

Abstract
CHD4 (chromodomain helicase DNA-binding protein 4) ATPase is a major subunit of the repressive NuRD (nucleosome remodelling and deacetylase) complex, which is involved in transcriptional regulation and development. CHD4 contains two PHD (plant homeodomain) fingers of unknown function. Here we show that the second PHD finger (PHD2) of CHD4 recognizes the N-terminus of histone H3 and that this interaction is facilitated by acetylation or methylation of Lys9 (H3K9ac and H3K9me respectively) but is inhibited by methylation of Lys4 (H3K4me) or acetylation of Ala1 (H3A1ac). An 18 μM binding affinity toward unmodified H3 rises to 0.6 μM for H3K9ac and to 0.9 μM for H3K9me3, whereas it drops to 2.0 mM for H3K4me3, as measured by tryptophan fluorescence and NMR. A peptide library screen further shows that phosphorylation of Thr3,Thr6 or Ser10 abolishes this interaction. A model of the PHD2–H3 complex, generated using a combination of NMR, data-driven docking and mutagenesis data, reveals an elongated site on the PHD2 surface where the H3 peptide is bound. Together our findings suggest that the PHD2 finger plays a role in targeting of the CHD4/NuRD complex to chromatin.
Keywords: chromodomain helicase DNA-binding protein 4 (CHD4), histone, methylation, plant homeodomain (PHD)
INTRODUCTION
CHD4 (chromodomain helicase DNA-binding protein 4), also known as Mi2β, is involved in a variety of fundamental cellular processes including chromatin remodelling, transcriptional repression, and DNA repair and recombination. CHD4 belongs to the class II subfamily of CHD ATPases and is a major subunit of the repressive NuRD (nucleosome remodelling and deacetylase) complex, which is required for regulating transcription and developmental signalling [1-4]. Along with CHD4, the NuRD complex contains MBD2/3, RbAp46/48, MTA1/2/3 and p66 components and HDAC1/2 (histone deacetylase 1/2) (Figure 1a). Coupling of the helicase and HDAC functions makes NuRD unique among chromatin remodelling complexes. Although the purpose of the dual enzymatic activity is unclear, it may be essential for the efficient formation of heterochromatin with densely packed hypoacetylated nucleosomes and the rapid shut-off of gene expression [5,6]. NuRD is broadly distributed throughout the body and is critical for the regulation of numerous genes, however the mechanism by which this complex associates with nucleosomes remains undefined.
Figure 1. The CHD4 PHD2 finger binds histone H3.
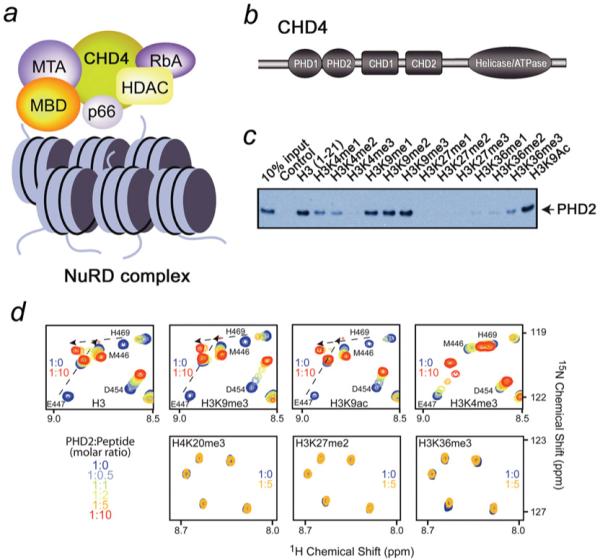
(a) The NuRD complex and its major components. The nucleosomes are shown as blue cylinders. (b) Architecture of CHD4: the N-terminal tandem of PHD fingers, the two adjacent chromodomains and a catalytic ATPase module. (c) Binding of the GST-fusion CHD4 PHD2 finger to biotinylated histone peptides. (d) Superimposed 1H,15N HSQC spectra of 0.2 mM PHD2, collected during titrating in the indicated histone peptides. The spectra are colour-coded according to the protein/peptide ratio, the position of low intensity peaks of E447 are marked by an asterisk.
CHD4 is a ~ 200 kDa protein, conserved throughout the animal and plant kingdoms. It contains tandem PHD (plant homeodomain) fingers, two chromodomains and a C-terminal ATPase module (Figure 1b). The ~400-residue SNF2-like helicase/ATPase domain provides the energy necessary for histone displacement and sliding during nucleosome remodelling [7]. Unlike other chromodomains that bind methylated histone marks, such as H3K9me3 and H3K27me3 [8-11], the CHD4 chromodomains do not recognize histone tails and instead show a unique DNA binding activity [12]. The function of the PHD fingers of CHD4 is unknown.
The PHD finger is a ~ 60-residue module characterized by a conserved C4HC3 sequence that co-ordinates two zinc ions in a cross-brace arrangement. The PHD finger was discovered over a decade ago and has since been found in a wide variety of eukaryotic, mostly nuclear, proteins. Recently, a subset of PHD fingers, including those of BPTF (bromodomain PHD finger transcription factor) and ING2 (inhibitor of growth family, member 2), were shown to recognize H3K4me3 (histone H3 trimethylated at Lys4) [13-16], whereas the PHD modules of BHC80 and AIRE (autoimmune regulator) were found to bind H3K4me0 (unmodified H3) [17-19]. However, the residues essential for binding to either H3K4me3 or H3K4me0 are not conserved in the majority of the PHD sequences. This raises the possibility of the existence of distinct subsets of PHD fingers that exhibit selectivity toward other histone modifications and/or are capable of recognizing non-histone ligands. Indeed, recent reports have suggested that PHD domains of ICBP90 and Lys4-specific demethylases SMCX and Lid2 may play a role in associating with the H3K9me3 mark [20-22].
