraphPad InStat. A p value of <0.05 was considered statistically significant.
Results
West Nile virus infection of fibroblasts and epithelial cells produces long, stable dsRNA molecules
While dsRNA was thought to be produced in virtually all virally-infected cells, a study using a dsRNA-specific antibody suggested that dsRNA can be detected following infection with positive-strand RNA, dsRNA or DNA viruses but not with negative-strand RNA viruses (26). However, a recent study showed that Vesicular stomatitis virus (VSV), a negative strand RNA virus, is capable of detectable dsRNA production (12), suggesting that viruses of all classification likely produce detectable amounts of dsRNA of various lengths. WNv (strain Kunjin) is a positive-strand RNA virus that has been shown by immunofluorescence microscopy to produce dsRNA following infection of Vero monkey kidney epithelial cells; however the length of the dsRNA produced not determined (27). In the present study abundant dsRNA was also detected by immunofluorescence microscopy in Kunjin virus infected wild type MEFs by 8 hours post infection using the J2 anti-dsRNA antibody (Figure 1A). As a control, poly IC was utilized. Furthermore, in both MEF and Vero cells, dsRNA of high molecular weight (>3000 bp) could be detected in extracts as late as 48 hours post infection by immunoblot analysis using the same antibody (Figure 1B). These data confirm the production of long, stable dsRNA molecules in WNv infected cells.
Figure 1. WNv infection leads to the generation of intracellular dsRNA.
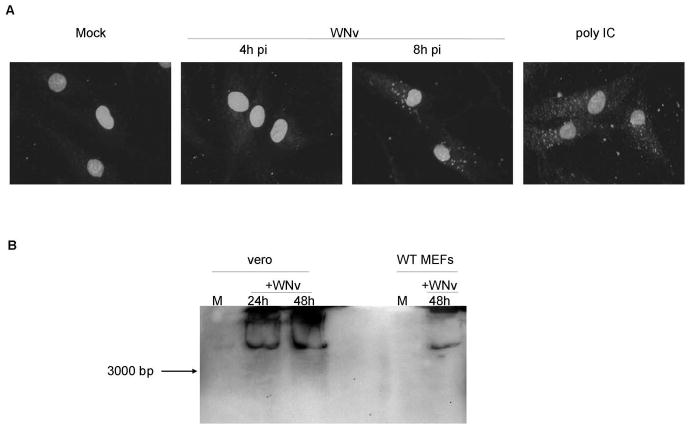
A, Immunofluorescence microscopy of wild type (WT) MEFs infected with WNv strain Kunjin (MOI=10) or treated with poly IC (100 μg/mL) using the J2 anti-dsRNA antibody. B, Immunoblot analysis of Vero cells and WT MEFs infected with Kunjin virus (MOI 5 and 10, respectively) using the J2 anti-dsRNA antibody. The arrow indicates the running distance of a 3000 bp sized in vitro transcribed dsRNA molecule.
In vitro transcribed dsRNA molecules of different lengths are stable within treated cells
To assess the biological consequence of dsRNA length, dsRNA molecules of specific lengths (200, 500, 1000 and 3000 base pairs) were in vitro transcribed from cloned fragments of the WNv genome. These particular lengths were chosen as similar sized dsRNA molecules have been observed in virally infected cells, such as those infected with reovirus, VSV, encephalomyocarditis virus (12) and WNv, as determined from the present study. Discrete bands of the appropriate size were detected on a 1% agarose gel (Figure 2A). The 3000 bp molecule consistently separated into two fragments. In contrast, poly IC ranged in length from 500 to 5000 bp, with the average length determined to be approximately 4000 bp. To ensure that the in vitro transcribed dsRNA molecules were stable within treated cells, similar to that observed with virus infection, immunoblot analysis using the J2 antibody was performed on extracts harvested 6 hours post dsRNA treatment. Intracellular dsRNA was relatively stable, particularly the 200, 500 and 1000 bp species (Figure 2B). While degradation of the 3000 bp species was evident at this time point, the predominant species observed were approximately 2000 to 3000 bp. Thus, in vitro transcribed dsRNA is relatively stable within treated cells, over a range of dsRNA lengths. Furthermore, dsRNA of all lengths entered into cells with equal efficiency (Supplementary Figure 2 and data not shown) and by one hour post-treatment, all cell-associated dsRNA had internalized (data not shown).
Figure 2. In vitro transcribed dsRNA of specific lengths derived from cloned fragments of the WNv genome is relatively stable within transfected cells.
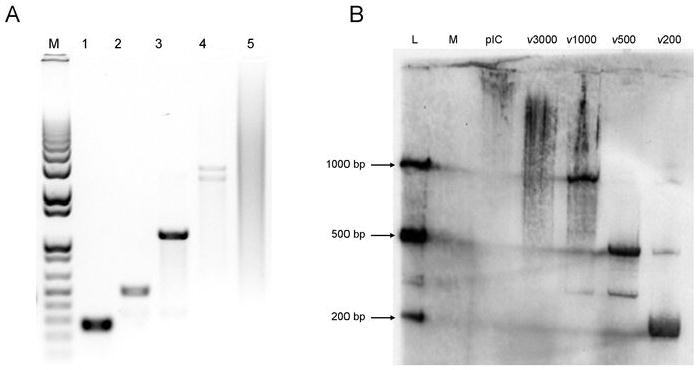
A, DsRNA of 200 (lane 1), 500 (lane 2), 1000 (lane 3) and 3000 (lane 4) bp lengths was generated using the WNv genome as a template. Approximately 500 ng of each dsRNA was separated on a 1% agarose gel and visualized by ethidium bromide staining. As dsRNA runs slower than DNA, all dsRNA appear to be of the appropriate length when compared to a 1kb DNA marker (lane M). 4 μg of poly IC was also run on the gel (lane 5), with the average length determined to be approximately 4000 bp. B, WT MEFs were mock treated (M) or treated with 1 μg/mL poly IC (pIC) or in vitro transcribed dsRNA of four lengths (ν200, ν500, ν1000 or ν3000) for 6h, after which RNA was extracted. A dsRNA immunoblot was performed using 20μg RNA and dsRNA was detected using the J2 anti-dsRNA antibody. A ladder of 25 ng in vitro transcribed dsRNA was included to determine size (L).
