

Abstract
Neuronal pentraxins (NPs) are hypothesized to play important roles in the recruitment of AMPA receptors (AMPARs) to immature synapses, yet a physiological role for NPs at nascent synapses in vivo has remained elusive. Here we report that the loss of NP1 and NP2 (NP1/2) leads to a dramatic and specific reduction in AMPAR-mediated transmission at developing visual system synapses. In thalamic slices taken from early postnatal mice (<P10) NP1/2 knock-out (KO) neurons displayed severely reduced AMPAR-mediated retinogeniculate transmission. The reduced currents reflected an increased number of silent synapses with no change in quantal amplitude or presynaptic release. These are the first data to demonstrate that NP1/2 are required in vivo for the normal development of AMPAR-mediated transmission. In addition, they suggest a novel role for NP1/2 in silent synapse conversion during a discrete developmental period when visual circuit connections are undergoing eye-specific refinement. After this period, retinogeniculate transmission not only recovered in the knock-outs but became excessive. The enhanced currents were attributable, at least in part, to a deficit in the characteristic elimination of functional inputs that occurs in the developing dLGN. These data indicate that the loss of NP1/2 disrupts several aspects of retinogeniculate development including the initial establishment of AMPAR transmission and the subsequent elimination of inappropriate circuit connections.
Introduction
AMPA receptor (AMPAR) recruitment is necessary to confer functionality to newly formed excitatory synapses and is a requirement for normal brain function; however, the mechanisms underlying the development of AMPAR signaling at nascent synapses remain controversial (Voronin and Cherubini, 2004; Xiao et al., 2004). Among the molecules hypothesized to act as early mediators of synaptic AMPAR recruitment are the neuronal pentraxins (NPs). This family consists of three members, NP1, NP2 (also known as neuronal activity-regulated protein or Narp), and NPR (neuronal pentraxin receptor). NPR is an integral membrane protein whereas NP1 and NP2 are secreted molecules, and the three form heteromultimers in which NP1 and NP2 can be tethered to the membrane through NPR (Kirkpatrick et al., 2000). NPs are multifunctional proteins expressed specifically at excitatory synapses throughout the brain. Recent evidence indicates that one member of this family, NPR, is required for mGluR1/5-dependent long-term depression (LTD) (Cho et al., 2008). In contrast, NP1 and NP2 are thought to act trans-synaptically to cluster AMPARs at postsynaptic sites and to promote excitatory synaptogenesis (O'Brien et al., 1999, 2002; Reti et al., 2002; Xu et al., 2003; Sia et al., 2007). In support of this, the C-terminal domain of NPs has been shown to interact with the N terminus of glutamate receptor subunits (Xu et al., 2003; Sia et al., 2007), and overexpression of NPs in cultured presynaptic neurons caused enhanced postsynaptic AMPAR clustering (O'Brien et al., 1999, 2002). In addition, expression of dominant negative NP2 in vitro reduces GluR1 clustering along dendrites of cultured spinal neurons (O'Brien et al., 2002), and both knockdown and knock-out of NPs were shown to reduce GluR4 clustering on hippocampal neurons (Sia et al., 2007). Despite this compelling evidence supporting a developmental role for NPs in synaptic AMPAR recruitment, a physiological function for NPs at nascent synapses in vivo has not been established (Bjartmar et al., 2006).
Here we investigate the function of NP1 and NP2 at retinogeniculate synapses in the developing mouse visual system. NP1/2 are expressed in retinal ganglion cells (RGCs) and in the visual thalamus where they are required for the eye-specific refinement of RGC axons during early postnatal life (Bjartmar et al., 2006); thus, NP1/2 are important molecules in this system during in vivo development. In addition, RGCs cultured from NP1/2 KO mice displayed a pronounced delay in the development of synaptic AMPAR-mediated currents (Bjartmar et al., 2006). These data suggested that NP1/2 might participate in the development of AMPAR-mediated currents at native retinogeniculate synapses, and that deficits in synaptic physiology could contribute to the failure of NP1/2 KO axons to segregate normally. Here we use acute slice physiology to characterize the role of NP1/2 at native retinogeniculate synapses over development. In addition, because NP1/2 KO mice displayed physiological phenotypes both in the retina and in the dLGN we used in vivo retinal activity manipulations and axon labeling to investigate the role of retinal activity in axon remodeling in NP1/2 KO mice.
Materials and Methods
Animals.
All experiments were performed in accordance with the animal handling guidelines put forward by the Institutional Animal Care and Use Committee. The generation of the NP1/2 KO animals has been previously described (Kirkpatrick et al., 2000; Bjartmar et al., 2006).
Slice preparation.
NP1/2 double knock-out mice were bred to C57BL/6 background for seven generations to make congenic NP1/2 C57BL/6 mice. Acute slices (325 μm) were prepared from wild-type (WT) C57BL/6 mice or from mice that were either knock-outs or heterozygous for NP1 and NP2 using the method of Chen and Regehr, which preserves the optic tract and dLGN (Chen and Regehr, 2000). Throughout the course of the experiments two different cutting solutions were used; either a high-sucrose-based solution containing the following (in mm): 50.0 NaCl, 25.0 NaHCO3, 150.0 Sucrose, 10.0 Dextrose, 2.5 KCl, 1.0 NaH2PO4, 0.5 CaCl2, 7.0 MgCl2*6H20, or a choline-based solution containing the following (in mm): 78.3 NaCl, 23.0 NaHCO3, 23.0 Dextrose, 33.8 Choline Chloride, 2.3 KCl, 1.1 NaH2PO4, 6.4 MgCl2, 0.45 CaCl2. Chemicals were obtained from Sigma. After cutting, slices were incubated in oxygenated 32–34°C cutting solution for 25–40 min and then transferred to oxygenated ACSF consisting of (in mm): 125.0 NaCl, 25.0 NaHCO3, 25.0 Dextrose, 2.5 KCl, 1.25 NaH2PO4, 2.0 CaCl2, 1.0 MgCl2*6H20. For recordings of quantal events calcium was omitted from the recording solution and replaced with 3 mm strontium. Slices were incubated in ACSF for 25–40 min during which time they were allowed to cool to room temperature. Slices were continuously perfused with oxygenated ACSF during the recording period.
Electrophysiology.
Whole-cell voltage-clamp recordings of geniculate neurons were obtained using 2.5–3.5 MOhm patch electrodes containing an internal solution consisting of the following (in mm): 35 CsF, 100 CsCl, 10 EGTA, and 10 HEPES. Inhibitory inputs were blocked with 20 μm bicuculline methobromide (Tocris Bioscience). Recordings were sampled at 10–20 kHz and filtered at 1 kHz. Access resistance was monitored throughout the recording period and was adjusted to 4–9 MOhms after 70% compensation. A stimulating electrode (either concentric bipolar or two glass electrodes filled with ACSF) was placed just touching the surface of the optic tract next to the ventral LGN, and 1 ms stimuli ranging from 0 to 40 μA were delivered every 30–60 s. NMDAR-mediated current amplitudes were measured at a holding potential of +40 mV by evoking synaptic currents and measuring the peak of the current trace at a time when the AMPAR-mediated currents no longer contributed to the response: 20–30 ms after the onset of the EPSC for the P6 through P9 recordings, and 10–15 ms after EPSC onset for the P17 through P20 recordings. Synaptic currents were analyzed using a variety of software programs including Igor Pro, Synaptosoft Mini Analysis, Microsoft Excel, and GraphPad Prism. Statistical comparisons were made using a Student unpaired t test unless otherwise stated.
Intraocular injections.