In the present study, we demonstrate that the second PHD finger of CHD4 is a reader of the histone H3 N-terminal tail. We use a combination of tryptophan fluorescence and NMR binding data, chemical shift perturbation analysis, immunoprecipitation data, peptide library screening, mutagenesis and data-driven docking to infer the molecular mechanism for H3 recognition. Our findings offer new insight into the role of the PHD2 finger in the CHD4/NuRD complex, suggesting that it plays a part in targeting the complex to chromatin.
MATERIALS AND METHODS
Protein purification
The wild-type CHD4 PHD2 finger and its mutants were expressed in Escherichia coli BL21(DE3) pLysS cells grown in LB (Luria–Bertani) broth or 15NH4Cl-supplemented minimal media. Bacteria were harvested by centrifugation after induction with IPTG (0.3–1.0 mM) and lysed by sonication. The unlabelled and 15N-labelled GST (glutathione transferase)-fusion proteins were purified on glutathione–Sepharose 4B beads (Amersham Biosciences). The GST tag was either cleaved with Prescission protease, or left for the purposes of Western blot analysis, in which case the GST-fusion protein was eluted off the glutathione–Sepharose beads using 0.05 M reduced l-glutathione (Sigma Aldrich). The proteins were concentrated into 20 mM Tris (or d10-Tris), pH 6.8–7.5, in the presence of 150 mM NaCl, 10 mM dithiothreitol and 1 mM NaN3.
PCR mutagenesis
Point mutants were prepared using the Stratagene QuikChange XL site-directed mutagenesis kit according to the manufacturer’s instructions.
Western blot analysis
GST-fusion CHD4 PHD fingers were incubated with C-terminal biotinylated peptides (Upstate Biotechnology) corresponding to the unmodified H3 (residues 1–21) and singly modified H3K4me1/2/3 (residues 1–21), H3K9me1/2/3 (histone H3 mono-/di-/tri-methylated at Lys9; residues 1–21), H3K9ac (histone H3 acetylated at Lys9; residues 1–21), H3K27me1/2/3 (histone H3 mono-di-/tri-methylated at Lys27; residues 21–44) and H3K36me1/2/3 (histone H3 mono-/di-/tri-methylated at Lys36; residues 21–44) histone tails in the presence of streptavidin–Sepharose beads (GE Healthcare) in binding buffer containing50 mM Tris, pH 7.5, 150 mM NaCl and 0.05 % Nonidet P-40. The beads were collected via centrifugation and washed five times with the peptide binding buffer. Bound protein was detected by Western blot using anti-GST HRP (horseradish peroxidase)-conjugate monoclonal antibodies (GE Healthcare). Negative controls using GST-fusion proteins in the absence of the peptides were run in parallel to ensure that the proteins did not bind to the streptavidin beads.
NMR spectroscopy
Chemical shift perturbation experiments were carried out using uniformly 15N-labelled wild-type or mutant PHD2. 1H,15NHSQC (heteronuclear single quantum coherence) spectra were recorded in the presence of increasing concentrations of 12-mer histone tail peptides (synthesized by the Peptide Core Facility, University of Colorado Denver, CO, U.S.A.) or 20-mer H3K9ac (Anaspec). NMR experiments were carried out on a 500 MHz Varian INOVA spectrometer in the on-site NMR Core Facility. Kd values were calculated by a nonlinear least-squares analysis in Kaleidagraph using the equation:
where [L] is concentration of the peptide, [P] is concentration of the protein, Δδ is the observed chemical shift change, and Δδmax is the normalized chemical shift change at saturation. Normalized chemical shift changes [23] were calculated using the equation:
where Δδ is the change in chemical shift in parts per million (ppm). NMR assignments of CHD4 PHD2 were taken from [24] (BMRB 5555).
Fluorescence spectroscopy
A Fluoromax-3 spectrofluorometer was used to carry out tryptophan fluorescence measurements on PHD2. Spectra were recorded at 25°C on samples containing 0.5 μM PHD2 and progressively increasing concentrations of histone peptides. Samples were excited at 295 nm and emission spectra were recorded between 305 and 405 nm with a 0.5 nm step size and a 1 s integration time, averaged over three scans. Kd values were determined by a nonlinear least-squares analysis in Kaleidagraph using the equation:
where [L] is the concentration of the peptide, [P] is the concentration of the protein,ΔI is the observed change of signal intensity, and ΔImax is the difference in signal intensity of the free and fully bound states of the CHD4 PHD finger. The Kd values were averaged over three or more separate experiments, with error calculated as the standard deviation between the runs.
Combinatorial on-bead screening assay
A 5000-member PTM (post-translational modification)-randomized combinatorial peptide library based on the first 10 residues of the histone H3 N-terminus was developed as described in (A.L. Garske, S.S. Oliver and J.M. Denu, unpublished work and [25]). The library was incubated first with the GST-tagged version of CHD4 PHD2, second with a GST-specific primary antibody, third with a biotinylated secondary antibody, and finally with streptavidin-conjugated alkaline phosphatase, catalysing the turnover of BCIP (5-bromo-4-chloroindol-3-yl phosphate), which results in formation of a turquoise precipitate on beads bearing sequences that bind to the target protein. The bead colour intensity is proportional to affinity of the interaction [25]. Peptides from individual beads were cleaved with cyanogen bromide and analysed by MALDI–TOF (matrix-assisted laser-desorption ionization–time-of-flight) MS. PTM patterns were determined from the resulting mass ladders. Discrimination factors were obtained by dividing the frequency of each modification observed in the intensely blue beads by the frequency of each corresponding modification from a random group of 100 library members. Discrimination factors represent the likelihood of observing a particular modification in a protein screening experiment relative to random chance.