Induction of IRFs, IFNs and ISGs are length dependent but IRF3 independent
Recent evidence suggests that recognition of dsRNA by PKR, TLR3, RIG-I and MDA5 is length dependent (9, 12, 28). However, in fibroblasts, signaling pathways activated by these proteins converge on IRF3 to mediate IFN and ISG induction. Little is understood regarding the biological significance of differential recognition of dsRNA molecules, particularly given that recognition of dsRNA by these four proteins leads to IRF3 activation. To specifically address this issue, we utilized equal molar amounts of dsRNA of various lengths, to ensure that the only variable was the dsRNA length and not the number of dsRNA molecules. However, when equal concentrations (by weight) of dsRNA were tested, length dependent gene induction as described below was still observed (data not shown). Wild type MEFs were treated with 1.5 nM of poly IC or dsRNA of 200, 1000 or 3000 bp lengths. This amount of dsRNA was determined to elicit a complete antiviral response in wild type MEFs (refer to Figure 4). RNA harvested 6 hours post treatment was reverse transcribed and the cDNAs were assayed using Mouse Interferons and Receptors RT2Profiler™ PCR Arrays (Supplementary Figure 3). In addition to housekeeping controls, this array contains 84 genes whose expression is controlled by or involved in signal transduction mediated by IFN ligands and receptors. Included on the array are 21 IFN and cytokine species, 35 IFN and cytokine receptors, 8 IRFs, 2 IRF binding proteins and 18 ISGs. Shown in Figure 3 are ISGs whose expression profile changed with treatment (with ADAR serving as a representative of an ISGs whose expression profile did not change), all IRFs, all type 1 IFN species and cytokines whose expression profile changed with treatment. No receptors are reported as none showed changes in expression profile with treatment. A length dependent induction of a subset of ISGs, IRFs and IFNs was observed, with dsRNA of 200 bp length eliciting a modest induction and poly IC demonstrating the most robust induction (Figure 3A). When IRF3-/- MEFs were treated with 3 nM of poly IC or dsRNA of various lengths (the concentration of dsRNA required to elicit a complete antiviral response in IRF3-/- MEFs; refer to Figure 4), the same subset of ISG, IRF and IFN transcripts were induced (Figure 3B), also in a length dependent fashion. However, dsRNA of 200 bp length was uniformly diminished in its capacity to stimulate transcription of most genes represented on the array, while treatment with poly IC and dsRNA of 3000 bp length dsRNA stimulated gene transcription to a similar capacity. Of note, IRF7 and IFNβ levels remained elevated in IRF3-/- MEFs, consistent with previous observations (4, 13).
Figure 4. Induction of Cxcl10, Ifit1, Irf7 and IFNb1 are more dependent on IRF3 following stimulation with short dsRNA molecules.
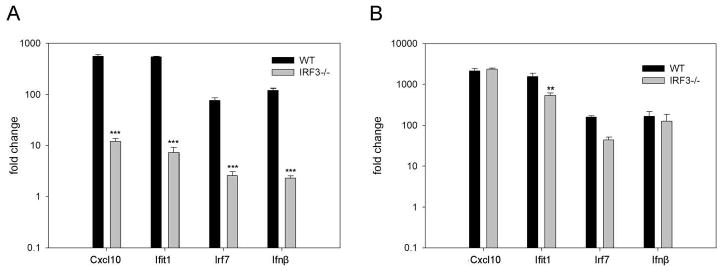
WT and IRF3-/- MEFs were treated with ν200 (A) or v3000 (B) for 6 hours. Quantitative RT-PCR was performed in triplicate with gene expression being normalized to the housekeeping gene (GAPDH) and expressed as fold change relative to mock treated cells. Results were analyzed using a one-way ANOVA with a tukey post test, ** p<0.01, *** p<0.001.
Figure 3. ISG, IRF and IFN genes are induced in a dsRNA length-dependent fashion.