Mice were anesthetized with isoflurane, and fused eyelids were gently separated with tweezers. A 30.5 gauge beveled syringe tip was used to pierce the eye at the corneal-scleral junction. This allowed some vitreal solution to escape, releasing some of the intraocular pressure. After the escaped vitreous was wicked away, a blunt-tipped Hamilton syringe was inserted into the hole and 0.5–1.0 μl of solution was injected (0.5 μl for P4 mice, 0.75 μl for P6 mice, and 1.0 μl for P8/9). To increase retinal activity a 2 mm solution of CPT-cAMP (Tocris Bioscience) was injected binocularly. For experiments in which spontaneous activity was blocked in one eye a 1 mm solution of epibatidine (Sigma) was injected into the right eye and a saline-only solution was injected into the left eye. For anterograde labeling of RGC axons a 0.5% solution (in saline) of the β subunit of choleratoxin conjugated to Alexa 594 (Invitrogen) was injected into the epibatidine-treated eye and an Alexa 488 conjugate was injected into the control-treated eye. Control animals received binocular saline injections followed by Ctβ-594 and Ctβ-488 in the right and left eyes, respectively.
Analysis of retinogeniculate projections.
Brains were removed 24 h after intraocular injection with the fluorescent tracers and fixed overnight in 4.0% PFA. Coronal sections (50 μm) were prepared using a Leica vibrating microtome and imaged on a Nikon Eclipse microscope. Images were taken using a Photometrics Coolsnap camera and imported into Adobe Photoshop for analysis. Grayscale images were converted into binary (black and white) images using a threshold that corresponded to a location in the grayscale histogram where there was a clear distinction between the label and background fluorescence signals, values typically corresponded ∼30 on the grayscale range of 0–255 (Muir-Robinson et al., 2002; Torborg and Feller, 2004). Thresholded images were imported into ImageJ. The boundaries of the dLGN were delineated to exclude label from the optic tract, intrageniculate leaflet, and ventral LGN. Measurements of the area of the dLGN occupied by the ipsilateral projections were calculated by selecting all white pixels within the delineated area.
Results
In the rodent dorsal lateral geniculate nucleus (dLGN), the bulk of synaptogenesis occurs over the first week of postnatal life and it is during this time that AMPAR-mediated currents can first be recorded from retinogeniculate synapses (Ziburkus and Guido, 2006). This period also corresponds to the peak of NP1/2 expression in this system (Bjartmar et al., 2006). Therefore, to assess the role of NP1/2 in the development of retinogeniculate synaptic function we recorded from dLGN neurons in acutely prepared brain slices between the ages of postnatal day 6 and 9 (P6–P9), when AMPAR-mediated currents could be reliably detected in all WT dLGN neurons. Slices containing the dLGN and the optic tract were prepared using the method of Chen and Regehr, which preserves the retinal inputs to the nucleus (Chen and Regehr, 2000). Optic tract stimulation was used to evoke transmission at retinogeniculate synapses and thalamic neurons were voltage clamped at either −70 mV or +40 mV to measure AMPAR-mediated and NMDAR-mediated currents, respectively. As is characteristic of early retinogeniculate transmission, this method produced robust evoked NMDAR-mediated currents and somewhat weaker AMPAR-mediated currents, and varying the stimulus intensity showed that the immature relay neurons were multi-innervated (Fig. 1A,B). Indeed all parameters of evoked WT transmission reported below are in good agreement with previous studies (Chen and Regehr, 2000; Liu and Chen, 2008).
Figure 1.

NP1/2 knock-out neurons have a specific deficit in AMPAR-mediated synaptic transmission during early postnatal development (P6–P9). A, Example traces of postsynaptic responses from WT, NP1/2 Het, and NP1/2 KO dLGN neurons voltage clamped at −70 mV (downward currents) and +40 mV (upward currents) where the optic tract was stimulated over a range of stimulus intensities. B, Example input–output curves showing EPSC amplitude as a function of stimulus intensity at −70 mV and +40 mV. C, Average amplitudes of maximal AMPAR and NMDAR-mediated currents. D, Average AMPA/NMDA ratios of maximal currents. C, D, n = 9 WT animals (26 cells), 6 NP1/2 Het animals (15 cells), and 6 NP1/2 KO animals (26 cells). Data displayed as mean ± SEM and compared by one-way ANOVA followed by Bonferroni post-tests. *p < 0.05. E, Example traces from a WT neuron (upper) and a NP1/2 KO neuron (lower) voltage clamped at +40 mV and stimulated with pairs of pulses at 5 different interstimulus intervals (50, 100, 200, 500, and 1000 ms). F, The average PPRs for WTs (3 animals, 8 cells) and NP1/2 KO neurons (2 animals, 4 cells) over the 5 ISIs tested. No differences were found by two-way ANOVA followed by Bonferroni post-tests.
Like WT neurons, neurons from NP1/2 heterozygous animals (NP1/2 Hets) and neurons from NP1/2 KO animals also displayed robust NMDAR-mediated evoked responses (Fig. 1A,B). However, in contrast to WTs and NP1/2 Hets, we observed little to no AMPAR-mediated transmission in recordings from NP1/2 KO neurons (Fig. 1A,B). When relay neurons were voltage clamped at −70 mV and a maximal stimulus was delivered to the optic tract, the peak amplitudes of the AMPAR-mediated responses were severely reduced in NP1/2 KO neurons compared to WT and NP1/2 Het neurons (Fig. 1C) (WT = −244.20 ± 31.97 pA, Het = −209.95 ± 42.32 pA, KO = −80.86 ± 19.48 pA; WT, KO p < 0.05, Het, KO p < 0.05 by one-way ANOVA followed by Bonferroni multiple-comparisons test). The AMPA/NMDA ratio, a measure of the relative strength of the two components of the evoked response, was also decreased in NP1/2 KO neurons (Fig. 1D) (WT = 0.44 ± 0.07, Het = 0.36 ± 0.04, KO = 0.14 ± 0.03; WT, KO p < 0.05, Het, KO p < 0.05 by one-way ANOVA followed by Bonferroni multiple-comparisons test). The reduced ratio reflected a specific deficit in the AMPAR-mediated currents because neither the average amplitude (Fig. 1C) (WT = 636.76 ± 129.98 pA, Het = 658.20 ± 115.49, KO = 657.97 ± 93.81 pA; WT, KO p > 0.05, Het, KO p > 0.05) nor time course (WT = 194.48 ± 16.66, Het = 220.92 ± 9.27, KO = 209.30 ± 19.55; WT, KO p > 0.05, Het, KO p > 0.05) of the NMDAR-mediated responses were affected by NP1/2 knock-out (one-way ANOVA followed by Bonferroni multiple-comparisons test). We reasoned that this specific reduction in AMPAR-mediated transmission with no accompanying change in NMDAR-mediated transmission was likely to have a postsynaptic origin, and we tested this using a common measure of presynaptic release probability, the paired pulse ratio (PPR). Because the NP1/2 KO AMPAR-mediated currents were small and sometimes absent we analyzed the PPR of the NMDAR-mediated currents. In this and subsequent experiments we focused on WT and KO neurons because heterozygous neurons displayed synaptic currents that were indistinguishable from WTs. PPRs were measured at 5 different interstimulus intervals (ISIs) (50, 100, 200, 500, and 1000 ms), and no differences were found between WT and KO PPRs at any of these intervals (Fig. 1E,F) (two-way ANOVA followed by Bonferroni post-tests), suggesting that presynaptic release is not reduced by the loss of NP1/2. Together, these data demonstrate that the absence of NP1/2 during in vivo development leads to a specific deficit in AMPAR-mediated transmission at newly formed synapses.
In the NP1/2 KO mouse RGC spiking was shown to be increased during early postnatal life (Bjartmar et al., 2006), raising the possibility that the severely reduced AMPAR-mediated currents in the KO dLGN could be attributable to altered retinal activity instead of to the loss of NP1/2 from retinogeniculate synapses (Shah and Crair, 2008a). To begin to distinguish between these possibilities we pharmacologically enhanced RGC spiking in WT animals in vivo and subsequently measured retinogeniculate currents to determine whether increased retinal activity can lead to diminished retinogeniculate transmission. Binocular injections of the cAMP analog, CPT-cAMP, were used to enhance RGC spiking because this treatment has been previously demonstrated to increase RGC activity in WT animals through enhancing presynaptic drive from cholinergic amacrine cells (however, we did not verify the effects of cAMP in this study) (Stellwagen et al., 1999). Retinal activity was increased for 3–4 d between the ages of P4 and P7 to P8 via repeat intraocular injections performed 48 h apart. Retinogeniculate currents were assessed on P7 and P8. No difference in retinogeniculate transmission was observed between CPT-cAMP and saline-treated animals (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). This finding shows that increased retinal spiking, per se, does not reduce AMPAR-mediated transmission at retinogeniculate synapses, which is consistent with a direct role for NP1/2 in retinogeniculate transmission. However, it remains possible that the unique features of retinal waves altered in the NP1/2 KOs might contribute to synaptic AMPAR-mediated recruitment.
