

Abstract
The structure of the sodium-benzylhydantoin transport protein, Mhp1, from Microbacterium liquefaciens comprises a 5-helix inverted repeat, which is widespread amongst secondary transporters. Here we report the crystal structure of an inward-facing conformation of Mhp1 at 3.8 Å resolution, complementing its previously-described structures in outward-facing and occluded states. From analyses of the three structures and molecular dynamics simulations we propose a mechanism for the transport cycle in Mhp1. Switching from the outward- to the inward- facing state, to effect the inward release of sodium and benzylhydantoin, is primarily achieved by a rigid body movement of transmembrane helices 3, 4, 8 and 9 relative to the rest of the protein. This forms the basis of an alternating access mechanism applicable to many transporters of this emerging superfamily.
Secondary-active transporters effect the cellular uptake and release of a wide range of substances across biological membranes in all organisms. They do this by coupling the uphill movement of the substrate against its concentration gradient with the energetically favourable downhill gradient of a second substrate, often a proton or a cation (1, 2). The kinetics and thermodynamics of these transporters can be explained by the alternating access model e.g. (3-6) but the structural details of the necessary conformational changes are only partly understood.
Recently the structures have been reported of a number of transporters of different families that have the same fold as LeuT (7), a bacterial homologue of the mammalian serotonin neurotransmitter transporter. These include the sodium-galactose symporter vSGLT (8), the sodium-benzylhydantoin transporter Mhp1 (9), the sodium-betaine symporter BetP (10), and two amino acid transporters, AdiC (11, 12) and ApcT (13). Dysfunction of members of this growing superfamily in humans can lead to diseases including neurological (14) and kidney (15) disorders. Other members are implicated in cancer as they can supply tumour cells with nutrients (16), cause drug resistance (17) and/or provide a means of treatment (18).
The fold shared by these transporters is an ‘inverted repeat’ motif with two sets of five transmembrane helices oppositely orientated with respect to the membrane (19, 20) (Fig. 1). The conformations observed for these transporters can be categorized into the following three classes: outward-facing, as observed in LeuT (7), Mhp1 (9) and AdiC (11, 12); occluded, where a trapped substrate is blocked from exiting on either side of the protein as seen in Mhp1 (9), BetP (10) and AdiC (21); and inward-facing, vSGLT (8) and ApcT (13). The alternating access model built up from these three conformations is accepted as a general concept for the LeuT superfamily of transporters (19, 20). However, the details of the transport mechanism remain largely controversial because the model has been derived by comparing transporters with divergent amino acid sequences that transport a wide variety of substrates.
Fig. 1.

Structure of the substrate-free inward-facing conformation of Mhp1. (A) Cartoon representation as seen from a viewpoint along the membrane. Each of the longer helices (TMs 3, 5, 8, 10) that show a distinct curvature are each shown as two shorter helices broken in the middle. The cartoon has been coloured with the ‘bundle’ (TMs 1, 2, 6, 7) in red, the ‘hash motif’ (TMs 3, 4, 8, 9) in yellow, the ‘flexible helices’ (TMs 5, 10) in blue, the C-terminal helices in light grey and the small extracellular and cytoplasmic surface helices (IN 2-3, OUT 7-8) in darker grey. To orientate the reader the positions of the sodium and benzylhydantoin that are observed in the occluded structure (9) are represented by a light grey sphere and sticks, respectively. (B) As (A) but viewed from the inside of the cell. (C) Topology diagram coloured as above, illustrating the pseudo twofold repeat unit.
Here, we present the substrate-free inward-facing structure of Mhp1, a sodium-hydantoin transporter from Microbacterium liquefaciens (9, 22, 23) at 3.8 Å resolution (Fig. 1). By comparing this with our previous structures of the outward-facing and occluded states (9) and using molecular dynamics simulations the transition from the outward-facing to the inward-facing conformations is seen primarily as a simple relative rotation of two rigid-body domains in combination with the bending of two TM helices. This straightforward mechanism synchronizes the opening and closing of the multiple ‘gates’ in the Mhp1 molecule to implement the flow of substrate and ions.