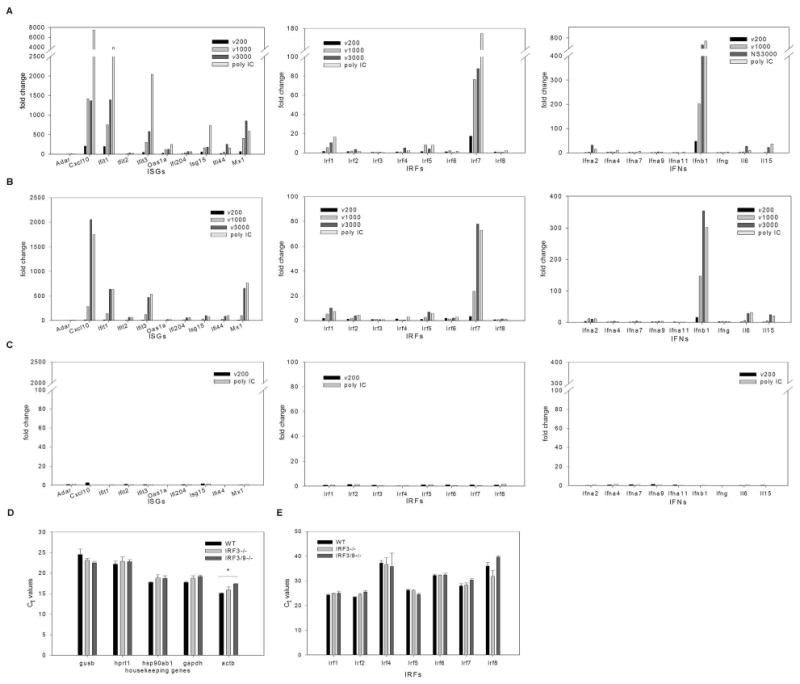
A, WT, B, IRF3-/- and C, IRF3&9-/- MEFs were treated with in vitro transcribed dsRNA of different lengths (ν200, ν1000, ν3000) or poly IC using concentrations shown to induce a maximal antiviral response (1.5nM, 3nM and 8.5 nM dsRNA for WT, IRF3-/- and IRF3&9-/- MEFs, respectively) for 6h (WT and IRF3-/- MEFs) or for 24h (IRF3&9-/- MEFs). The fold change in transcript expression levels compared to levels in mock treated cells were measured using real time PCR arrays. These results are representative of two independent experiments. Transcript levels for the PCR array housekeeping genes (D) and IRF family members (E) were compared between mock treated WT, IRF3-/- and IRF3&9-/- MEFs. Differences in Ct values were not statistically significant, with the exception of β actin in IRF3&9-/- MEFs, which was found to be expressed at lower levels when compared to WT, but not IRF3-/-, MEFs. For each gene a one-way ANOVA was performed with a tukey post test, * p<0.05.
When IRF3&9-/- MEFs were treated with 8.5 nM poly IC or dsRNA of various lengths (the highest concentration of dsRNA possible under these experimental conditions), ISG, IRF or IFN transcript accumulation was not observed, even after 24 hours of treatment (Figure 3C). The levels of housekeeping genes in mock-treated cells were similar in all cell types (Figure 3D), confirming the integrity of the RNA. Furthermore, the levels of the remaining IRF family members were similar in all cell types (Figure 3E). These data suggest that while IRF3 is not essential for dsRNA-mediated ISG, IRF and IFN induction per se, there appears to be a length-dependent requirement for IRF3, particularly with smaller lengths of dsRNA. Furthermore, IFN signaling (via IRF9 associated pathways) is required for dsRNA mediated ISG, IRF and IFN induction in the absence of IRF3.
To confirm that the expression of ISGs, IRFs and IFN was more dependent on IRF3 following stimulation with short dsRNA molecules as opposed to long dsRNA molecules, we performed quantitative RT-PCR analysis of CXCL10, Ifit1, IRF7 and IFNb1 in WT and IRF3-/- MEFs following treatment with short (200 bp) and long (3000 bp) dsRNA molecules. As shown in Figure 4A, induction of all four transcripts was significantly reduced in IRF3-/- MEFs relative to WT MEFs following stimulation with short dsRNA molecules. In contrast, a similar level of induction was observed following stimulation with long dsRNA molecules in both cell types (Figure 4B).
The dependence on dsRNA length is exacerbated in the absence of IRF3
To confirm the dsRNA length dependent nature of ISG and IFN induction and to determine the biological outcome in the presence and absence of IRF3, we performed standard antiviral assays to ascertain the effective concentration of dsRNA that prevents viral replication by 50% (EC50). Wild type and IRF3-/- MEFs were treated with serially diluted nM concentrations of in vitro transcribed dsRNA of specific lengths or poly IC for 6 hours, followed by challenge with VSV expressing green fluorescent protein under the viral promoter (VSVgfp). GFP fluorescence was monitored 24 hours post infection and the EC50 values determined. VSVgfp replication was similar in untreated WT and IRF3-/- cells, indicating that these cells are equally susceptible to VSVgfp infection (Supplementary Figure 1). A statistically significant difference in the EC50 values for dsRNA molecules of different lengths was observed in both wild type and IRF3-/- MEFs (Figure 5 A & B). A length dependent antiviral response was also observed in both WT and IRF3-/- MEFs when induced by equal weights of dsRNA (data not shown). When the EC50 values for a given length of dsRNA were compared between wild type and IRF3-/- MEFs, there was a statistically significant difference in the ability of dsRNA of 200 and 500 bp to elicit an antiviral response, whereas dsRNA of 1000 and 3000 bp and poly IC elicited a similar antiviral response in the two cell types (Figure 5C). Furthermore, polyI:C was able to limit the replication of HSV-1, a large DNA virus, indicating that this antiviral response is not unique to VSV (Supplementary Figure 4). These data support what was observed at the transcript level; the antiviral response to dsRNA is length dependent and the antiviral response to shorter dsRNA molecules is more reliant on IRF3 whereas the antiviral response to larger dsRNA molecules is predominantly IRF3-independent.
Figure 5. The dependence on dsRNA length for antiviral state induction is exacerbated in the absence of IRF3.