What synaptic mechanisms might account for the reduced AMPAR-mediated currents in NP1/2 KO neurons? Because neuronal pentraxins have been reported to cluster glutamate receptors at hippocampal and spinal synapses (O'Brien et al., 1999, 2002; Xu et al., 2003; Sia et al., 2007), we hypothesized that the decrease in AMPAR-mediated transmission was attributable to the presence of fewer AMPARs clustered at retinogeniculate synapses. To test this we measured quantal size, which should be reduced in NP1/2 KO neurons if each synapse contains fewer AMPARs. Because dLGN neurons receive excitatory input from both retinal and cortical inputs it is impossible to know the origin of spontaneously released vesicles; consequently, we used optic tract stimulation to evoke quantal events specifically at retinogeniculate synapses. This was accomplished by replacing extracellular calcium with strontium, which promotes the asynchronous release of synaptic vesicles (Miledi, 1966). Accordingly, we found that strontium both reduced the EPSC (Fig. 2A, arrows) and allowed for the resolution of unitary events (Fig. 2A, asterisks) at a frequency that was 5.9-fold greater than the background rate (event frequency without stimulation compared to the frequency measured during the first 300 ms after stimulation). This suggests that ∼83% of the analyzed events were evoked from RGC synapses, whereas the remaining 17% were a mixed population arising from both retinal and cortical inputs; therefore, at least 83% of the events occurred at retinogeniculate synapses. Although the initial responses were greatly reduced in NP1/2 KO neurons compared to WT neurons (Fig. 2A), the average size of the quantal events did not differ between the genotypes (Fig. 2B, example traces) (average quantal size for WTs and NP1/2 KOs were −9.49 ± 0.42 pA and −9.89 ± 0.64 pA, respectively; p > 0.05). Because quantal size at young retinogeniculate synapses is quite small (Chen and Regehr, 2000) decrements in quantal size may be difficult to detect as reduced events could fall into the noise. To address this issue we first confirmed that the baseline signals did not differ between the WT and NP1/2 KO recordings by comparing their average SDs (WT = 2.76 ± 0.30 pA, NP1/2 KO = 2.71 ± 0.33 pA; p > 0.05). To then determine whether the two populations of event amplitudes differed in profile we analyzed the cumulative probability of event amplitudes using 1 pA bins (Fig. 2C). If the loss of NP1/2 leads to many quantal events falling below the detection limit then we would expect this distribution to be left-shifted; however, the distributions were very similar in profile and were not statistically different from one another (Kolmogorov–Smirnov test, p > 0.05). These surprising data strongly suggest that reduced quantal size does not underlie the deficit in AMPAR-mediated transmission in NP1/2 KO neurons.
Figure 2.

Quantal size is not reduced in early postnatal NP1/2 KO dLGN neurons (P6–P9). A, Examples of evoked responses in the absence and presence of Sr+2. Top left traces shows a WT recording in our regular Ca2+-containing external. The middle trace shows the WT response to the Sr2+ external. The bottom right trace is an example from an NP1/2 KO recording in Sr2+. Traces display an initial fast response (arrows) followed by delayed release of quantal events (asterisks). B, Overlay of the average miniature even from a WT neuron (black) and an NP1/2 KO neuron (gray). C, Cumulative probability histogram shows the average of the individual cumulative probability histograms. Distributions were not different by a Kolmogorov–Smirnov test. Data displayed as mean ± SEM. For WTs n = 4 animals (13 cells), and for KOs n = 3 animals (10 cells).
If quantal size is unaltered in NP1/2 KO, neurons what accounts for their diminished AMPAR-mediated currents? One possibility is that loss of NP1/2 results in fewer AMPAR-containing synapses, that is, an increased number of silent synapses. Analysis of the currents arising from the stimulation of presumably single retinal fibers has previously provided evidence for silent synapses at this developing connection (Chen and Regehr, 2000; Liu and Chen, 2008). Early in development individual retinal inputs form relatively few AMPAR-containing synapses with dLGN neurons; therefore, sometimes all the synapses associated with an individual retinal input are silent. These silent inputs can be detected using the protocol of Chen and Regehr (2000) in which reduced stimulus intensity is used to presumably activate just one retinal axon. Based on these studies, a retinal input is considered silent when this stimulus, which produces the smallest reliable NMDAR-mediated response when held at +40 mV, fails to elicit an AMPAR-mediated current when the neuron is held at −70 mV. We compared the number of these silent inputs observed in the WT and NP1/2 KO populations as an indicator of the relative abundance of silent synapses present on WT and KO neurons (Fig. 3). Using this protocol most WT neurons responded when held at either +40 mV or −70 mV; however, 38% (9 of 24) showed no AMPAR-mediated current creating a bimodal histogram of response amplitudes at −70 mV (Fig. 3A,C). In contrast, most NP1/2 KO neurons had detectable responses only at +40 mV with 69% (18 of 26) showing no AMPAR-mediated current; therefore, NP1/2 KO neurons had approximately twice as many silent inputs as WT neurons (Fig. 3A,D). Because single fiber stimulation either did or did not result in a postsynaptic response at −70 mV we used the binomial test to determine whether the probability of failure was higher in the NP1/2 KOs compared to WTs. We found that, given the probability of failure in the WT population, the likelihood of obtaining the specific distribution of responses and failures that was observed in the NP1/2 KO population (that is, 8 responses and 18 failures) was 0.0003, a highly significant reduction from WT (p < 0.001).
Figure 3.

Young NP1/2 KO dLGN neurons (P6 through P9) have an increased number of silent synapses. A, Example traces from WT (left) and NP1/2 KO (right) neurons at +40 and −70 mV showing responses to minimal stimulation (7.5 μA for both cells). WT neuron shows responses at both +40 and −70 mV, whereas NP1/2 KO neuron shows a response only at +40 mV. B, Average amplitudes of presumed single fiber responses at −70 mV and +40 mV. C, Histogram of single fiber responses of WT and NP1/2 KO neurons held at −70 mV. Note the increase in the number of NP1/2 KO neurons that showed no response. D, Histogram of single fiber responses of WT and NP1/2 KO neurons held at +40 mV. B–D, Data displayed as mean ± SEM. **p < 0.01. Dataset included 24 cells from 8 WT animals and 26 cells from 6 KO animals.
Because there are twice as many silent inputs in the NP1/2KOs compared to the WTs this suggests that KO neurons have at least twice as many silent synapses as WT neurons, and the average amplitudes of the successful single fiber responses recorded at −70 mV were in good agreement with this interpretation. On average the single fiber responses of the KOs at −70 mV were approximately half the size of the single fiber WT responses (Fig. 3B) (WT = −31.06 ± 4.03 pA, NP1/2KO = −16.29 ± 0.78 pA; p < 0.01). Similarly, the average AMPA/NMDA ratios were also reduced in NP1/2 KO neurons (WT = 0.44 ± 0.12, KO = 0.13 ± 0.05; p < 0.05). In WT animals the average quantal response at this age is −9.49 pA and the average single fiber response is −31.06 pA, suggesting that a typical WT RGC forms ∼3 fully functional AMPAR-containing synapses with a given WT dLGN neuron. In contrast, the average quantal and single fiber responses of the NP1/2 KOs were −9.89 pA and −16.29 pA, respectively, suggesting 1.5 AMPAR-containing synapses per RGC input; a reduction of approximately one half. Finally, the average single fiber responses at +40 mV were identical between WTs and KOs (Fig. 3B,D) (WT = 54.49 ± 7.06, NP1/2KO = 61.20 ± 10.07, p > 0.05). Because both the maximal and presumed single fiber NMDAR-mediated responses were the same for WTs and KOs, this suggests that the total number of synapses formed between individual RGCs and dLGN neurons was not altered by the loss of NP1/2 (Chen and Regehr, 2000). Based on these data we conclude that the deficit in synaptic transmission in the NP1/2 KO animals is most likely attributable to an increased number of synapses lacking functional AMPARs. These surprising findings suggest a novel role for NP1/2 in silent synapse conversion during early postnatal development; however, they do not rule out the possibility that NP1/2 may modulate the functional properties of AMPARs.