HADDOCK docking calculations
Modelling of the PHD2–H3 complex was performed using the flexible docking program HADDOCK (www.haddocking.org). The protein co-ordinates were taken from the structure of the ligand-free CHD4 PHD2 finger (1MM2) [24]. Co-ordinates for the unmodified H3 (residues 1–10) ligand were taken from the structure of the BHC80–H3 complex (2PUY) [17]. The modified peptides, H3K9me3 and H3K9ac, were generated using the Insight II molecular modelling system. NMR chemical shift perturbation data were used to restrain the calculation. Specifically, residues Glu447, Gly455, Gly456, Leu458, Cys460, Thr463, Ser467, His469 and Trp485 in PHD2 were identified as active residues, and residues Gly483 and Glu484 were identified as passive residues. For the peptides, residues 1–9 were identified as active residues and residue 10 was identified as passive. The four lowest energy structures from the top cluster were analysed for each run. The ensemble of the four best structures for PHD2–H3 and the plot of the HADDOCK score versus rmsd (root-mean-squared deviation) for the top cluster of all three calculations are shown in Supplementary Figure S1 (at http://www.BiochemJ.org/bj/423/bj4230179add.htm).
RESULTS AND DISCUSSION
CHD4 PHD2 recognizes histone H3
The function of the CHD4 PHD2 finger as a histone H3-binding module was initially identified by pull-down experiments (Figure 1c). GST-tagged PHD2 was first incubated with biotinylated histone peptides corresponding to unmodified H3 (residues 1–21) and singly modified H3K4me1/2/3 (residues 1–21), H3K9me1/2/3 (residues 1–21), H3K9ac (residues 1–21), H3K27me1/2/3 (residues 21–44) and H3K36me1/2/3 (residues 21–44), and then with streptavidin–Sepharose beads. After collecting the beads by centrifugation, the histone peptide-bound protein was detected by Western blot analysis. As shown in Figure 1(c), the PHD2 finger was able to recognize unmodified H3, H3K9me1/2/3 and H3K9ac. Binding to the H3K4me1/2/3 peptides was significantly weaker, gradually diminishing with accumulation of each additional methyl group at Lys4. Little to no interaction was observed with methylated H3K27 and H3K36 peptides. Together, these results suggested that the CHD4 PHD2 finger binds to a region within the first 21 residues of the H3 tail and that methylation at Lys4 disrupts this interaction.
Recognition of the shorter H3 peptides was characterized by NMR titration experiments. 1H,15N HSQC spectra of the uniformly 15N-labelled PHD2 finger were recorded as unmodified H3, H3K4me3, H3K9me3, H3K9ac, H3K27me2, H3K36me3 or H4K20me3 peptides were gradually added to the NMR samples (Figure 1d). Substantial chemical-shift changes in the PHD2 finger were detected during titration with either unmodified H3, H3K9me3 or H3K9ac. These interactions were in the intermediate-to-fast exchange regime on the NMR time scale, characterized by the progressive shifting and broadening of amide resonances, indicative of strong binding.
Addition of H3K4me3 induced small chemical shift changes in the CHD4 PHD2 spectrum, and the observed fast exchange regime pointed to a weak association with this peptide (Figure 1d). A lack of significant resonance perturbations upon titrating in H3K27me2, H3K36me3 or H4K20me3 indicated that the PHD2 finger does not recognize these epigenetic marks (Figure 1d). The NMR data were in agreement with pull-down experiments, corroborating the fact that methylated Lys4 inhibits the interaction of CHD4 PHD2 with histone H3.
Selectivity of the CHD4 PHD2 finger for histone modifications
To determine whether other post-translational histone modifications influence the interaction with H3, the CHD4 PHD2 finger was tested in an on-bead combinatorial H3 tail library assay (Figure 2). Construction of the library involved a split-pool approach, generating a library with 5000-member diversity [25]. Using the ten most N-terminal residues of histone H3, the library encompassed the possible modification states for 7 residues within the peptide. Sites of modification included positions 2 (R, Rme1, Rme2s, Rme2a, Cit), 3 (T, Tph), 4 (K, Kme1, Kme2, Kme3, Kac), 6 (T, Tph), 8 (R, Rme1, Rme2s, Rme2a, Cit), 9 (K, Kme1, Kme2, Kme3, Kac) and 10 (S, Sph). We screened the library with GST-tagged CHD4 PHD2, employing a colorimetric assay to identify beads that bound the protein with high affinity. To determine the preferred modification state at different positions, 32 beads that indicated positive binding were selected and the PTM state was revealed by MALDI MS. Positional discrimination factors for each site were determined by dividing the observed frequency of the positive beads by the frequency determined among a random set of 100 beads.
Figure 2. Binding specificity of the CHD4 PHD2 finger was established using positional discrimination factors.
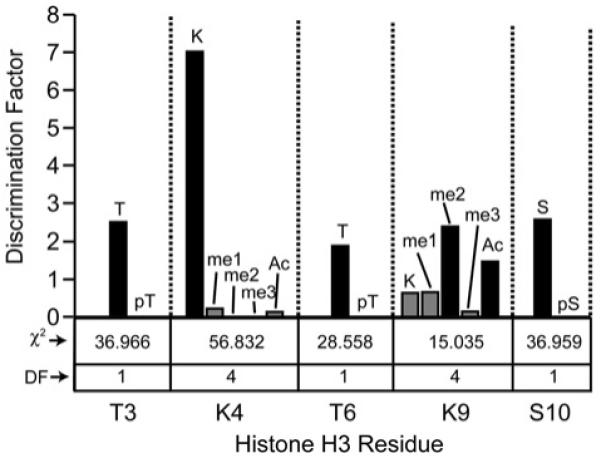
The discrimination factor for a single modification at any one position is calculated as a mathematical ratio of frequencies between the results of screening the library against CHD4 PHD2 and a random library sampling (50 sequences). A larger discrimination factor (a larger bar in graph) represents a modification favouring binding. A discrimination factor of 1 denotes no difference between the CHD4 screen and a random bead sampling. χ2 analysis was performed at each position. For positions with 4 (n–1) degrees of freedom (DF), a χ2 value greater than 13.28 denotes statistical significance at a 99% confidence level. Positions with 1 degree of freedom (DF) must have a value greater than 6.63 in order to be considered statistically significant at the 99% confidence level. A non-significant result means that no effects were discovered and chance could explain the observed differences at that position. The modification states at positions Thr3 (T, pT), Lys4 (K, Kme1, Kme2, Kme3, Kac), Thr6 (T, pT), Lys9(K, Kme1, Kme2, Kme3, Kac) and Ser10 (S, pS) are shown. ac, acetylated; me, methylated; p, phosphorylated. We note that K9me3 is not well represented in the screen. This could be due to the sample size, which was not large enough to resolve all tendencies at this position and because any of the three modifications (acetylation, di- and tri-methylation) results in enhanced binding. Additionally, based on χ2 analysis, PTM discrimination at Lys9 is less pronounced compared with positions 3, 4 and 6.