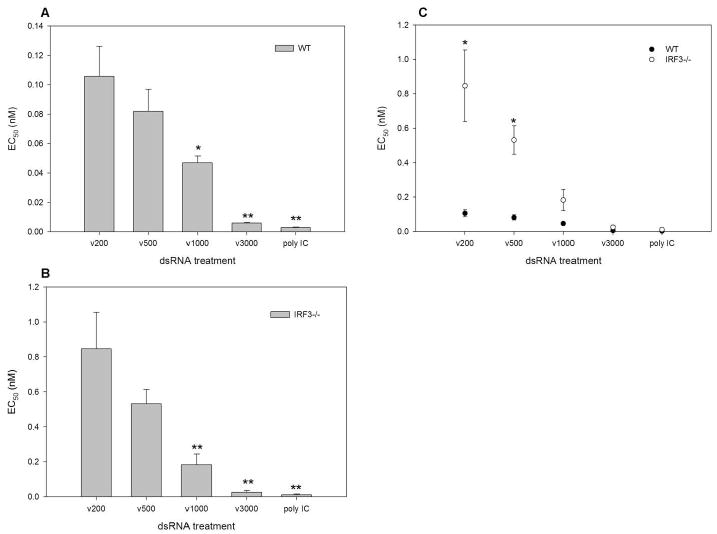
A, WT and B, IRF3-/- MEFs were treated with serially diluted nM concentrations of in vitro transcribed dsRNA of specific lengths (ν200, ν500, ν1000 and ν3000 bp) or poly IC for 6h. Cells were then challenged with VSVgfp and GFP fluorescence intensity was measured at 24h pi. EC50 values were determined as the dsRNA concentration providing 50% protection against VSVgfp. Length dependent antiviral responses were determined by performing a one- way ANOVA and a Dunnett's post test with ν200 being the control comparison. C, IRF-3 dependence was demonstrated by performing an unpaired T test, comparing WT to IRF3-/- EC50 values for each dsRNA length individually. * p<0.05, ** p<0.01.
Length dependent induction of an antiviral response in the absence of IRF3 and type I IFN
We failed to detect significant IRF, ISG or IFN induction in response to dsRNA or poly IC in IRF3&9-/- MEFs (Figure 3C), consistent with previous reports (4, 13). To determine if these cells were capable of eliciting an antiviral response in the absence of IFN or ISG induction, we performed a standard antiviral assay using VSVgfp following treatment with dsRNA and poly IC. Although we were unable to determine EC50 values for each length of dsRNA, given that a complete antiviral response was not observed at the highest concentration of dsRNA permissible, we did observe a significant reduction in the ability of VSVgfp to replicate in cells treated for 24 hours (Figure 6). No significant antiviral activity was observed following a 6-hour treatment (data not shown). At dsRNA lengths of 200, 500 and 1000 bp, VSVgfp replication was reduced by approximately 60% (56.7% ± 2.2, 58.0% ± 1.7 and 60.8% ± 1.1 respectively), whereas dsRNA of 3000 bp length and poly IC reduced VSVgfp replication by approximately 70% and 90% (67.4% ± 3.4 and 85.7% ± 1.3, respectively). A statistically significant difference was observed with the latter two treatments. These data suggest that dsRNA can elicit an antiviral response in the absence of the type I IFN system, including IRF3. As observed in IRF3-/- MEFs, treatment with polyI:C significantly inhibited the replication of HSV-1 (Supplementary Figure 4). Since dsRNA also activates NFκB and elicits the induction of cytokines and chemokines, we assessed induction of cytokines and chemokines using pathway specific PCR arrays. However, we did not detect significant induction of any genes under conditions that elicit an antiviral response (data not shown). The nature of this cellular response is unknown, but is currently under investigation. While dsRNA length dependence was observed, this dependence was not as robust as observed for the cellular antiviral response mediated by IRF3 and type 1 IFNs.
Figure 6. Evidence of a dsRNA length dependent antiviral response in the absence of IRF3 and type I IFN.
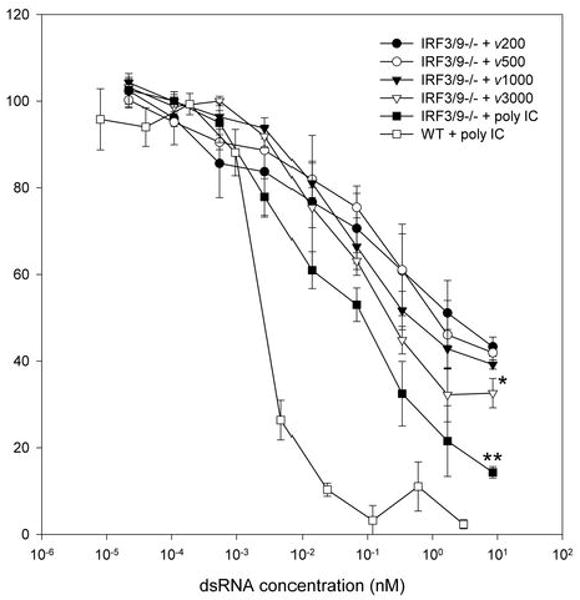
IRF3&9-/- MEFs were treated with 8.5 nM of the indicated dsRNA for 24h. Cells were then challenged with VSVgfp, and GFP fluorescence intensity was measured at 24h pi. DsRNA length dependence was observed with ν3000 and poly IC but not with ν500 and ν1000 when a one way ANOVA was performed using a Dunnett's post test with ν200 being the control comparison. Results with poly IC in WT MEFs are shown for comparison purposes. * p<0.05, ** p<0.01.