In WT mice AMPAR-mediated responses at retinogeniculate synapses strengthen dramatically between P10 and P30, which leads to approximately a fivefold increase in the AMPA/NMDA ratio and shifts retinogeniculate transmission from being NMDAR to AMPAR-dominated (Chen and Regehr, 2000). To determine whether this characteristic phase of synaptic strengthening is prevented in the absence of NP1/2 we measured synaptic transmission during this period, between the ages of P17 and P20. At this later age we found robust AMPAR-mediated responses in both WT and NP1/2 KO neurons (Fig. 4A) with AMPA/NMDA ratios that no longer differed between the two groups (WT = 1.17 ± 0.21; KO = 1.53 ± 0.29; p > 0.05), indicating that at this age NP1/2 are no longer required for the strengthening of AMPAR-mediated transmission. Although these data do not rule out a role for NP1/2 in the strengthening of AMPAR-mediated transmission later in development, they highlight the importance of NP1/2 at immature synapses because at this early stage retinogeniculate synapses cannot easily compensate for the loss of NP1/2.
Figure 4.

P17 to P20 NP1/2 KO animals strengthen AMPAR-mediated currents by an NP1/2-independent mechanism and develop aberrantly large current amplitudes. A, Example traces at +40 mV and −70 mV from WT and NP1/2 KO neurons at age P19 showing maximal evoked responses (50 μA stimulus). B, Average saturating AMPAR and NMDAR-mediated responses. C, Example traces of presumed single fiber responses from a WT neuron (10 μA stimulus) and a KO neuron (7.5 μA stimulus) recorded at +40 mV and −70 mV. D, Average minimal AMPAR and NMDAR-mediated responses. B, D, Data displayed as mean ± SEM. For WTs n = 8 animals and 17 cells. For KOs n = 9 animals and 17 cells. *p < 0.05. E, Examples of PPRs at −70 mV in cyclothiazide. F, Quantification of PPRs recorded at −70 mV and +40 mV. For −70 mV recordings n = 3 WT animals (8 cells) and n = 2 KO animals and 6 cells. For +40 mV recordings n = 2 animals and 4 cells for both genotypes. Data displayed as mean ± SEM.
A surprising aspect of our P17-P20 recordings was that although the AMPA/NMDA ratios of NP1/2 KO neurons were not significantly different from WTs, the absolute amplitudes of both the AMPAR and NMDAR-mediated currents were increased in NP1/2 KO neurons compared to WT neurons (Fig. 4B) (WT AMPA = −1102.92.08 ± 173.16 and NMDA = 1057.03 ± 220.92 pA; KO AMPA = −2993.84 ± 535.27 and NMDA = 1900.29 ± 184.48; p < 0.05 for both current components). We next asked whether an increase in the strength of individual inputs might be contributing to the enhanced maximal KO currents. Using minimal stimulation to measure the currents arising from presumably single fibers we found that WTs and KOs did not differ in the amplitudes of their presumed single fiber AMPAR-mediated currents (Fig. 4C,D) (WT = −195.77 ± 53.86, NP1/2KO = −193.13 ± 73.80; p > 0.05), the amplitude of their single fiber NMDAR-mediated currents (Fig. 4C,D) (WT = 184.33 ± 84.64, NP1/2KO = 144.27 ± 25.22; p > 0.05), or in their AMPA/NMDA ratios (WT = 1.16 ± 0.18, NP1/2KO = 1.46 ± 0.49; p > 0.05).
Because the presumed single fiber currents were not increased in the KOs, this suggests that enhanced presynaptic release may not contribute to their excessive total synaptic drive; however, to investigate the contribution of altered release probability to the aberrantly large synaptic currents in the KOs we measured the paired pulse ratios of P17-P20 WT and NP1/2 KO neurons. Using an ISI of 100 ms and found that there was no difference between the two groups when recorded at either −70 mV in the presence of cyclothiazide to eliminate the effects of AMPAR desensitization (Chen and Regehr, 2003) (Fig. 4E,F) (WT = 0.50 ± 0.06; KO = 0.51 ± 0.09; p > 0.05) or at +40 mV (Fig. 4F) (WT = 0.51 ± 0.05, NP1/2KO = 0.49 ± 0.06; p > 0.05). These findings suggest that enhanced presynaptic release does not account for the increased synaptic currents in the NP1/2 KOs.
Because the maximal currents were enhanced in the KOs while their single fiber currents were unchanged, this suggests individual dLGN neurons may be innervated by an increased number of RGC axons. While AMPAR-mediated transmission is dramatically strengthened at “winning” retinogeniculate synapses between P10 and P30, another prominent feature of this phase of retinogeniculate development is the removal of inappropriate functional connections. This phase is often referred to as “within-eye segregation” because eye-specific segregation is largely completed but dLGN neurons are still refining excessive inputs originating from the same eye. During this period the number of RGC inputs onto individual dLGN neurons is reduced from around a dozen to just one or two (Chen and Regehr, 2000; Ziburkus et al., 2003). Because both synaptic maturation and eye-specific segregation are severely delayed in the NP1/2 KOs (Bjartmar et al., 2006), we reasoned that within-eye segregation might also be delayed in the absence of NP1/2 leading to more highly innervated dLGN neurons. Two methods were used to determine whether P17-P20 NP1/2 KO neurons might receive input from a greater number of RGCs than WT neurons. First, we divided the maximal currents by the minimal currents to estimate the number of inputs received by individual dLGN neurons. This method suggested that on average WT neurons between the ages of P17 and P20 receive approximately four to five inputs (Fig. 5B) (input number = 4.71 ± 1.80 and 4.94 ± 1.91 when neurons were held at −70 mV and +40 mV, respectively), an estimate similar to previous reports (Hooks and Chen, 2006). In contrast, NP1/2 KO neurons were estimated to receive input from around a dozen RGCs, a significant increase (Fig. 5B) (input number = 12.99 ± 5.64 at −70 mV and 13.14 ± 3.58 at +40 mV; p < 0.05 for both).
Figure 5.

NP1/2 KOs display abnormal input elimination during development. A, Example traces showing a “refined” P18 WT neuron (top) and an “unrefined” P18 NP1/2 KO neuron (bottom). B, The total current divided by the presumably single fiber current for WT and NP1/2 KO neurons held at −70 mV and +40 mV. Dataset includes 17 WT cells from 6 animals and 17 KO cells from 7 animals. *p < 0.05. C, WT and KO neurons were classified into unrefined, resolving, and refined categories based on the number of stepwise increases in synaptic current amplitudes observed in response in increasing stimulus intensity. WT dataset includes 16 cells from 6 animals and KO dataset includes 14 cells from 6 animals.
To gain further evidence regarding whether input elimination might be aberrant in the NP1/2 KOs we counted functional inputs onto dLGN neurons using a common method in which the stimulus amplitude is adjusted to elicit a single fiber response and then gradually increased to systematically recruit all additional inputs to the neuron (Fig. 5A,C) (Chen and Regehr, 2000). The visibly discernable “steps” in the current traces were counted and cells were characterized by sorting them into three categories: “refined” cells had just one or two large inputs, “resolving” cells had three to six easily discernable inputs, and “unrefined” cells had more than six inputs with current steps that were less well separated (Hooks and Chen, 2006; Stevens et al., 2007). Inputs were counted blind to genotype to prevent experimenter bias. At this age, dLGN neurons are in the process of becoming refined and they typically receive one or two strong inputs and sometimes a few additional weak inputs (Jaubert-Miazza et al., 2005; Hooks and Chen, 2006; Ziburkus and Guido, 2006). As expected for this age, we found that all WT cells were either refined or resolving (Fig. 5C). However, we found no fully refined NP1/2 KO neurons at this age while many KO neurons remained unrefined (Fig. 5C), which is consistent with the increased input number estimated above. Together these data indicate that NP1/2 KO dLGN neurons fail to prune their retinal inputs on a normal time course and suggest that collectively the excess inputs are capable of driving aberrantly large currents.