The screening results substantiated the strong preference of CHD4 PHD2 for unmodified Lys4 (discrimination factor of 7.1) compared with methylated or acetylated Lys4 (Figure 2). A significant preference for K9me2 and K9ac (discrimination factors of 2.4 and 1.5, respectively) over unmodified Lys9 (discrimination factor of 0.6) was also observed. Phosphorylation at Thr3, Thr6 or Ser10 was not tolerated at all (discrimination factors of 2.5, 1.9 and 2.6 respectively for the unphosphorylated threonine and serine residues); not a single phosphorylation mark was identified at any of these positions in the positive hits. In contrast, no significant preferences were observed for Arg2 and Arg8 with regard to either citrullation or mono- or di-methylation (see Supplementary Figure S2 at http://www.BiochemJ.org/bj/423/bj4230179add.htm). Collectively, the on-bead combinatorial library data points to the high selectivity of the CHD4 PHD2 finger, which shows clear preference for unmodified Thr3, Lys4, Thr6 and Ser10, and favours modified Lys9 (methylated or acetylated) over the unmodified residue.
Methylation or acetylation at Lys9 increases the binding affinity of CHD4 PHD2, whereas methylation at Lys4 decreases it
The histone H3-binding affinities of the PHD2 finger were measured using tryptophan fluorescence and NMR. We found that CHD4 PHD2 associates with unmodified H3 with a Kd of 18 μM (Figure 3). However, this binding was gradually diminished by mono-, di- and tri-methylation of Lys4, with the affinities for H3K4me1, H3K4me2 and H3K4me3 dropping to ~300 μM, 1 mM and 2 mM respectively. In contrast, the affinity of PHD2 was augmented as the number of methyl groups attached to Lys9 was increased, ultimately reaching 0.9 μM for H3K9me3. Likewise, acetylation of Lys9 enhanced the binding, yielding a Kd of 0.6 μM. Overall, the CHD4 PHD2 finger bound ~100-fold stronger to unmodified H3 than to H3K4me3, but 20- and 30-fold stronger to H3K9me3 and H3K9ac than to H3. Consistent with these observations, it has previously been shown that association of the entire CHD4-containing NuRD complex with H3 is disrupted by methylation of Lys4 but is tolerated when Lys9 is methylated [26].
Figure 3. Histone H3 binding affinities of the CHD4 PHD2 finger.

(a) Representative binding curves used to determine the Kd values of the PHD2–H3, PHD2–H3K9me3 and PHD2–H3K9ac interactions by tryptophan fluorescence. (b) The binding affinities of the CHD4 PHD2 finger, wild-type and mutants for indicated peptides, measured by tryptophan fluorescence (a) and NMR (b).
The H3-binding site of the CHD4 PHD2 finger
The histone-binding site of the PHD2 finger was defined by analysing NMR resonance perturbations in PHD2 caused by the unmodified H3 peptide. Plotting chemical shift changes for each backbone amide allowed us to identify the residues most affected due to interaction with H3 (Figure 4a). The largest perturbations or disappearance of signals were observed for Glu447, Gly455, Gly456, Leu458, Cys460, Thr463, Ser467, His469, Gly483 and Trp485, suggesting that these residues are directly or indirectly involved in the binding. Mapping these residues onto the surface of the ligand-free CHD4 PHD2 finger [24] revealed a well-defined, elongated site where the histone peptide is bound (Figure 4c).
Figure 4. Identification of the H3-, H3K9ac- and H3K9me3-binding sites of the CHD4 PHD2 finger and models of the PHD2-H3, PHD2-H3K9ac and PHD2-H3K9me3 complexes.

(a, d and e) The histograms show normalized 1H, 15N chemical shift changes in backbone amides of PHD2 upon addition of the H3 (a), H3K9ac (d) and H3K9me3 (e) peptides. An asterisk indicates the disappearance of the resonances, and P indicates a proline residue. (b, f and g) Models of the PHD2–H3, PHD2–H3K9ac and PHD2–H3K9me3 complexes as obtained using the flexible docking program HADDOCK. Residues of PHD2 and H3 are labelled in black and green, respectively. Hydrogen bonds and salt bridges are shown by red dotted lines. (c) Residues that exhibit significant H3-induced resonance perturbations [more than average plus one quarter (yellow), one half (orange) and one (red) standard deviation] in (a) are mapped onto the surface of the ligand-free CHD4 PHD2 [24] and labelled.