The mitochondria-associated adaptor IPS-1 is dispensable for the antiviral response mediated by long dsRNA molecules
Previous studies in MEFs harvested from IPS-1 null mice demonstrated that IPS-1 is essential for IFNβ production following treatment with virus or dsRNA (29). The lack of IFNβ production was attributed to impaired IRF3 and NFκB activation in these cells. Given that neither IRF3 nor type 1 IFN are necessary for cellular antiviral activity, particularly in response to long dsRNA molecules, we tested whether IPS-1 is necessary under similar conditions. Similar to results observed in IRF3&9-/- MEFs, dsRNA of any length failed to elicit a significant antiviral response following a 6-hour treatment (data not shown). However, following a 24-hour treatment, dsRNA of 3000 bp and poly IC induced an antiviral response with EC50 values of 0.51 nM ± 0.16 and 0.23 nM ± 0.04, respectively (Figure 7). Short dsRNA molecules of 200 bp lengths failed to induce an antiviral response in the absence of IPS-1. Conversely, dsRNA of 200 bp, 3000 bp lengths and poly IC all induced an antiviral response in IPS-1+/+ littermate controls with EC50 values of 0.64 +/- 0.23 nM, 0.22 +/- 0.05 nM and 0.17 +/- 0.04 nM, respectively (Figure 7). DsRNA entry was similar between IPS-1+/+ and IPS-1-/- MEFs (data not shown) as was VSVgfp infectivity in untreated cells (Supplementary Figure 1).
Figure 7. IPS-1 is dispensable for induction of an antiviral state in response to long dsRNA molecules.
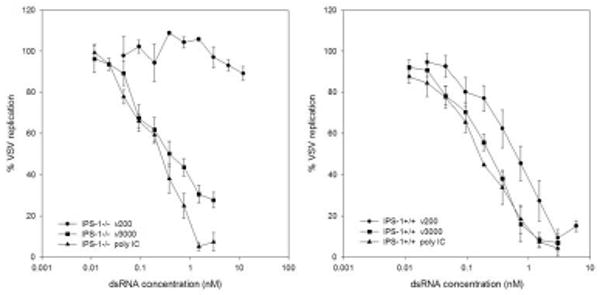
IPS-1-/- and IPS-1 +/+ littermate controls were treated with the indicated amounts of dsRNA for 24h. Cells were then challenged with VSVgfp and GFP fluorescence intensity was measured at 24h pi.
Discussion
It is well established that type 1 IFNs produced early following virus infection play a critical role in the immune defense against most viruses by limiting virus replication and spread (30). While various structural components of a virus can elicit type 1 IFN production, the most potent viral inducer is dsRNA, particularly in non-professional innate immune cells such as fibroblasts. Although it is recognized that the majority of viral dsRNA is bound by viral proteins, suggesting that little “free” viral dsRNA exists within a cell, the characterization of multiple cellular dsRNA binding proteins predicts that either entire dsRNA molecules, or portions of these molecules, are available for recognition and binding by cellular proteins. At least four cellular proteins recognize viral dsRNA and mediate type 1 IFN production: TLR3, PKR, RIG-I and MDA-5. While these proteins preferentially bind dsRNA based on its cellular localization, viral origin and/or length, the outcome of dsRNA binding is signaling through the adaptors TRIF (TLR3) or IPS-1 (PKR, RIG-I and MDA-5) to activate IRF3. Activated IRF3 can induce ISGs directly, in the absence of IFN production, or co-operate with additional transcription factors such as IRF1, IRF7 and NF-κB to elicit type 1 IFN production.
Although length plays a role in determining which cellular proteins preferentially bind a given dsRNA molecule, the biological significance of dsRNA length is unclear, particularly given that IRF3 activation ensues regardless of the dsRNA binding protein. To investigate this issue, we produced dsRNA of various lengths from the WNv genome, as the antiviral response to WNv involves TLR3, PKR, RIG-I and MDA-5 (31-33) and WNv produces stable dsRNA in fibroblasts following infection, thus confirming the biological relevance of dsRNA species from this viral origin. Here, we provide evidence that dsRNA induces a length-dependent antiviral response, with long dsRNA molecules inducing significantly higher levels of ISG, IRF and IFN transcripts than short dsRNA molecules when equal nM amounts were compared. The length dependence was confirmed upon determination of EC50 values in a standard antiviral assay. Previous studies have shown that activation of TLR3 and PKR requires a minimal length of dsRNA, either to span multiple dsRNA binding sites within a single TLR3 molecule (5-8) or to elicit dimerization of two PKR molecules (9, 10). While activation of RIG-I and MDA5 following dsRNA binding does not appear to have similar requirements, it has been established that MDA-5 preferentially binds long dsRNA molecules and poly IC (11, 12). Accordingly, a simple explanation for the data presented in this manuscript is that MEFs express elevated levels of MDA-5 relative to RIG-I and thus long dsRNA molecules induce a greater biological response. However, untreated MEFs express low to undetectable levels of both MDA-5 and RIG-I, but rapidly up-regulate the expression of both ISGs following virus or dsRNA treatment (34), similar to what is seen with PKR. Another possible explanation is that MDA5 binds dsRNA with a higher affinity than RIG-I. To date, comprehensive studies to specifically address this possibility have not been performed. An alternative explanation is that longer dsRNA molecules facilitate better multimerization of MDA5 compared to shorter dsRNA lengths with RIG-I. There is a general precedence with cellular signal transduction that multimerization of pathway components leads to enhanced signal transduction and downstream biological activity. Regardless, the focus of the current study was not to determine which dsRNA binding protein(s) mediates an antiviral response, but rather to delineate the biological outcome of differential dsRNA binding, particularly given that all characterized pathways converge on IRF3. Indeed, it is likely that per dsRNA molecule, long dsRNA molecules have an increased capacity to activate multiple dsRNA binding proteins (of the same or different species), thus potentiating the downstream biological response.