Overall Structure
The substrate-free inward-facing structure of Mhp1 was solved by the Se-SAD method and modelled using the high-resolution structure of the outward-facing conformation as a reference (9). Details are in the supplementary online material (24). It was refined at a resolution of 3.8 Å to an R-factor of 27.3% and a corresponding R-free of 31.3% (Table S1) (25). Despite the relatively low resolution of the data, the protein from residues 6-470 is well defined (Fig. S1A) enabling ready comparisons with the outward-facing and occluded structures of Mhp1 (9).
Mhp1 is composed of 12 transmembrane helices (TMs). The first 10 constitute the conserved motif in which TMs 1-5 are related to TMs 6-10 by a pseudo two-fold axis along the plane of the membrane (Fig. 1C). It is convenient to describe the structure as being made of two major parts. The first is a four-helix bundle comprising TMs 1-2 and their pseudo two-fold equivalents TMs 6-7 (residues 23-86 and residues 193-278). The second is another motif of four helices formed from TMs 3-4 and their pseudo two-fold equivalents, TMs 8-9 (residues 101-159 and 296-355). TMs 4 and 9 pack at 60° onto one surface of TMs 3 and 8 giving the appearance of the hash sign (#) when viewed from a point in the plane of the membrane (Fig. 1A). These two motifs will be referred to as the bundle and the hash motif. TM 5 and two short surface helices (IN 2-3, OUT 7-8) link the two motifs together and TM10 connects the hash motif with the two C-terminal TMs (Fig. 1C). We have named TM5 and TM10 the ‘flexible helices’ because they bend during the state transitions described below.
Substrate Binding Site
A large cavity on the inward facing side of the protein is made up of the neighbouring surfaces of TMs 1, 3, 5, 6 and 8 (Figs. 1B and S2). This cavity connects the substrate and cation binding sites that were identified in the occluded and outward-facing conformations (9) with the inside of the cell. The substrate binding site is located between TMs 1, 3, 6 and 8. In the occluded structure the substrate is sandwiched between the side chains of Trp 117 on TM3 and Trp 220 on TM6 (Fig. 2). Gln 121 on TM3 and Asn 318 on TM8 are also in close proximity to the ligand. In changing to the inward-facing structure large scale conformational changes result in Trp 117 moving slightly into the binding site and Gln 121 and Asn 318 moving away. These changes would significantly perturb the site and potentially impair the ligand binding ability.
Fig. 2.

Comparison of the sodium and substrate binding sites in the occluded and inward-facing structures of Mhp1. (A) The occluded state (9). The sodium is coloured magenta and the benzylhydantoin is represented with carbon atoms in cyan. The carbon atoms of the amino acids are coloured as in Fig. 1. The interactions between the sodium and neighbouring residues are shown with dotted black lines. (B) The inward-facing conformation. The amino acids are coloured as in (A). The positions of the sodium and benzylhydantoin are represented as in Fig. 1. In this conformation Thr 313 and Ser 312 would not be within liganding distance of a sodium at this position.
Sodium Binding Site
The Na+ binding site is located at the interface of TMs 1 and 8. In the outward-facing structure the cation interacts with the carbonyl oxygen atoms of Ala 38 and Ile 41 of TM1 and the carbonyl oxygen of Ala 309 and the side chains of Ser 312 and Thr 313 of TM8 (Fig. 2). In the inward-facing structure, on the other hand, TM8 has moved ~4.5Å away from TM1 so that the Na+ binding site is no longer intact (Fig. 2), which is reminiscent of the situation in the inward-facing vSGLT structure (8). In the electron density maps, we observe an elongated feature at this site (Fig. S3). The density is unlikely to be a Na+ ion because of its shape and its peak height (8σ). The use of anomalous diffraction failed to help identify the molecule: no significant peaks were observed in an X-ray fluorescence spectrum (26) (excited at 14keV) and no peaks were observed at this position in an X-ray anomalous difference Fourier map from data collected at 0.9791Å. One interesting hypothesis is that this molecule is trapping Mhp1 in the inward-facing conformation. This site could be a potential binding site for inhibitors of the ‘LeuT superfamily’ transporters, the subject of further studies.