To establish the contribution of each binding site residue in the PHD2-H3 interaction, Glu447, Gly455, Gly456, Leu458, Cys460 and Trp485 were mutated in turn, and the mutant proteins were tested by NMR. The 1H,15N HSQC experiments showed that substitution of the core residues Gly456, Leu458 and Trp485 with alanine, and Gly455 with valine, resulted in collapse of the tertiary structure of the protein, thus pointing to the importance of these residues for the structural stability of PHD2. In contrast, the E447A and C460W mutants remained folded. We note that Glu447 and Cys460 of CHD4 correspond to Asp489 and Met502 of BHC80 [17] and to Asp297/Asp299 and Cys310/Cys312 of human/murine AIRE [18,19]. The conserved aspartate residues of BHC80 and AIRE form a salt bridge with Lys4, a hallmark of the K4me0 recognition, whereas the hydrophobic side chains of methionine/cysteine insert between Arg2 and Lys4 of the peptide [17-19]. Replacement of the aspartate residues with alanine or the hydrophobic residues with a bulky tryptophan in the PHD fingers of BHC80 and AIRE abolish interactions with H3K4me0 [17,27]. Likewise, Glu447 and Cys460 of CHD4 are required for the interaction with unmodified H3. NMR titrations demonstrate that E447A and C460W mutants bind to the H3 peptide significantly weaker than the wild-type protein (Kd = 0.9 mM and 3.7 mM respectively), suggesting that these residues play critical roles in the binding energetics (Figure 3b).
A model of the PHD2–H3 complex
Employing the NMR data as restraints in the flexible docking algorithm HADDOCK, a model of the PHD2–H3 complex was generated. The modelling revealed that the H3 peptide is bound to PHD2 through a number of hydrogen bonds and salt bridges (Figure 4b). The peptide forms a third antiparallel β-strand, pairing with the first β-strand of PHD2, and is stabilized through backbone interactions between Arg2, Lys4 and Thr6 of H3 and the protein residues Cys460, Leu458 and either Gly455 or Gly456. The peptide is further restrained through the co-ordination of the amino group of Ala1 by Gly483 and Glu484, the formation of a salt bridge between Arg2 and Asp462, and hydrogen bonding interactions involving Lys4,Thr6 and Arg8 (Figure 4b).
The significance of the Ala1 recognition was demonstrated by examining binding of the PHD2 finger to a histone H3 peptide in which the primary amino group of Ala1 was acetylated (H3A1ac). The Kd value of the PHD2–H3A1ac interaction was found to be ~400 μM, which is ~ 20-fold lower than the affinity of PHD2 for unmodified H3 (Figure 3b). These results suggest that the proper co-ordination of unmodified Ala1 is necessary for the formation of the complex. In addition, the backbone amide cross peaks of Asn482, Gly483, Glu484 and Trp485 exhibited different perturbation patterns upon binding to H3A1ac as compared with unmodified H3, providing further evidence that these residues are involved in the recognition of Ala1 (see Supplementary Figure S3 at http://www.BiochemJ.org/bj/423/bj4230179add.htm).
To define further the role of the N-terminus of H3, a peptide comprising residues Thr3–Ser10 but lacking Ala1 and Arg2 [H3(3–10)], was tested in NMR titration experiments. Even a 10-fold excess of H3(3-10) induced no changes in the NMR spectra of PHD2, implying that CHD4 PHD2 does not recognize H3 lacking the first two N-terminal residues (Figure 3b). Given that the loss of binding was substantially greater than the reduction in binding to H3A1ac, where Ala1 is N-terminally blocked, these data suggest that both Ala1 and Arg2 and/or their backbone contacts are required for robust interaction with CHD4 PHD2.
The H3-binding mode of CHD4 PHD2 in the model is similar to that observed in the BHC80-PHD–H3 and AIRE-PHD1–H3 complexes [17-19], indicating that the molecular mechanism of the H3 recognition by this subset of PHD fingers is conserved.
Increasing hydrophobicity of Lys9 facilitates the PHD2–H3 interaction
To obtain insight into the role of Lys9, we modelled the complexes of PHD2 with H3K9ac and H3K9me3 (Figures 4f and 4g). A superimposition of these complexes with the PHD2–H3 complex showed that the first seven residues of the H3 peptide interacts with PHD2 similarly. However, methylated or acetylated Lys9 is positioned more closely to the protein surface in the H3K9ac and H3K9me3 complexes, allowing Arg8 to form a salt bridge with Glu447.
The position of Arg8 and modified Lys9 in the complexes was corroborated by NMR data. The majority of chemical shift changes in PHD2 induced by H3K9ac or H3K9me3 were paralleled to those caused by unmodified H3, indicating that the modified and unmodified peptides occupy the same binding pocket. However, the amide cross peaks of Glu447/Phe448/Cys449/Lys450/Gly455/Gly456/Ser467 and Asp454/Gly455/Gly456 were shifted to a larger extent in the PHD2–H3K9ac and PHD2–H3K9me3 complexes respectively (Figure 5). The solvent-exposed Glu447, Phe448, Asp454, Gly455 and Gly456 residues form a contiguous patch on the surface, with Cys449 and Ser467 lying underneath this patch. Interestingly, in the AIRE PHD–H3K4me0 complex, the Glu298 and Asp304 residues, equivalent to Phe448 and Asp454 of CHD4, are involved in transient electrostatic interactions with unmodified Lys9 of the peptide [19]. Mutations of Glu298 or Asp304 in the AIRE PHD finger cause a 5- to 16-fold decrease in the binding affinity, revealing a role of Lys9 in the formation of the complex [19]. Likewise, when Asp454 of CHD4 was substituted with valine, binding of the CHD4 PHD2 finger to H3K9me3 was significantly compromised (Figure 3b).
Figure 5. Differences in NMR resonance perturbations observed in the CHD4 PHD2 finger upon binding to H3K9me3 and H3K9ac as compared with unmodified H3.