Of particular interest, the dsRNA length dependence was exacerbated in the absence of IRF3. When comparing either individual transcript induction or EC50 values for a given length of dsRNA, a significant difference was observed between the ability of short, but not long, dsRNA molecules to induce ISGs, IRFs and IFN, or to protect wild type versus IRF3-/- MEFs from virus infection. These results confirm that IRF3 is not essential per se for ISG and IFN production (4, 13), but suggest that IRF3 plays a more critical role in response to recognition of short dsRNA molecules. Long dsRNA molecules (3000 bp and poly IC) were equally capable of preventing virus replication in the presence and absence of IRF3. These data suggest that either a novel transcription factor with a similar activity to IRF3 exists, or that in the absence of IRF3, another member of the IRF family can compensate for the loss of IRF3. Recent biochemical and structural studies of the IFNβ promoter show that IRF binding is critical for co-operative association of the IFNβ enhanceosome components and IFNβ transcription (35, 36). While IRF1, IRF5 and IRF7 have been implicated in type 1 IFN production, it is unlikely that these IRFs are responsible for the effects seen in IRF3-/- MEFs. We failed to detect differences in the basal expression of IRF mRNA species in the various MEFs, and previous studies showed that ectopic expression of IRF1 fails to restore type 1 IFN induction in the absence of IRF3 (4) and IRF5 activation does not occur in response to treatment with poly IC (37). Furthermore, IRF7 expression in fibroblasts is dependent on type 1 IFN signaling [ref. (4) and Figure 3] and IRF7 predominantly activates IFNα promoters, while robust IFNβ expression was observed in the absence of IRF3 following treatment with dsRNA or poly IC. Little is known regarding alternative pathways of dsRNA-mediated type 1 IFN induction involving proteins other than IRFs. While NLRs have been shown to bind to dsRNA (38) and are important in mediating antiviral immune responses (39), activation of type 1 IFN has not been observed. Furthermore, is possible that pattern recognition receptors capable of binding dsRNA that signal through alternative transcription factors exist. Thus it remains to be elucidated how dsRNA, particularly large species, mediate type 1 IFN production in the absence of IRF3.
A further surprising observation from these data is the ability of dsRNA to control virus replication in the absence of IPS-1, IRF3 and type 1 IFN production. While IRF3-dependent, IFN-independent and IRF3-independent, IFN-dependent antiviral responses have been observed, to our knowledge this is the first observation of antiviral activity mediated by dsRNA in fibroblasts under conditions where we fail to detect induction of IRFs, IFNs or ISGs. Furthermore, we failed to detect induction of cytokines and chemokines under similar conditions. While we were unable to elicit a complete antiviral response in IRF3&9-/- MEFs at the highest concentration of dsRNA permissible within our experimental system (8.5 nM), we observed a reduction in virus replication ranging from approximately 60% to 90%, depending on dsRNA length. Consistent with data generated in IRF3-/- MEFs, the antiviral response was more robust following treatment with long dsRNA molecules. This antiviral response reduced virus replication of both an RNA virus (VSV) and a DNA virus (HSV-1), albeit with different efficiency. While a 6hr treatment with dsRNA was sufficient to significantly inhibit HSV-1 replication, a 24hr treatment was required to significantly inhibit VSV replication. These data highlight the complex nature of virus-host interactions.
A similar reduction in virus replication was made in MEFs deficient for the mitochondria-associated adaptor IPS-1. While previous studies have shown that IPS-1 is important for activation of IRF3 and NFκB and the subsequent induction of IFNβ, we observed that treatment with long dsRNA molecules was able to induce an antiviral response capable of controlling VSV infection. In both IRF3&9-/- and IPS-1-/- MEFs, however, an extended treatment time (24 hours) was required to elicit a biological response. These data suggest that the cellular pathway(s) that function independently of IPS-1, IRF3 and type 1 IFN preferentially respond to long dsRNA molecules and require sufficient stimulation to accumulate and/or activate constituent signaling components. It is not clear at this time whether a novel dsRNA binding protein participates in this antiviral response or whether the known dsRNA proteins signal through alternative pathways to block virus replication. Studies are underway to address this issue and determine the cellular genes that are induced by dsRNA under these conditions. Overall, these finding further characterize the requirement (or lack thereof) of IRF3 and the type 1 IFN system in the innate antiviral immune response following recognition of dsRNA.
Supplementary Material
Acknowledgments
We thank A. Thompson and D. Cummings for technical assistance and B. Lichty and M. Diamond for reagents.