Molecular Dynamics Simulations of Sodium Binding
To analyze if this perturbed site in the inward-facing conformation can still bind Na+, multi-scale molecular dynamics (MD) simulations were performed (Fig. 3, (24)). For the simulations, the Mhp1 molecules in three different conformations were placed in the native-like membrane composed of a 4:1 molar ratio of 1-palmitoyl-2-oleoylphosphatidylethanolamine (POPE) and 1-palmitoyl-2-oleoylglycero-3-phosphoglycerol (POPG) lipids (Fig. 3). The observed atomic Cα root mean square (r.m.s.) fluctuations range from 0.5 Å for rigid secondary structural elements to 4 Å for mobile loops (Fig. S4). Likewise the r.m.s. deviations from the starting structure range from 1.5 Å to 3 Å after 100 ns. These values are comparable with those for simulations of other high resolution membrane proteins (see supporting online text). In the multiple MD simulations of the inward-facing structure, sodium initially located in the binding site, very rapidly (< 2 ns) diffuses into the bulk water on the intracellular side of the protein through a large aqueous pathway between the bundle and the flanking region (Fig. 3C). Such rapid Na+ release is in marked contrast to similar simulations carried out on the occluded structure where the Na+ remained in its binding site for over 100 ns with only small variations in bond lengths to the ligating carbonyl oxygen atoms. In the outward-facing state without benzylhydantoin, an exit event was observed only after 18 ns in just one of three 100-ns simulations. We conclude from both structural and dynamic considerations that the Na+ binding site is intact in the outward-facing and occluded forms and disrupted in the inward-facing form. The presence of benzylhydantoin in its binding site blocks the pathway of the sodium ion to the extracellular side in the simulations and thus couples Na+ binding with substrate binding as shown in our earlier fluorescence quenching experiments (9). Recent simulations of sodium release from vSGLT arrive at similar conclusions for this structural homologue of Mhp1 (27).
Fig. 3.
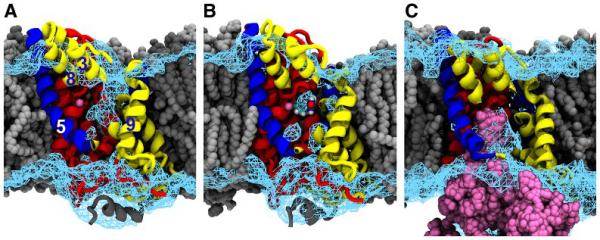
Accessibility of the sodium and substrate binding sites in different states of the transport cycle as defined by equilibrium MD simulations. Mhp1, coloured as in Fig. 1, is shown embedded in the lipid bilayer (grey) with part of the hash motif removed (TM4, parts of TM3 and TM8) to afford a view of the central cavity. The cyan wire mesh represents the solvent contoured at 20% of the bulk density. Sodium is shown in magenta. (A) In the outward-facing state, the central cavity is accessible from the extracellular side. (B) In the occluded state, the bound benzylhydantoin and the sodium ion together with a few water molecules are isolated in the central cavity. (C) In the inward-facing state, the pathway to the extracellular side is sealed and the sodium ion is released into the cytosol as shown by an overlay of the sodium ion positions from six independent MD trajectories (magenta).
Conformational Differences
Previously (9), we reported that, in going from the outward-facing to the occluded state, only TM10 and the preceding loop show any large scale difference in conformation. Between the occluded and inward-facing conformations, however, we observe much more dramatic changes (Fig. 4A) resulting in a root mean square deviation (r.m.s.d.) in the positions of all pairs of Cα atoms of the two structures of 3.3Å so that it resembles much more the conformation of the inward-facing structure of vSGLT (8) rather than the outward facing LeuT (7). The major changes are primarily due to a rigid body movement of the hash motif relative to the bundle. The C-terminal helices (TMs 11 and 12) move only slightly with respect to the bundle. Consistent with a rigid body movement, the respective residues of the bundle from the three conformations superpose very well with an r.m.s.d. of 0.7Å (Fig. S5B). Likewise the residues in the hash motif from the three conformational states are very similar (r.m.s.d. 0.9 Å, Fig. S5C) as are the C-terminal helices (r.m.s.d. 0.9Å, Fig. S5D).
Fig. 4.