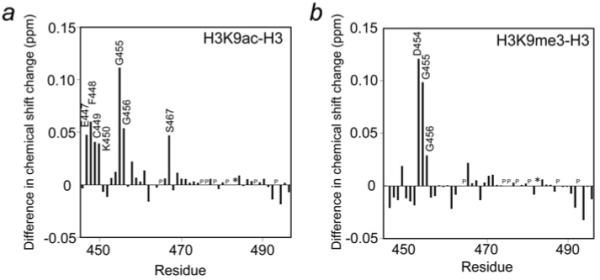
An asterisk indicates the disappearance of the resonances, and P indicates a proline residue.
Another key acidic residue of AIRE (Glu298) is replaced with a hydrophobic Phe488 in the CHD4 PHD2 sequence. The aromatic ring of Phe488 lies orthogonal to the protein surface and may favourably interact with K9ac or form hydrophobic and cation-π contacts with methylated Lys9. Methylation of a lysine residue is known to enhance interactions with aromatic residues due to the increased hydrophobic contacts involving the aromatic side chain and both the lipophilic methyl groups and the εCH2 of lysine [28]. Furthermore, the loss or shielding of the positive charge in K9ac and K9me3 may prevent repulsion between Lys9 and Lys453, a residue adjacent to Phe488. These factors could lead to a higher affinity for the CHD4 PHD2–H3K9ac/me interaction compared with the interaction between PHD2 and unmodified H3. In agreement with this, the F448A mutant associates with the H3K9ac and H3K9me3 peptides more weakly than the wild-type protein (Figure 3c), and displays chemical shift changes in 1H,15N HSQC spectra almost identical to those observed for binding of wild-type PHD2 to the unmodified H3 peptide (results not shown). Since both methylation and acetylation of Lys9 enhance the affinity, we concluded that increased hydrophobicity of the side chain most likely draws this residue toward the hydrophobic surface of the N-terminal region of the protein, further facilitating the interaction of Arg8 with PHD2.
Comparison of the subsets of PHD fingers
Recent studies suggest that the large family of PHD fingers can be separated into subsets based on the specificity of this module toward post-translational histone modifications. At least four subsets have been identified, including PHD fingers that recognize (i) H3K4me3 [BPTF, ING/YNG, Pygopus, RAG2 (recombination-activating gene 2) and TAF3] [13-16,29-37], (ii) unmodified H3 (AIRE, BHC80 and the cysteine-rich domain of DNMT3L) [17-19,38], (iii) H3K9me3 (ICBP90, Lid2 and SMCX) [20-22], and (iv) H3K36me3 (Ecm5 and Nto1) [34]. In addition, the RAG2 PHD finger has been recently shown to bind a double modification, H3R2me2/K4me3 [33]. A comparison of the amino acid sequences reveals that PHD fingers which bind H3K4me3 must contain an aromatic residue preceding the first cysteine, a hydrophobic residue upstream from the third cysteine, and a tryptophan residue preceding the zinc-co-ordinating histidine to form a (semi)aromatic/hydrophobic cage around trimethylated Lys4. The PHD fingers that recognize unmodified H3 are characterized by the presence of an acidic patch at the N-terminus (NEDE in AIRE and HEDF in BHC80) and a hydrophobic residue (methionine/cysteine) preceding the third cysteine. In the CHD4 PHD2 finger, the N-terminal patch (HMEF), being significantly more hydrophobic, favours acetylated or methylated Lys9 of H3K4me0. The signature motif(s) responsible for the recognition of other modifications remain to be determined.
In summary, we demonstrate that the second PHD finger of CHD4 recognizes the N-terminus of histone H3 and that methylation at Lys4 abolishes this interaction, whereas methylation or acetylation of Lys9 facilitates it. A model of the complex reveals that the H3 tail occupies an elongated binding site of PHD2 and is held in place through a conserved set of salt bridges and hydrogen bonds. The preference of the CHD4 PHD2 finger for modified Lys9 is most likely attributed to the N-terminal sequence, which is more hydrophobic compared with the acidic sequences of other H3K4me0-binding PHD fingers. Our results suggest that CHD4 may be involved in stabilization of the CHD4–NuRD complex at chromatin through binding of the PHD2 finger to the histone H3 tail carrying unmodified, acetylated or methylated Lys9. It is intriguing that the same domain is able to bind two distinct histone marks, which are often associated with opposite processes such as gene activation and repression. One possible explanation of these findings is in the multiple and dynamic function of the CHD4–NuRD–HDAC repressive complex, which may require initial association with H3K9ac and later with H3K9me3, promoting both the deacetylase activity of HDAC and methylation of Lys9 in the pathway from an active to inactive state of chromatin [26]. Another important question concerns the interplay between the activities of two PHD fingers of CHD4 and two chromodomains of this protein. Future studies are required to establish the relationship and functional significance of multiple interactions of the CHD4 modules with chromatin.
Supplementary Material
ACKNOWLEDGEMENTS
We thank A.M.J.J. Bonvin, S. Roy and K. Champagne for discussions and help with the experiments.
FUNDING
This research is supported by grants from the NIH (National Institutes for Health) [grant numbers CA113472, GM071424 (to T.G.K.) and GM059785 (to J.M.D.)], the NHMRC (National Health and Medical Research Council) and the NSW Cancer Institute (to J.P.M.), and the Cancer League of Colorado (to T.G.K.). C.A.M. is a recipient of an American Heart Association postdoctoral fellowship.