Footnotes
This study was funded by the National Institutes of Health and the Canadian Institute for Health Research (MOP-57669)
Abbreviations: IPS-1, IFNβ promoter stimulator 1; IRF, IFN regulatory factor; ISG, IFN-stimulated gene; MDA-5, melanoma differentiation-associated gene 5; MEF, mouse embryo fibroblast; NLR, nucleotide oligomerization domain-like receptors; poly IC, polyinosinic/polycytidylic acid; PKR, protein kinase regulated by RNA; RIG-I, retinoic acid-inducible gene-I; RLR, RIG-I-like receptor; TRIF, Toll/Interleukin-1 receptor domain-containing adaptor inducing IFNβ; VSV, vesicular stomatitis virus; WNv, West Nile virus; WT, wild type.
References
- 1.Jacobs BL, Langland JO. When two strands are better than one: the mediators and modulators of the cellular responses to double-stranded RNA. Virology. 1996;219:339–349. doi: 10.1006/viro.1996.0259. [DOI] [PubMed] [Google Scholar]
- 2.Takeuchi O, Akira S. Innate immunity to virus infection. Immunol Rev. 2009;227:75–86. doi: 10.1111/j.1600-065X.2008.00737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang P, Samuel CE. Induction of protein kinase PKR-dependent activation of interferon regulatory factor 3 by vaccinia virus occurs through adapter IPS-1 signaling. J Biol Chem. 2008;283:34580–34587. doi: 10.1074/jbc.M807029200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, Taniguchi T. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity. 2000;13:539–548. doi: 10.1016/s1074-7613(00)00053-4. [DOI] [PubMed] [Google Scholar]
- 5.Fukuda K, Watanabe T, Tokisue T, Tsujita T, Nishikawa S, Hasegawa T, Seya T, Matsumoto M. Modulation of double-stranded RNA recognition by the N-terminal histidine-rich region of the human toll-like receptor 3. J Biol Chem. 2008;283:22787–22794. doi: 10.1074/jbc.M802284200. [DOI] [PubMed] [Google Scholar]
- 6.Liu L, Botos I, Wang Y, Leonard JN, Shiloach J, Segal DM, Davies DR. Structural Basis of Toll-Like Receptor 3 Signaling with Double-Stranded RNA. Science. 2008;320:379–381. doi: 10.1126/science.1155406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pirher N, Ivicak K, Pohar J, Bencina M, Jerala R. A second binding site for double-stranded RNA in TLR3 and consequences for interferon activation. Nat Struct Mol Biol. 2008;15:761–763. doi: 10.1038/nsmb.1453. [DOI] [PubMed] [Google Scholar]
- 8.de Bouteiller O, Merck E, Hasan UA, Hubac S, Benguigui B, Trinchieri G, Bates EEM, Caux C. Recognition of Double-stranded RNA by Human Toll-like Receptor 3 and Downstream Receptor Signaling Requires Multimerization and an Acidic pH. J Biol Chem. 2005;280:38133–38145. doi: 10.1074/jbc.M507163200. [DOI] [PubMed] [Google Scholar]
- 9.Lemaire PA, Anderson E, Lary J, Cole JL. Mechanism of PKR Activation by dsRNA. J Mol Biol. 2008;381:351–360. doi: 10.1016/j.jmb.2008.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McAllister CS, Samuel CE. The RNA-activated protein kinase enhances the induction of interferon-beta and apoptosis mediated by cytoplasmic RNA sensors. J Biol Chem. 2009;284:1644–1651. doi: 10.1074/jbc.M807888200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Sousa C Reis e, Matsuura Y, Fujita T, Akira S. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 12.Kato H, Takeuchi O, Mikamo-Satoh E, Hirai R, Kawai T, Matsushita K, Hiiragi A, Dermody TS, Fujita T, Akira S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J Exp Med. 2008;205:1601–1610. doi: 10.1084/jem.20080091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hata N, Sato M, Takaoka A, Asagiri M, Tanaka N, Taniguchi T. Constitutive IFN-alpha/beta signal for efficient IFN-alpha/beta gene induction by virus. Biochem Biophys Res Commun. 2001;285:518–525. doi: 10.1006/bbrc.2001.5159. [DOI] [PubMed] [Google Scholar]
- 14.Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 15.Andersen J, VanScoy S, Cheng TF, Gomez D, Reich NC. IRF-3-dependent and augmented target genes during viral infection. Genes Immun. 2008;9:168–175. doi: 10.1038/sj.gene.6364449. [DOI] [PubMed] [Google Scholar]
- 16.Prescott JB, Hall PR, Bondu-Hawkins VS, Ye C, Hjelle B. Early innate immune responses to Sin Nombre hantavirus occur independently of IFN regulatory factor 3, characterized pattern recognition receptors, and viral entry. J Immunol. 2007;179:1796–1802. doi: 10.4049/jimmunol.179.3.1796. [DOI] [PubMed] [Google Scholar]
- 17.Ali S, Kukolj G. Interferon regulatory factor 3-independent double-stranded RNA-induced inhibition of hepatitis C virus replicons in human embryonic kidney 293 cells. J Virol. 2005;79:3174–3178. doi: 10.1128/JVI.79.5.3174-3178.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Daffis S, Samuel MA, Keller BC, Gale M, Jr, Diamond MS. Cell-specific IRF-3 responses protect against West Nile virus infection by interferon-dependent and -independent mechanisms. PLoS Pathog. 2007;3:e106. doi: 10.1371/journal.ppat.0030106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bourne N, Scholle F, Silva MC, Rossi SL, Dewsbury N, Judy B, De Aguiar JB, Leon MA, Estes DM, Fayzulin R, Mason PW. Early production of type I interferon during West Nile virus infection: role for lymphoid tissues in IRF3-independent interferon production. J Virol. 2007;81:9100–9108. doi: 10.1128/JVI.00316-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Noyce RS, Collins SE, Mossman KL. Identification of a novel pathway essential for the immediate-early, interferon-independent antiviral response to enveloped virions. J Virol. 2006;80:226–235. doi: 10.1128/JVI.80.1.226-235.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo J, Peters KL, Sen GC. Induction of the human protein P56 by interferon, double-stranded RNA, or virus infection. Virology. 2000;267:209–219. doi: 10.1006/viro.1999.0135. [DOI] [PubMed] [Google Scholar]