Finally, the striking temporal correlation between the delayed development of AMPAR-mediated currents observed in this study and the delay in eye-specific segregation in the KOs that was previously reported begs the question of whether these processes might be linked. As a first step toward answering this question we sought to determine whether eye-specific segregation might be blocked at the level of retinogeniculate synapses in the NP1/2 KOs. Alternatively, as mentioned earlier, NP1/2 KOs also displayed slightly altered retinal activity during this period. Although none of the parameters of spontaneous RGC activity that are thought to be required for eye-specific segregation were altered in the NP1/2 KOs (Torborg et al., 2005; Bjartmar et al., 2006) it remains possible that increased spike frequency within bursts prevents eye-specific segregation in the KOs. To investigate the level of the circuit at which segregation is blocked in the absence of NP1/2 we used in vivo drug administration and axon labeling. In WT animals intraocular drug administration was previously used to show that eye-specific segregation is a competitive process (Penn et al., 1998; Stellwagen and Shatz, 2002). In these studies correlated retinal waves were either disrupted or enhanced in just one eye. The authors showed that reducing activity in one eye causes the axon arbors from the blocked eye to lose territory in the dLGN while the arbors from the untreated eye expand, and the converse is true when activity is enhanced in one eye. Here we used this protocol to determine the level of the circuit at which eye-specific segregation is blocked in the absence of NP1/2. If in the KOs retinal axons fail to refine because these animals experience slightly altered retinal activity, then pharmacological manipulations that dramatically perturb the balance of activity between the two eyes (for example, inhibiting activity in one eye) may be able to overcome this and drive axon remodeling in the KO dLGN. However, if in the NP1/2 KOs eye-specific segregation is blocked downstream of retinal activity (for instance, at the level of retinogeniculate synapses) then one would predict that axon refinement would remain blocked even when activity is reduced in one eye. For these experiments one eye was injected with epibatidine on P4, P6, and P8 to abolish correlated retinal waves and to reduce overall retinal spiking (Sun et al., 2008) between the ages of P4 and P10, the normal period for eye-specific segregation (Godement et al., 1984). The other eye was treated with saline. Fluorescent dyes were injected into the eyes on P9 and brains were removed for analysis at P10. In addition, control groups of WT and KO animals received binocular saline injections. Our assumption in these experiments is that epibatidine blocks correlated retinal activity in KOs as well as in WTs, though we did not test this directly.
As was previously shown, we observed that in WT animals, epibatidine treatment led to a reduction in the size of the ipsilateral projection arising from the drug-treated eye and an expansion of the arbors from the saline-treated eye (Fig. 6A,B) (one-way ANOVA followed by Bonferroni multiple-comparisons test). In contrast to these WT findings, when performed on NP1/2 KO mice monocular injections of epibatidine failed to reduce the ipsilateral projection arising from the treated eye (Fig. 6A,B). Animals treated with binocular saline injections showed no imbalance between the sizes of their ipsilateral projections regardless of genotype. These data indicate that when NP1/2 are absent axon remodeling in the dLGN is insensitive to manipulations of retinal activity, which strongly suggests that eye-specific segregation is blocked downstream of retinal activity in young NP1/2 KO mice. These data argue against altered retinal waves as being the primary cause of the abnormal eye-specific segregation in the NP1/2 KOs and are consistent with the idea that the loss of NP1/2 disrupts activity-dependent axon segregation at the level of the dLGN.
Figure 6.
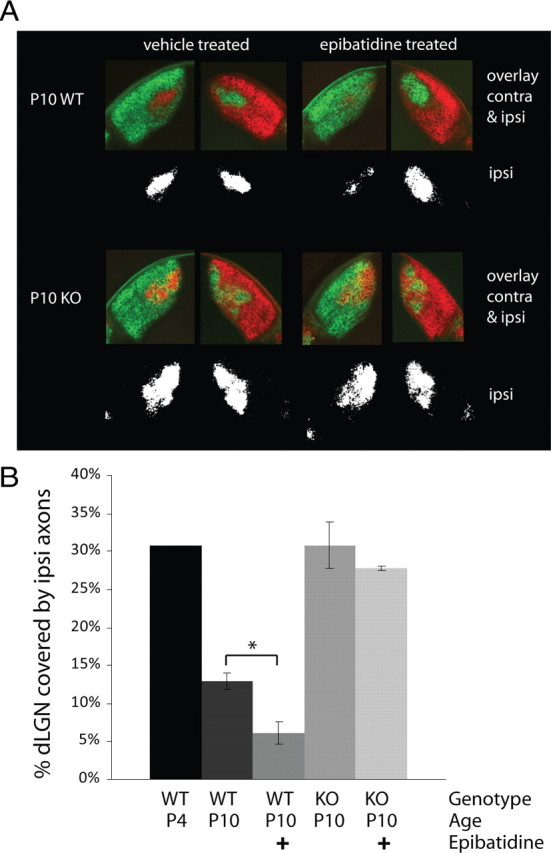
Monocular retinal activity blockade with epibatidine between the ages of P4 and P10 does not drive axon remodeling in the NP1/2 KO dLGN. A, Examples of RGC axon labeling in the dLGN of P10 vehicle-treated (left column) and epibatidine-treated (right column) WT (top row) and NP1/2 KO (bottom row) animals. Fluorescent images show the arborization patterns of both the contralateral and ipsilateral afferents, whereas the black-and-white images below display the thresholded ipsilateral projections. For the epibatidine-treated animals, the drug-treated eye received red label. Note the diminished ipsilateral projection from the epibatidine-treated eye in the WT example, whereas epibatidine failed to diminish the ipsilateral projection in the KO example. B, Bar graph quantifying the percentage of dLGN area over which the ipsilateral axons extend. In WT animals epibatidine treatment reduced the area covered by the ipsilateral axons (n = 3 WT vehicle-treated animals and n = 4 WT drug-treated animals), whereas epibatidine had no effect on the ipsilateral axons in the NP1/2 KO animals (n = 4 KO animals in each group). Note that the ipsilateral projection is larger in the P10 KOs than in the WTs and is similar in size to the ipsi projection of an unrefined P4 WT mouse. B, Data displayed as mean ± SEM. Data analyzed by one-way ANOVA followed by Bonferroni multiple-comparisons test. *p < 0.05.
Discussion
NP1/2 loss leads to reduced synaptic AMPAR-mediated currents in vivo
Here we report that in mice lacking NP1/2 AMPAR-mediated synaptic transmission is severely reduced in the visual thalamus during early postnatal life. This reduction can be explained by an increase in the number of synapses lacking detectable AMPAR-mediated currents with no accompanying changes in quantal size, paired pulse ratio, or NMDAR-mediated currents. These data are the first to demonstrate a requirement for NP1/2 in the establishment of AMPAR-mediated synaptic transmission in vivo.
Our finding that NPs are key factors mediating the early development of AMPAR-mediated transmission is well supported by previous studies of NP function in cell culture systems (O'Brien et al., 1999, 2002; Xu et al., 2003; Sia et al., 2007). However, that NP1/2 are specifically required for early silent synapse conversion is novel and not predicted from the earlier in vitro studies. For instance, Sia et al. recorded from HEK293T cells that were transfected with GluR4 and neuroligin and cultured on NP triple knock-out hippocampal neurons and found a reduction in both the amplitude and frequency of their spontaneous currents (Sia et al., 2007). Similarly, based on biochemical and immunocytochemical data, Xu et al. put forth a model in which NP1/2 modulate both the number and size of glutamate receptor clusters at excitatory synapses with NP1 stimulating modest increases in glutamate receptor aggregation over baseline and NP2 synergizing with NP1 to more dramatically increase this clustering (Xu et al., 2003). In contrast to these in vitro findings that suggest graded models of NP-dependent glutamate receptor recruitment, our in situ data are more consistent with a model in which NP1/2 are required for the synaptic localization of clusters of glutamate receptors in an all-or-none fashion.