Abbreviations used
- AIRE
autoimmune regulator
- BPTF
bromodomain PHD finger transcription factor
- CHD4
chromodomain helicase DNA-binding protein 4
- GST
glutathione transferase
- H3
histone H3
- H3A1ac
histone H3 acetylated at Ala1
- H3K9ac
histone H3 acetylated at Lys9
- H3K9me1/2/3
histone H3 mono-/di-/tri-methylated at Lys9
- H3K4me0
unmodified histone H3
- H3K4me1/2/3
histone H3 mono-/di-/tri-methylated at Lys4
- H3K27me1/2/3
histone H3 mono-/di-/tri-methylated at Lys27
- H3K36me1/2/3
histone H3 mono-/di-/tri-methylated at Lys36
- HDAC
histone deacetylase
- HSQC
heteronuclear single quantum coherence
- ING
inhibitor of growth family
- MALDI
matrix-assisted laser-desorption ionization
- NuRD
nucleosome remodelling and deacetylase
- PHD
plant homeodomain
- PTM
post-translational modification
- RAG2
recombination-activating gene 2
REFERENCES
- 1.Tong JK, Hassig CA, Schnitzler GR, Kingston RE, Schreiber SL. Chromatin deacetylation by an ATP-dependent nucleosome remodelling complex. Nature. 1998;395:917–921. doi: 10.1038/27699. [DOI] [PubMed] [Google Scholar]
- 2.Wade PA, Jones PL, Vermaak D, Wolffe AP. A multiple subunit Mi-2 histone deacetylase from Xenopus laevis cofractionates with an associated Snf2 superfamily ATPase. Curr. Biol. 1998;8:843–846. doi: 10.1016/s0960-9822(98)70328-8. [DOI] [PubMed] [Google Scholar]
- 3.Xue Y, Wong J, Moreno GT, Young MK, Cote J, Wang W. NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Mol. Cell. 1998;2:851–861. doi: 10.1016/s1097-2765(00)80299-3. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Y, LeRoy G, Seelig HP, Lane WS, Reinberg D. The dermatomyositis-specific autoantigen Mi2 is a component of a complex containing histone deacetylase and nucleosome remodeling activities. Cell. 1998;95:279–289. doi: 10.1016/s0092-8674(00)81758-4. [DOI] [PubMed] [Google Scholar]
- 5.Denslow SA, Wade PA. The human Mi-2/NuRD complex and gene regulation. Oncogene. 2007;26:5433–5438. doi: 10.1038/sj.onc.1210611. [DOI] [PubMed] [Google Scholar]
- 6.Wang HB, Zhang Y. Mi2, an auto-antigen for dermatomyositis, is an ATP-dependent nucleosome remodeling factor. Nucleic Acids Res. 2001;29:2517–2521. doi: 10.1093/nar/29.12.2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hall JA, Georgel PT. CHD proteins: a diverse family with strong ties. Biochem. Cell Biol. 2007;85:463–476. doi: 10.1139/O07-063. [DOI] [PubMed] [Google Scholar]
- 8.Jacobs SA, Khorasanizadeh S. Structure of HP1 chromodomain bound to a lysine 9-methylated histone H3 tail. Science. 2002;295:2080–2083. doi: 10.1126/science.1069473. [DOI] [PubMed] [Google Scholar]
- 9.Nielsen PR, Nietlispach D, Mott HR, Callaghan J, Bannister A, Kouzarides T, Murzin AG, Murzina NV, Laue ED. Structure of the HP1 chromodomain bound to histone H3 methylated at lysine 9. Nature. 2002;416:103–107. doi: 10.1038/nature722. [DOI] [PubMed] [Google Scholar]
- 10.Fischle W, Wang Y, Jacobs SA, Kim Y, Allis CD, Khorasanizadeh S. Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev. 2003;17:1870–1881. doi: 10.1101/gad.1110503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Min J, Zhang Y, Xu RM. Structural basis for specific binding of Polycomb chromodomain to histone H3 methylated at Lys 27. Genes Dev. 2003;17:1823–1828. doi: 10.1101/gad.269603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bouazoune K, Mitterweger A, Langst G, Imhof A, Akhtar A, Becker PB, Brehm A. The dMi-2 chromodomains are DNA binding modules important for ATP-dependent nucleosome mobilization. EMBO J. 2002;21:2430–2440. doi: 10.1093/emboj/21.10.2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li H, Ilin S, Wang W, Duncan EM, Wysocka J, Allis CD, Patel DJ. Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature. 2006;442:91–95. doi: 10.1038/nature04802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peña PV, Davrazou F, Shi X, Walter KL, Verkhusha VV, Gozani O, Zhao R, Kutateladze TG. Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2. Nature. 2006;442:100–103. doi: 10.1038/nature04814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi X, Hong T, Walter KL, Ewalt M, Michishita E, Hung T, Carney D, Pena P, Lan F, Kaadige MR, et al. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature. 2006;442:96–99. doi: 10.1038/nature04835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wysocka J, Swigut T, Xiao H, Milne TA, Kwon SY, Landry J, Kauer M, Tackett AJ, Chait BT, Badenhorst P, et al. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature. 2006;442:86–90. doi: 10.1038/nature04815. [DOI] [PubMed] [Google Scholar]
- 17.Lan F, Collins RE, De Cegli R, Alpatov R, Horton JR, Shi X, Gozani O, Cheng X, Shi Y. Recognition of unmethylated histone H3 lysine 4 links BHC80 to LSD1-mediated gene repression. Nature. 2007;448:718–722. doi: 10.1038/nature06034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Org T, Chignola F, Hetenyi C, Gaetani M, Rebane A, Liiv I, Maran U, Mollica L, Bottomley MJ, Musco G, Peterson P. The autoimmune regulator PHD finger binds to non-methylated histone H3K4 to activate gene expression. EMBO Rep. 2008;9:370–376. doi: 10.1038/embor.2008.