Conformational changes in Mhp1. A) Overall superposition of the two structures. The outward-facing structure has been coloured as in Fig 1. The inward-facing structure is shown in salmon for the bundle, light green for the hash motif and light blue for TMs 5 and 10. Panels B-G show two views of the three crystal structures (outward-facing (B,E), occluded (C,F) and inward-facing (D,G)) with arrows representing the conformational changes in moving from one to the other. TMs 11 and 12 and loop regions have been omitted for clarity. In going from outward to occluded the N-terminal part of TM 10 packs over the substrate (arrow A). From occluded to inward a 30° rotation of the hash motif relative to the bundle (arrow B) effectively switches the protein from outward to inward-facing. At the same time helix OUT 7-8 moves into the extracellular cavity (arrow C) and the N-terminal part of TM 5 (complementary to the N-terminal part of TM 10) moves out to open the intracellular cavity further. The rotation axis is shown by a black line in B-D. The bulge at the tip of this line on the outward face of the protein is the black dot in E-G where the viewpoint is along the rotation axis. The sodium and benzylhydantoin have been coloured as in the previous figures.
The transition from outward-facing to inward-facing conformations has been analysed for the LeuT family of transporters in terms of two gating models (20). ‘Thin gates’ involving only a few residues control the opening and closing of the substrate binding site to the exterior or interior of the cell and a ‘thick gate’, so called because of the 20Å barrier blocking the substrate binding site from the cytoplasm in the outward facing structure, controls the transition of the protein from the outward to the inward states. Based on the three structures of Mhp1 the conformational changes involved in these transitions can be described in terms of these gating models as follows.
The first step is the change of the outward-facing state to the occluded state (Figs. 4 and 5). In Mhp1, the outward-facing cavity is formed by TMs 1, 3, 6, 8 and 10 (Fig. 4). The N-terminal end of a flexible helix, TM10, acts as an extracellular ‘thin’ gate folding over the substrate as it binds to close the outward-facing cavity, partially sealing it from the exterior. This is followed by a change of the occluded state to the inward-facing state. Here the hash motif (TMs 3, 4, 8 and 9) acts as the thick gate undergoing a rotation of 30° with respect to the bundle (concomitant with a translation of 3 Å) around an axis 40° to the plane of the membrane (Fig. 4). With this movement of the hash motif, the part of the outward-facing cavity that still remains after one side is closed by the extracellular flexible TM10, is completely filled by the C-termini of TMs 3 and 9 and a small extracellular helix (OUT 7-8, see Fig1); meanwhile the movement of TMs 4 and 8 opens the ion and substrate binding sites towards the cytoplasm, forming an inward-facing cavity. At the same time, the intracellular ‘thin gate’ made of the flexible helix TM5, bends to open this cavity further (Fig. 4). TM5 is the pseudo two-fold equivalent of the extracellular ‘thin gate’ TM10.
Fig 5.

Schematic representation of the alternating access model in Mhp1 highlighting the thick and thin gates. The three diagrams coloured as in Fig. 1 represent the outward-facing, occluded and inward-facing crystal structures. The black and white diagrams show another possible state. Upon sodium and substrate binding the extracellular thin gate (TM 10) closes to form the occluded state. The thick gate then opens with a rigid body rotation of TMs 3, 4, 8 and 9 (the hash motif) relative to TMs 1, 2, 6, 7 (the bundle). Either independently or concomitantly with this the intracellular thin gate (TM 5) also opens to allow the substrates to exit towards the cytoplasm. See also the online movies (S1 and S2).