Our results are also somewhat surprising given that AMPAR-mediated transmission was measured in NP1/2 KO hippocampal slices and was not found to be reduced (Bjartmar et al., 2006; Cho et al., 2008). There are at least three potential explanations for this discrepancy. First, the mechanisms for AMPAR recruitment may vary across brain regions. Second, in this study we found that the requirement for NP1/2 was restricted to a specific developmental period, suggesting that deficits in hippocampal transmission may have been missed in the aforementioned studies because those recordings were performed at later ages. Finally, it has been suggested that NPs may act preferentially at excitatory synapses located on dendritic shafts (as opposed to spine synapses) (Mi et al., 2002). Because thalamic relay neurons are largely nonspiny (Rafols and Valverde, 1973; Kriebel, 1975) they receive nearly all of their excitatory input on dendritic shafts, and therefore the loss of NP1/2 may more profoundly impact relay cell transmission (and that of other nonspiny neurons) than pyramidal cell transmission.
Are the affects of NP1/2 on retinogeniculate transmission direct?
NP1/2 are synaptic molecules that cluster AMPARs at synapses in vitro; therefore, it is reasonable to speculate that their loss from retinogeniculate synapses may alter AMPAR-mediated transmission. Alternatively, the effects of NP1/2 loss on retinogeniculate transmission could be an indirect consequence of alterations in upstream visual circuitry. Specifically, developing RGCs fire spontaneous bursts of action potentials, and spike frequency during bursts was enhanced in the NP1/2 KOs. To begin to examine the affect of altered retinal spiking on retinogeniculate transmission we pharmacologically increased retinal spiking in WT animals in vivo over several days to determine whether enhanced RGC spiking can reduce retinogeniculate transmission. Because increased retinal spiking had no affect on WT transmission, this result argues against the idea that the enhanced spiking in the KOs is the major cause of their reduced AMPAR-mediated currents and is consistent with NP1/2 playing a direct role at developing retinogeniculate synapses. However, though cAMP treatment and NP1/2 loss both enhance retinal spiking, their effects on retinal activity are not identical; therefore, it remains possible that the specific aspects of retinal activity that are altered in the KOs may play a role in recruiting AMPARs to retinogeniculate synapses. An additional possibility is that the KO RGCs increase their spiking in an effort to compensate for their reduced ability to transmit signals to their postsynaptic partners. In any case, NP1/2 are clearly required for the normal development of AMPAR-mediated currents at retinogeniculate synapses in vivo.
The requirement for NP1/2 is restricted to a specific developmental period
An important finding from this study is that the absence of NP1/2 leads to a deficit in silent synapse conversion during a specific developmental period, and that after P9 an NP1/2-independent mechanism arises which can compensate for the loss of NP1/2. This observation supports the emerging notion that nascent and more mature synapses use differing mechanisms for synaptic AMPAR recruitment. Studies have begun to demonstrate fundamental differences in the plasticity rules, signaling pathways, and expression mechanisms that govern synaptic AMPAR recruitment at immature and mature synapses. For example, immature and mature hippocampal synapses display differing signaling pathways mediating long-term potentiation (Yasuda et al., 2003). The involvement of NMDAR activation in synaptic AMPAR expression also appears to change over development (Kim et al., 2005; Hall and Ghosh, 2008), and early and late synapses throughout the brain differ in a variety of molecular components including both NMDAR and AMPAR subunits (Zhu et al., 2000; Barria and Malinow, 2002; Ritter et al., 2002) as well as synaptic adhesion, signaling, and scaffolding molecules (Petralia et al., 2005). Though our results certainly do not exclude the possibility that NP1/2 may strengthen AMPAR-mediated transmission at more mature synapses, they strongly indicate that NP1/2 play an important role during the initial establishment of functional synaptic transmission and thereby contribute to our understanding of the mechanisms orchestrating the development of AMPAR-mediated transmission.
What is the basis of the excessive synaptic currents observed in the P17-P20 NP1/2 knock-out animals?
We have previously reported that in the NP1/2 KO dLGN eye-specific segregation does not occur during the normal period for this process, but instead ensues later during the period that normally corresponds to within-eye segregation and functional input elimination (Bjartmar et al., 2006). Here we show that in addition to delayed eye-specific segregation, functional input elimination is also altered in the absence of NP1/2. At P17 NP1/2 KO neurons received input from more RGCs than WT neurons at this age. This finding is likely to reflect a delay in input elimination rather than a permanent prevention of functional refinement because we observed some KO dLGN neurons that were clearly beginning to resolve, and from our previous study we know that eye-specific axons eventually sort themselves out in the KOs (Bjartmar et al., 2006). However, it remains possible that within-eye inputs may not completely segregate in the KOs.
Our finding that the extra RGC inputs were collectively capable of driving larger currents at retinogeniculate synapses is unique because other manipulations that have been shown to delay or prevent input elimination at this synapse do not appear to alter total current amplitudes. For instance, Hooks and Chen reported that retinal activity block with tetrodotoxin between the ages examined here similarly prevents input elimination but does not increase the total AMPAR or NMDAR-mediated currents (Hooks and Chen, 2006), and Stevens et al. found a similar phenotype in the absence of the classical immune molecule C1q (Stevens et al., 2007). These data suggest that excessive numbers of retinal inputs do not necessarily lead to excessive currents. In fact they suggest quite the opposite, that total current amplitude may be homeostatically regulated in dLGN neurons. This type of homeostasis has also been reported in the superior colliculus, another retinorecipient region (Shah and Crair, 2008a,b). In light of these previous findings, the enhanced synaptic currents observed in the older NP1/2 KO neurons may be attributable to the failure of such a homeostatic mechanism. For example, the early loss of AMPAR-mediated transmission in the NP1/2 KO mouse could lead to an elevation in the homeostatic set-point resulting in abnormally large AMPAR-mediated currents once these cells become capable of strengthening them. Interestingly, the AMPA/NMDA ratios of the older NP1/2 KO neurons were identical to their WT counterparts, suggesting the amplitudes of the two current components may be mechanistically linked. Whether NP1/2 play a role in homeostasis at this connection and what the nature of that role might be remain open questions for future studies.
In addition, it is worth noting that the third NP, NPR, has recently been shown to be required for mGluR1/5-dependent LTD in slices from P21 to P29 hippocampus (Cho et al., 2008); therefore, an alternative explanation could be that although NP1/2 clearly strengthen AMPAR-mediated transmission early on, as development proceeds they may take on a new role in either limiting or weakening synaptic currents. However, it is unlikely that defective LTD can explain the excessive retinogeniculate currents observed in this study for two reasons: first, the Cho et al. study found no further deficit in LTD when NP1/2 were deleted in addition to NPR, and second, retinogeniculate synapses are not thought to express mGluRs (Godwin et al., 1996; Salt, 2002).
Finally, if NP1/2 play a role in the generation of stage III (glutamatergic) retinal waves then the KOs may experience abnormal retinal activity during within eye segregation, which could contribute to their altered input elimination during this period. In the future, the basis of the excessive synaptic currents and the role of NP1/2 during within eye segregation will be interesting topics for additional studies.
What mechanisms underlie the eye-specific segregation defect in the NP1/2 KOs?