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chignola F, Gaetani M, Rebane A, Org T, Mollica L, Zucchelli C, Spitaleri A, Mannella V, Peterson P, Musco G. The solution structure of the first PHD finger of autoimmune regulator in complex with non-modified histone H3 tail reveals the antagonistic role of H3R2 methylation. Nucleic Acids Res. 2009;37:2951–2961. doi: 10.1093/nar/gkp166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karagianni P, Amazit L, Qin J, Wong J. ICBP90, a novel methyl K9 H3 binding protein linking protein ubiquitination with heterochromatin formation. Mol. Cell Biol. 2008;28:705–717. doi: 10.1128/MCB.01598-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iwase S, Lan F, Bayliss P, de la Torre-Ubieta L, Huarte M, Qi HH, Whetstine JR, Bonni A, Roberts TM, Shi Y. The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell. 2007;128:1077–1088. doi: 10.1016/j.cell.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 22.Li F, Huarte M, Zaratiegui M, Vaughn MW, Shi Y, Martienssen R, Cande WZ. Lid2 is required for coordinating H3K4 and H3K9 methylation of heterochromatin and euchromatin. Cell. 2008;135:272–283. doi: 10.1016/j.cell.2008.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grzesiek S, Stahl SJ, Wingfield PT, Bax A. The CD4 determinant for downregulation by HIV-1 Nef directly binds to Nef. Mapping of the Nef binding surface by NMR. Biochemistry. 1996;35:10256–10261. doi: 10.1021/bi9611164. [DOI] [PubMed] [Google Scholar]
- 24.Kwan AHY, Gell DA, Verger A, Grossley M, Matthews JM, Mackay JP. Engineering a protein scaffold from a PHD finger. Structure. 2003;11:803–813. doi: 10.1016/s0969-2126(03)00122-9. [DOI] [PubMed] [Google Scholar]
- 25.Garske AL, Craciun G, Denu JM. A combinatorial H4 tail library for exploring the histone code. Biochemistry. 2008;47:8094–8102. doi: 10.1021/bi800766k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zegerman P, Canas B, Pappin D, Kouzarides T. Histone H3 lysine 4 methylation disrupts binding of nucleosome remodeling and deacetylase (NuRD) repressor complex. J. Biol. Chem. 2002;277:11621–11624. doi: 10.1074/jbc.C200045200. [DOI] [PubMed] [Google Scholar]
- 27.Koh AS, Kuo AJ, Park SY, Cheung P, Abramson J, Bua D, Carney D, Shoelson SE, Gozani O, Kingston RE, et al. Aire employs a histone-binding module to mediate immunological tolerance, linking chromatin regulation with organ-specific autoimmunity. Proc. Natl. Acad. Sci. U.S.A. 2008;105:15878–15883. doi: 10.1073/pnas.0808470105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hughes RM, Wiggins KR, Khorasanizadeh S, Waters ML. Recognition of trimethyllysine by a chromodomain is not driven by the hydrophobic effect. Proc. Natl. Acad. Sci. U.S.A. 2007;104:11184–11188. doi: 10.1073/pnas.0610850104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martin DG, Baetz K, Shi X, Walter KL, MacDonald VE, Wlodarski MJ, Gozani O, Hieter P, Howe L. The Yng1p plant homeodomain finger is a methyl-histone binding module that recognizes lysine 4-methylated histone H3. Mol. Cell Biol. 2006;26:7871–7879. doi: 10.1128/MCB.00573-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taverna SD, Ilin S, Rogers RS, Tanny JC, Lavender H, Li H, Baker L, Boyle J, Blair LP, Chait BT, et al. Yng1 PHD finger binding to H3 trimethylated at K4 promotes NuA3 HAT activity at K14 of H3 and transcription at a subset of targeted ORFs. Mol. Cell. 2006;24:785–796. doi: 10.1016/j.molcel.2006.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Y, Subrahmanyam R, Chakraborty T, Sen R, Desiderio S. A plant homeodomain in RAG-2 that binds hypermethylated lysine 4 of histone H3 is necessary for efficient antigen-receptor-gene rearrangement. Immunity. 2007;27:561–571. doi: 10.1016/j.immuni.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matthews AG, Kuo AJ, Ramon-Maiques S, Han S, Champagne KS, Ivanov D, Gallardo M, Carney D, Cheung P, Ciccone DN, et al. RAG2 PHD finger couples histone H3 lysine 4 trimethylation with V(D)J recombination. Nature. 2007;450:1106–1110. doi: 10.1038/nature06431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramon-Maiques S, Kuo AJ, Carney D, Matthews AG, Oettinger MA, Gozani O, Yang W. The plant homeodomain finger of RAG2 recognizes histone H3 methylated at both lysine-4 and arginine-2. Proc. Natl. Acad. Sci. U.S.A. 2007;104:18993–18998. doi: 10.1073/pnas.0709170104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi X, Kachirskaia I, Walter KL, Kuo JH, Lake A, Davrazou F, Chan SM, Martin DG, Fingerman IM, Briggs SD, et al. Proteome-wide analysis in Saccharomyces cerevisiae identifies several PHD fingers as novel direct and selective binding modules of histone H3 methylated at either lysine 4 or lysine 36. J. Biol. Chem. 2007;282:2450–2455. doi: 10.1074/jbc.C600286200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vermeulen M, Mulder KW, Denissov S, Pijnappel WW, van Schaik FM, Varier RA, Baltissen MP, Stunnenberg HG, Mann M, Timmers HT. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell. 2007;131:58–69. doi: 10.1016/j.cell.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 36.Fiedler M, Sanchez-Barrena MJ, Nekrasov M, Mieszczanek J, Rybin V, Muller J, Evans P, Bienz M. Decoding of methylated histone H3 tail by the Pygo-BCL9 Wnt signaling complex. Mol. Cell. 2008;30:507–518. doi: 10.1016/j.molcel.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Ingen H, van Schaik FM, Wienk H, Ballering J, Rehmann H, Dechesne AC, Kruijzer JA, Liskamp RM, Timmers HT, Boelens R. Structural insight into the recognition of the H3K4me3 mark by the TFIID subunit TAF3. Structure. 2008;16:1245–1256. doi: 10.1016/j.str.2008.04.015. [DOI] [PubMed] [Google Scholar]
- 38.Ooi SK, Qiu C, Bernstein E, Li K, Jia D, Yang Z, Erdjument-Bromage H, Tempst P, Lin SP, Allis CD, et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448:714–717. doi: 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.