- 22.Peters KL, Smith HL, Stark GR, Sen GC. IRF-3-dependent, NFkappa B- and JNK-independent activation of the 561 and IFN-beta genes in response to double-stranded RNA. Proc Natl Acad Sci U S A. 2002;99:6322–6327. doi: 10.1073/pnas.092133199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grandvaux N, Servant MJ, tenOever B, Sen GC, Balachandran S, Barber GN, Lin R, Hiscott J. Transcriptional profiling of interferon regulatory factor 3 target genes: direct involvement in the regulation of interferon-stimulated genes. J Virol. 2002;76:5532–5539. doi: 10.1128/JVI.76.11.5532-5539.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Minaker RL, Mossman KL, Smiley JR. Functional inaccessibility of quiescent herpes simplex virus genomes. Virol J. 2005;2:85. doi: 10.1186/1743-422X-2-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Minarova E, Spurna V, Keprtova J. Interaction of DEAE-dextran with mammalian cells cultivated in vitro. Experientia. 1972;28:333–334. doi: 10.1007/BF01928723. [DOI] [PubMed] [Google Scholar]
- 26.Weber F, Wagner V, Rasmussen SB, Hartmann R, Paludan SR. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J Virol. 2006;80:5059–5064. doi: 10.1128/JVI.80.10.5059-5064.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mackenzie JM, Kenney MT, Westaway EG. West Nile virus strain Kunjin NS5 polymerase is a phosphoprotein localized at the cytoplasmic site of viral RNA synthesis. J Gen Virol. 2007;88:1163–1168. doi: 10.1099/vir.0.82552-0. [DOI] [PubMed] [Google Scholar]
- 28.Liu L, Botos I, Wang Y, Leonard JN, Shiloach J, Segal DM, Davies DR. Structural basis of toll-like receptor 3 signaling with double-stranded RNA. Science. 2008;320:379–381. doi: 10.1126/science.1155406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar H, Kawai T, Kato H, Sato S, Takahashi K, Coban C, Yamamoto M, Uematsu S, Ishii KJ, Takeuchi O, Akira S. Essential role of IPS-1 in innate immune responses against RNA viruses. J Exp Med. 2006;203:1795–1803. doi: 10.1084/jem.20060792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van den Broek MF, Muller U, Huang S, Zinkernagel RM, Aguet M. Immune defence in mice lacking type I and/or type II interferon receptors. Immunol Rev. 1995;148:5–18. doi: 10.1111/j.1600-065x.1995.tb00090.x. [DOI] [PubMed] [Google Scholar]
- 31.Fredericksen BL, Keller BC, Fornek J, Katze MG, Gale M., Jr Establishment and maintenance of the innate antiviral response to West Nile Virus involves both RIG-I and MDA5 signaling through IPS-1. J Virol. 2008;82:609–616. doi: 10.1128/JVI.01305-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Daffis S, Samuel MA, Suthar MS, Gale M, Jr, Diamond MS. Toll-Like Receptor 3 Has a Protective Role against West Nile Virus Infection. J Virol. 2008;82:10349–10358. doi: 10.1128/JVI.00935-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Samuel MA, Whitby K, Keller BC, Marri A, Barchet W, Williams BRG, Silverman RH, Gale M, Jr, Diamond MS. PKR and RNase L Contribute to Protection against Lethal West Nile Virus Infection by Controlling Early Viral Spread in the Periphery and Replication in Neurons. J Virol. 2006;80:7009–7019. doi: 10.1128/JVI.00489-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Loo YM, Fornek J, Crochet N, Bajwa G, Perwitasari O, Martinez-Sobrido L, Akira S, Gill MA, Garcia-Sastre A, Katze MG, Gale M., Jr Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J Virol. 2008;82:335–345. doi: 10.1128/JVI.01080-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dragan AI, Carrillo R, Gerasimova TI, Privalov PL. Assembling the human IFN-beta enhanceosome in solution. J Mol Biol. 2008;384:335–348. doi: 10.1016/j.jmb.2008.09.015. [DOI] [PubMed] [Google Scholar]
- 36.Panne D, Maniatis T, Harrison SC. An atomic model of the interferon-beta enhanceosome. Cell. 2007;129:1111–1123. doi: 10.1016/j.cell.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paun A, Pitha PM. The IRF family, revisited. Biochimie. 2007;89:744–753. doi: 10.1016/j.biochi.2007.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tattoli I, Carneiro LA, Jehanno M, Magalhaes JG, Shu Y, Philpott DJ, Arnoult D, Girardin SE. NLRX1 is a mitochondrial NOD-like receptor that amplifies NF-kappaB and JNK pathways by inducing reactive oxygen species production. EMBO Rep. 2008;9:293–300. doi: 10.1038/sj.embor.7401161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med. 2009;206:79–87. doi: 10.1084/jem.20081667. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.