Molecular Dynamics Simulations of the Conformational Changes
To evaluate these proposed conformational changes, the transition from the outward-facing conformation through the occluded state to the inward-facing structure has been simulated using dynamic importance sampling (DIMS) MD (24, 28, movie S1, Fig. S6). The simulations demonstrate that no large steric or energetic barriers occur to hamper this transition (Fig. S7). Equilibrium simulations, on the other hand, show that the thin gates can sample conformations near their open and closed states regardless of the starting conformations (Fig S8). It is likely, therefore, that the action of the thin gates is partially a stochastic process, occurring on the timescale of a few hundred nanoseconds. In the simulations, movement of the intracellular thin gates was largely independent of the movement of the thick gate. The conformation of the extracellular thin gate, however, is coupled to the thick gate via the movement of TM9 towards the bundle, which locks the extracellular gate into its closed position (Fig S9). MD simulations started in the inward-facing conformation (closed thick gate) only sample the extracellular-closed gate region, in contrast to simulations beginning with the occluded state during which the extracellular gate can partially open (Fig S8). In an analogous manner, the movement of the intracellular gate (TM5) is restricted when the thick gate is open (Fig S8). No spontaneous transition was observed for the thick gate within a total of 1.6 μs of simulations. Thus it appears that the action of the thick gate becomes the rate limiting step of the transport process, with the thin gates modulating much faster access/egress of substrates.
Transport Mechanism
With the recent elucidation of the various structures belonging to the LeuT superfamily there has been much speculation on the conformational changes necessary for the protein to switch from the outward to the inward conformations. As discussed recently (19, 20), essentially two mechanisms have been proposed. The first (7, 10) involves the flexing of the extended TMs 1 and 6 of the bundle. These helices have been seen to bend in going from the occluded structure of LeuT (29) to an open inhibitor-bound structure and between the outward open and occluded forms of AdiC (21). In the second mechanism (9, 30, 31) these helices would remain rigid and instead the bundle and flanking helices would move relative to one another. In the structures of Mhp1 there is no significant change in conformation of TMs 1 and 6. Instead the structures are much more consistent with the rocking bundle mode first suggested by Forrest et al (30), where the 10 TM core of the protein could be split into the ‘bundle’ (TMs 1, 2, 6 and 7) and the ‘scaffold’ (TMs 3, 4, 5, 8, 9, 10) with the bundle and scaffold rocking relative to each other to alter the conformation from outward to inward. There are, however, some differences between that model and the observations on Mhp1. Analyses of which helices differ amongst the three structures of Mhp1, together with the MD simulations in a native-like bilayer, show that the bundle (TMs 1, 2, 6, and 7) and hash motif (TMs 3, 4, 8, 9) move relative to each other as approximately rigid bodies. The helices that form the thin gates (TM5 and 10), on the other hand, can move independently although the simulations suggest that they can be locked in place depending on the position of the thick gate. This is shown schematically in Fig. 5 and Movie S2.
It is evident that the details of substrate transport will vary amongst the many transporters of the LeuT superfamily. The occlusion mechanism shown for AdiC, for example, differs substantially from that of Mhp1 (21). However, the similarities of the outward-facing structure of LeuT and the inward-facing structure of vSGLT with the corresponding structures of Mhp1, together with the consistency of our model with accessibility measurements on the serotonin transporter (30), suggest that the switch between the two conformations is not just limited to Mhp1. It seems, therefore, that the molecular basis of the alternating access model revealed by this study is likely to be common amongst this expanding superfamily.
One Sentence Summary.
Three crystal structures together with molecular dynamics reveal the mechanism of alternating access of a transport protein.
Supplementary Material
Acknowledgments
This work was funded by the BBSRC (grant no. BB/C51725), the EU (EMeP grant and LSHG-CT-2004-504601 and the EDICT grant 201924). The authors are grateful for the use of the Membrane Protein Laboratory (MPL) funded by the Wellcome Trust (grant no. 062164/Z/00/Z) at the Diamond Light Source Limited. PJFH received personal funding from the Leverhulme Trust, SW from a European Molecular Biology Organization long-term fellowship, TS from a Grant-in-Aid for Scientific Research (B) (grant no. 21370043) and JH from the BBSRC MPSi (grant no. BBS/B/14418). We appreciate the additional support of Dr Shun'ichi Suzuki and Ajinomoto Company Incorporated. A part of this work was also supported by a grant from the ERATO Iwata human receptor crystallography project from the Japan Science and Technology Agency (JST). Data were collected at the European Synchrotron Radiation Facility and further experiments were carried out at Diamond Light Source. We are grateful to Dr David Drew for critical reading of the manuscript. The coordinates and the structure factors for Mhp1 have been deposited in the Protein Data Bank (entry 2X79).