In our previous study we demonstrated that in the NP1/2 KO mouse retinal axons fail to undergo eye-specific refinement during the normal period for this process; however, the mechanistic basis of this anatomical defect remained unclear (Bjartmar et al., 2006). Eye-specific segregation requires spontaneous spiking activity in the retina (Shatz and Stryker, 1988; Sretavan et al., 1988), and the structure of the this activity is thought to instruct synaptic competition in the dLGN (Penn et al., 1998; Rossi et al., 2001; Muir-Robinson et al., 2002; Stellwagen and Shatz, 2002; but see Huberman et al., 2003; Stafford et al., 2009). Like WT RGCs, NP1/2 KO RGCs fired periodic bursts of action potentials, and KO retinas displayed propagating waves of retinal activity during eye-specific segregation. In addition, many of the features of retinal waves that are thought to be important for driving segregation were intact in the KOs (KO RGCs fired action potentials within high-frequency bursts, bursting among neighboring KO RGCs was highly correlated, correlated firing diminished with distance, and interburst intervals were identical between WTs and KOs). Despite this, some differences between WT and KO waves were apparent and could contribute to the delayed segregation (Bjartmar et al., 2006). Namely, NP1/2 KO RGCs exhibited increased spike frequencies during bursts which resulted in both neighboring and more distant KO RGCs firing greater numbers of synchronous spikes. Consequently, we performed an experiment to begin to investigate the impact of altered retinal activity on eye-specific segregation in the NP1/2 KOs. Retinal waves were pharmacologically blocked in one eye over the period of eye-specific segregation using epibatidine (our presumption is that epibatidine also blocks waves in the KO retinas). Similar to previous reports, we found that in WT animals monocular blockade of retinal activity caused the axons from the drug-treated eye to lose territory in the dLGN while the axons from the saline-treated eye expanded; however, in the absence of NP1/2 monocular activity blockade had no effect on the arborization patterns of RGC axons. The insensitivity of NP1/2 KO axons to the manipulation of retinal activity suggests that eye-specific refinement may be blocked downstream of retinal activity when NP1/2 are absent, perhaps at the level of retinogeniculate synapses. Thus, NP1/2 may be among a small number of molecules currently shown to sit at the crossroads between structural and functional plasticity in this developing circuit (Upton et al., 1999; Huh et al., 2000; Menna et al., 2003; Stevens et al., 2007). However, additional experiments will clearly be required to clarify the relationships between the functional and structural phenotypes in the NP1/2 KO mouse.
In summary, the findings presented here demonstrate that NP1/2 are required in vivo for the normal development of AMPAR-mediated synaptic transmission and specifically implicate NP1/2 in silent synapse conversion. These data provide new insight into the molecular mechanisms for AMPAR recruitment at nascent versus mature synapses. In addition, the requirement for NP1/2 was restricted to an early developmental period when neural circuits are undergoing dramatic remodeling, suggesting that NP1/2-dependent synaptic strengthening could contribute to developmental circuit refinement.
Footnotes
This work was supported by a Research to Prevent Blindness Young Investigator Award, March of Dimes Basil O'Connor Award, That Man May See (to E.M.U.), National Institutes of Health-National Eye Institute Grant T32 EY07120, and Achievement Rewards for College Scientists Foundation Scholarship (to S.M.K.). We thank David Copenhagen, Andrew Huberman, and the members of the Ullian laboratory for their thoughtful comments on this manuscript. We thank Dr. Mark Perin for providing NP1/2 KO mice.
References
- Barria A, Malinow R. Subunit-specific NMDA receptor trafficking to synapses. Neuron. 2002;35:345–353. doi: 10.1016/s0896-6273(02)00776-6. [DOI] [PubMed] [Google Scholar]
- Bjartmar L, Huberman AD, Ullian EM, Rentería RC, Liu X, Xu W, Prezioso J, Susman MW, Stellwagen D, Stokes CC, Cho R, Worley P, Malenka RC, Ball S, Peachey NS, Copenhagen D, Chapman B, Nakamoto M, Barres BA, Perin MS. Neuronal pentraxins mediate synaptic refinement in the developing visual system. J Neurosci. 2006;26:6269–6281. doi: 10.1523/JNEUROSCI.4212-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Regehr WG. Developmental remodeling of the retinogeniculate synapse. Neuron. 2000;28:955–966. doi: 10.1016/s0896-6273(00)00166-5. [DOI] [PubMed] [Google Scholar]
- Chen C, Regehr WG. Presynaptic modulation of the retinogeniculate synapse. J Neurosci. 2003;23:3130–3135. doi: 10.1523/JNEUROSCI.23-08-03130.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho RW, Park JM, Wolff SB, Xu D, Hopf C, Kim JA, Reddy RC, Petralia RS, Perin MS, Linden DJ, Worley PF. mGluR1/5-dependent long-term depression requires the regulated ectodomain cleavage of neuronal pentraxin NPR by TACE. Neuron. 2008;57:858–871. doi: 10.1016/j.neuron.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godement P, Salaün J, Imbert M. Prenatal and postnatal development of retinogeniculate and retinocollicular projections in the mouse. J Comp Neurol. 1984;230:552–575. doi: 10.1002/cne.902300406. [DOI] [PubMed] [Google Scholar]
- Godwin DW, Van Horn SC, Eriir A, Sesma M, Romano C, Sherman SM. Ultrastructural localization suggests that retinal and cortical inputs access different metabotropic glutamate receptors in the lateral geniculate nucleus. J Neurosci. 1996;16:8181–8192. doi: 10.1523/JNEUROSCI.16-24-08181.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall BJ, Ghosh A. Regulation of AMPA receptor recruitment at developing synapses. Trends Neurosci. 2008;31:82–89. doi: 10.1016/j.tins.2007.11.010. [DOI] [PubMed] [Google Scholar]
- Hooks BM, Chen C. Distinct roles for spontaneous and visual activity in remodeling of the retinogeniculate synapse. Neuron. 2006;52:281–291. doi: 10.1016/j.neuron.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Huberman AD, Wang GY, Liets LC, Collins OA, Chapman B, Chalupa LM. Eye-specific retinogeniculate segregation independent of normal neuronal activity. Science. 2003;300:994–998. doi: 10.1126/science.1080694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh GS, Boulanger LM, Du H, Riquelme PA, Brotz TM, Shatz CJ. Functional requirement for class I MHC in CNS development and plasticity. Science. 2000;290:2155–2159. doi: 10.1126/science.290.5499.2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaubert-Miazza L, Green E, Lo FS, Bui K, Mills J, Guido W. Structural and functional composition of the developing retinogeniculate pathway in the mouse. Vis Neurosci. 2005;22:661–676. doi: 10.1017/S0952523805225154. [DOI] [PubMed] [Google Scholar]
- Kim MJ, Dunah AW, Wang YT, Sheng M. Differential roles of NR2A- and NR2B-containing NMDA receptors in Ras-ERK signaling and AMPA receptor trafficking. Neuron. 2005;46:745–760. doi: 10.1016/j.neuron.2005.04.031. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick LL, Matzuk MM, Dodds DC, Perin MS. Biochemical interactions of the neuronal pentraxins. Neuronal pentraxin (NP) receptor binds to taipoxin and taipoxin-associated calcium-binding protein 49 via NP1 and NP2. J Biol Chem. 2000;275:17786–17792. doi: 10.1074/jbc.M002254200. [DOI] [PubMed] [Google Scholar]
- Kriebel RM. Neurons of the dorsal lateral geniculate nucleus of the albino rat. J Comp Neurol. 1975;159:45–67. doi: 10.1002/cne.901590105. [DOI] [PubMed] [Google Scholar]
- Liu X, Chen C. Different roles for AMPA and NMDA receptors in transmission at the immature retinogeniculate synapse. J Neurophysiol. 2008;99:629–643. doi: 10.1152/jn.01171.2007. [DOI] [PubMed] [Google Scholar]