References
- 1.Crane RK, Miller D, Bihler I. In: Membrane Transport and Metabolism. Kleinzeller A, Kotyk A, editors. Czech Academy of Sciences; Prague: 1961. pp. 439–449. [Google Scholar]
- 2.Mitchell P. Adv Enzymol. 1967;29:33. doi: 10.1002/9780470122747.ch2. [DOI] [PubMed] [Google Scholar]
- 3.Jardetzky O. Nature. 1966;211:969. doi: 10.1038/211969a0. [DOI] [PubMed] [Google Scholar]
- 4.Tanford C. Proc Natl Acad Sci U S A. 1983;80:3701. doi: 10.1073/pnas.80.12.3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaback HR, et al. Proc Natl Acad Sci U S A. 2007;104:491. doi: 10.1073/pnas.0609968104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Loo DD, Hirayama BA, Karakossian MH, Meinild AK, Wright EM. J Gen Physiol. 2006;128:701. doi: 10.1085/jgp.200609643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Nature. 2005;437:215. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- 8.Faham S, et al. Science. 2008;321:810. doi: 10.1126/science.1160406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weyand S, et al. Science. 2008;322:709. doi: 10.1126/science.1164440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ressl S, Terwisscha AC, van Scheltinga, Vonrhein C, Ott V, Ziegler C. Nature. 2009;458:47. doi: 10.1038/nature07819. [DOI] [PubMed] [Google Scholar]
- 11.Fang Y, et al. Nature. 2009;460:1040. doi: 10.1038/nature08201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao X, et al. Science. 2009;324:1565. doi: 10.1126/science.1173654. [DOI] [PubMed] [Google Scholar]
- 13.Shaffer PL, Goehring A, Shankaranarayanan A, Gouaux E. Science. 2009;325:1010. doi: 10.1126/science.1176088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen NH, Reith ME, Quick MW. Pflugers Arch. 2004;447:519. doi: 10.1007/s00424-003-1064-5. [DOI] [PubMed] [Google Scholar]
- 15.Wright EM, Hirayama BA, Loo DF. J Intern Med. 2007;261:32. doi: 10.1111/j.1365-2796.2006.01746.x. [DOI] [PubMed] [Google Scholar]
- 16.Yanagida O, et al. Biochim Biophys Acta. 2001;1514:291. doi: 10.1016/s0005-2736(01)00384-4. [DOI] [PubMed] [Google Scholar]
- 17.Harada N, et al. Acta Haematol. 2000;103:144. doi: 10.1159/000041037. [DOI] [PubMed] [Google Scholar]
- 18.Dohan O, et al. Endocr Rev. 2003;24:48. doi: 10.1210/er.2001-0029. [DOI] [PubMed] [Google Scholar]
- 19.Abramson J, Wright EM. Curr Opin Struct Biol. 2009;19:425. doi: 10.1016/j.sbi.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krishnamurthy H, Piscitelli CL, Gouaux E. Nature. 2009;459:347. doi: 10.1038/nature08143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao X, et al. Nature. 463:828. doi: 10.1038/nature08741. [DOI] [PubMed] [Google Scholar]
- 22.Suzuki S, Henderson PJ. J Bacteriol. 2006;188:3329. doi: 10.1128/JB.188.9.3329-3336.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shimamura T, et al. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2008;64:1172. doi: 10.1107/S1744309108036920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Materials and Methods are available as supporting material in Science online.
- 25.Adams PD, et al. Acta Crystallogr. D. 2002;58:1948. [Google Scholar]
- 26.Leonard GA, et al. Journal of Applied Crystallography. 2009;42:333. [Google Scholar]
- 27.Li J, Tajkhorshid E. Biophys J. 2009;97:L29. doi: 10.1016/j.bpj.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beckstein O, Denning EJ, Perilla JR, Woolf TB. J Mol Biol. 2009;394:160. doi: 10.1016/j.jmb.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Singh SK, Piscitelli CL, Yamashita A, Gouaux E. Science. 2008;322:1655. doi: 10.1126/science.1166777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Forrest LR, et al. Proc Natl Acad Sci U S A. 2008;105:10338. doi: 10.1073/pnas.0804659105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Forrest LR, Rudnick G. Physiology. 2009;24:377. doi: 10.1152/physiol.00030.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.