- Menna E, Cenni MC, Naska S, Maffei L. The anterogradely transported BDNF promotes retinal axon remodeling during eye specific segregation within the LGN. Mol Cell Neurosci. 2003;24:972–983. doi: 10.1016/s1044-7431(03)00258-6. [DOI] [PubMed] [Google Scholar]
- Mi R, Tang X, Sutter R, Xu D, Worley P, O'Brien RJ. Differing mechanisms for glutamate receptor aggregation on dendritic spines and shafts in cultured hippocampal neurons. J Neurosci. 2002;22:7606–7616. doi: 10.1523/JNEUROSCI.22-17-07606.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miledi R. Strontium as a substitute for calcium in the process of transmitter release at the neuromuscular junction. Nature. 1966;212:2. doi: 10.1038/2121233a0. [DOI] [PubMed] [Google Scholar]
- Muir-Robinson G, Hwang BJ, Feller MB. Retinogeniculate axons undergo eye-specific segregation in the absence of eye-specific layers. J Neurosci. 2002;22:5259–5264. doi: 10.1523/JNEUROSCI.22-13-05259.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien R, Xu D, Mi R, Tang X, Hopf C, Worley P. Synaptically targeted narp plays an essential role in the aggregation of AMPA receptors at excitatory synapses in cultured spinal neurons. J Neurosci. 2002;22:4487–4498. doi: 10.1523/JNEUROSCI.22-11-04487.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien RJ, Xu D, Petralia RS, Steward O, Huganir RL, Worley P. Synaptic clustering of AMPA receptors by the extracellular immediate-early gene product Narp. Neuron. 1999;23:309–323. doi: 10.1016/s0896-6273(00)80782-5. [DOI] [PubMed] [Google Scholar]
- Penn AA, Riquelme PA, Feller MB, Shatz CJ. Competition in retinogeniculate patterning driven by spontaneous activity. Science. 1998;279:2108–2112. doi: 10.1126/science.279.5359.2108. [DOI] [PubMed] [Google Scholar]
- Petralia RS, Sans N, Wang YX, Wenthold RJ. Ontogeny of postsynaptic density proteins at glutamatergic synapses. Mol Cell Neurosci. 2005;29:436–452. doi: 10.1016/j.mcn.2005.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafols JA, Valverde F. The structure of the dorsal lateral geniculate nucleus in the mouse. A Golgi and electron microscopic study. J Comp Neurol. 1973;150:303–332. doi: 10.1002/cne.901500305. [DOI] [PubMed] [Google Scholar]
- Reti IM, Reddy R, Worley PF, Baraban JM. Prominent Narp expression in projection pathways and terminal fields. J Neurochem. 2002;82:935–944. doi: 10.1046/j.1471-4159.2002.01051.x. [DOI] [PubMed] [Google Scholar]
- Ritter LM, Vazquez DM, Meador-Woodruff JH. Ontogeny of ionotropic glutamate receptor subunit expression in the rat hippocampus. Brain Res Dev Brain Res. 2002;139:227–236. doi: 10.1016/s0165-3806(02)00572-2. [DOI] [PubMed] [Google Scholar]
- Rossi FM, Pizzorusso T, Porciatti V, Marubio LM, Maffei L, Changeux JP. Requirement of the nicotinic acetylcholine receptor beta 2 subunit for the anatomical and functional development of the visual system. Proc Natl Acad Sci U S A. 2001;98:6453–6458. doi: 10.1073/pnas.101120998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salt TE. Glutamate receptor functions in sensory relay in the thalamus. Philos Trans R Soc Lond B Biol Sci. 2002;357:1759–1766. doi: 10.1098/rstb.2002.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah RD, Crair MC. Retinocollicular synapse maturation and plasticity are regulated by correlated retinal waves. J Neurosci. 2008a;28:292–303. doi: 10.1523/JNEUROSCI.4276-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah RD, Crair MC. Mechanisms of response homeostasis during retinocollicular map formation. J Physiol. 2008b;586:4363–4369. doi: 10.1113/jphysiol.2008.157222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shatz CJ, Stryker MP. Prenatal tetrodotoxin infusion blocks segregation of retinogeniculate afferents. Science. 1988;242:87–89. doi: 10.1126/science.3175636. [DOI] [PubMed] [Google Scholar]
- Sia GM, Béïque JC, Rumbaugh G, Cho R, Worley PF, Huganir RL. Interaction of the N-terminal domain of the AMPA receptor GluR4 subunit with the neuronal pentraxin NP1 mediates GluR4 synaptic recruitment. Neuron. 2007;55:87–102. doi: 10.1016/j.neuron.2007.06.020. [DOI] [PubMed] [Google Scholar]
- Sretavan DW, Shatz CJ, Stryker MP. Modification of retinal ganglion cell axon morphology by prenatal infusion of tetrodotoxin. Nature. 1988;336:468–471. doi: 10.1038/336468a0. [DOI] [PubMed] [Google Scholar]
- Stafford BK, Sher A, Litke AM, Feldheim DA. Spatial-temporal patterns of retinal waves underlying activity-dependent refinement of retinofugal projections. Neuron. 2009;64:200–212. doi: 10.1016/j.neuron.2009.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen D, Shatz CJ. An instructive role for retinal waves in the development of retinogeniculate connectivity. Neuron. 2002;33:357–367. doi: 10.1016/s0896-6273(02)00577-9. [DOI] [PubMed] [Google Scholar]
- Stellwagen D, Shatz CJ, Feller MB. Dynamics of retinal waves are controlled by cyclic AMP. Neuron. 1999;24:673–685. doi: 10.1016/s0896-6273(00)81121-6. [DOI] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- Sun C, Speer CM, Wang GY, Chapman B, Chalupa LM. Epibatidine application in vitro blocks retinal waves without silencing all retinal ganglion cell action potentials in developing retina of the mouse and ferret. J Neurophysiol. 2008;100:3253–3263. doi: 10.1152/jn.90303.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torborg CL, Feller MB. Unbiased analysis of bulk axonal segregation patterns. J Neurosci Methods. 2004;135:17–26. doi: 10.1016/j.jneumeth.2003.11.019. [DOI] [PubMed] [Google Scholar]
- Torborg CL, Hansen KA, Feller MB. High frequency, synchronized bursting drives eye-specific segregation of retinogeniculate projections. Nat Neurosci. 2005;8:72–78. doi: 10.1038/nn1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton AL, Salichon N, Lebrand C, Ravary A, Blakely R, Seif I, Gaspar P. Excess of serotonin (5-HT) alters the segregation of ispilateral and contralateral retinal projections in monoamine oxidase A knock-out mice: possible role of 5-HT uptake in retinal ganglion cells during development. J Neurosci. 1999;19:7007–7024. doi: 10.1523/JNEUROSCI.19-16-07007.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voronin LL, Cherubini E. ‘Deaf, mute and whispering’ silent synapses: their role in synaptic plasticity. J Physiol. 2004;557:3–12. doi: 10.1113/jphysiol.2003.058966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao MY, Wasling P, Hanse E, Gustafsson B. Creation of AMPA-silent synapses in the neonatal hippocampus. Nat Neurosci. 2004;7:236–243. doi: 10.1038/nn1196. [DOI] [PubMed] [Google Scholar]
- Xu D, Hopf C, Reddy R, Cho RW, Guo L, Lanahan A, Petralia RS, Wenthold RJ, O'Brien RJ, Worley P. Narp and NP1 form heterocomplexes that function in developmental and activity-dependent synaptic plasticity. Neuron. 2003;39:513–528. doi: 10.1016/s0896-6273(03)00463-x. [DOI] [PubMed] [Google Scholar]
- Yasuda H, Barth AL, Stellwagen D, Malenka RC. A developmental switch in the signaling cascades for LTP induction. Nat Neurosci. 2003;6:15–16. doi: 10.1038/nn985. [DOI] [PubMed] [Google Scholar]
- Zhu JJ, Esteban JA, Hayashi Y, Malinow R. Postnatal synaptic potentiation: delivery of GluR4-containing AMPA receptors by spontaneous activity. Nat Neurosci. 2000;3:1098–1106. doi: 10.1038/80614. [DOI] [PubMed] [Google Scholar]
- Ziburkus J, Guido W. Loss of binocular responses and reduced retinal convergence during the period of retinogeniculate axon segregation. J Neurophysiol. 2006;96:2775–2784. doi: 10.1152/jn.01321.2004. [DOI] [PubMed] [Google Scholar]
- Ziburkus J, Lo FS, Guido W. Nature of inhibitory postsynaptic activity in developing relay cells of the lateral geniculate nucleus. J Neurophysiol. 2003;90:1063–1070. doi: 10.1152/jn.00178.2003. [DOI] [PubMed] [Google Scholar]