

Abstract
The importance of T cell-mediated immunity for resistance to the disease (cryptococcal disease) caused by Cryptococcus neoformans is incontrovertible, but whether Ab immunity also contributes to resistance remains uncertain. To investigate the role of IgM in resistance to C. neoformans, we compared the survival, fungal burden, lung and brain inflammatory responses, and lung phagocytic response of sIgM−/− mice, which lack secreted IgM, to that of IgM sufficient C57BL6×129Sv (heretofore, control) mice at different times after intranasal infection with C. neoformans (24067). sIgM−/− mice had higher mortality and higher blood and brain CFUs 28 d postinfection, but lung CFUs were comparable. Lungs of control mice manifested exuberant histiocytic inflammation with visible C. neoformans, findings that were not observed in sIgM−/− mice, whereas in brain sections, sIgM−/− mice had marked inflammation with visible C. neoformans that was not observed in control mice. Cytokine responses were significant for higher levels of lung IL-1β and IL-12 24 h postinfection in control mice and higher levels of lung and brain IL-17 28 d postinfection in sIgM−/− mice. Alveolar macrophage phagocytosis was significantly higher for control than for sIgM−/− mice 24 h postinfection; however, phagocytic indices of sIgM−/− mice increased after reconstitution of sIgM−/− mice with polyclonal IgM. These data establish a previously unrecognized role for IgM in resistance to intranasal infection with C. neoformans in mice and suggest that the mechanism by which it mediates a host benefit is by augmenting Th1 polarization, macrophage recruitment and phagocytosis of C. neoformans.
Infection with Cryptococcus neoformans first occurs in childhood and is generally followed by a state of latency that is established without a clinically evident state of disease (1, 2). Cryptococcosis (cryptococcal disease [CD]) reflects a transition from the state of latency to that of disease, generally, but not exclusively in the setting of immunosuppression (3-5). Hence, immunologically normal humans exhibit natural resistance to CD. The pathogenesis of C. neoformans has been investigated extensively in murine models, which have established the central importance of T cells in host resistance to pulmonary infection with C. neoformans (6). However, the role that B cells or naturally occurring (naive) Abs plays in the (natural) resistance of naive hosts to C. neoformans requires further study. B cell-deficient (μMT knock-out) mice were more susceptible to intratracheal challenge with C. neoformans than normal mice, and B cells were required for glucuronoxylomannan (GXM)-specific MAbs to protect against C. neoformans in this model (7), perhaps owing to the ability of B cells and/or natural Ab to enhance the antifungal inflammatory response (8, 9) and/or to dampen the inflammatory response that can be induced by Ab action (7, 10).
Recent human studies have established a link between depletion of peripheral IgM memory (IgM+CD27+IgD−) B cells and HIV-associated CD (11) and found lower levels of GXM-binding IgM in sera from HIV-infected than HIV-uninfected patients (11, 12) and solid organ transplant recipients who subsequently developed CD than those who did not (13). In humans, IgM memory B cells are the source of naturally occurring (naive) IgM Abs (14). Naturally occurring (naive) IgM is produced spontaneously, without antigenic stimulation, and has an inherent ability to bind carbohydrate and polysaccharide Ags (15). As such, it is considered to constitute a rapidly responsive, innate, pathogen-reactive repertoire that can provide immediate defense in a naive host. The homolog of human IgM memory B cells and source of naturally occurring (naive) IgM in mice is B-1 B cells (14). B-1 B cells, which are identified by CD19+CD23−IgMhiIgDlo cell surface expression (9, 14), consist of two subsets: B-1a and B-1b, which are distinguished by surface expression of CD5 (16). B cells have been observed in lung infiltrates of C. neoformans-infected mice (17), and B-1 B cells, which reside predominantly in the peritoneal and pleural cavities, can differentiate into mononuclear phagocytes and migrate to sites of inflammation (9, 14), and kill C. neoformans in vitro (8).
It has been established by different groups using i.p. infection models that specific, acquired (GXM-reactive) IgM MAbs confer protection against i.p. challenge in mice (18-21). Recently, it was shown that mice that lack IgM, sIgM−/− mice, were more resistant to i.p. infection with C. neoformans than IgM-sufficient mice (22). This finding might appear to be at variance with human data showing that levels of GXM-binding IgM are lower in patients at high risk for CD, namely, HIV-infected patients (12, 23-25), than those at low risk, such as solid organ transplant recipients who subsequently developed CD (13). However, given that sIgM−/− mice have an expanded B-1 B cell repertoire (22, 26) and normal levels of IgG2a, which is highly opsonic for C. neoformans, B-1 B cells and/or acquired Ab immunity are likely to protect sIgM−/− mice against i.p. infection (22). Nonetheless, because C. neoformans infection occurs in the lungs, the i.p. infection model might not sufficiently recapitulate natural infection to evaluate the role that naturally occurring IgM plays in resistance to CD. To investigate this question, we developed an intranasal (i.n.) infection model with C. neoformans in sIgM−/− mice and compared their susceptibility to death, cellular recruitment and cytokine profiles in the lungs and brain, and ability to phagocytose C. neoformans in vivo to the same parameters in wild-type C57BL/6×129Sv (designated hereafter as control) mice. Our data show that sIgM−/− mice were more susceptible to death after i.n. challenge than control mice and exhibited less of an ability to phagocytose C. neoformans in the lungs. We also show that i.n. administration of polyclonal IgM increased the phagocytic index of sIgM−/− mice, providing mechanistic evidence that IgM enhances resistance to C. neoformans in the lungs by enhancing the phagocytic capacity of alveolar macrophages.
Materials and Methods
Mouse strains
Age-matched sIgM−/− mice (provided by Marianne Boes, Harvard Medical School, Boston, MA) (26) and C57BL/6×129Sv mice (provided by Louis Weiss, Albert Einstein College of Medicine, [AECOM], Bronx, NY) were used for this study. C57BL/6×129Sv mice were used as controls, because sIgM−/− mice were created using 129 embryonic stem cells and the mice were not fully backcrossed at the start of our studies. Mixed background mice have been used by other investigators, including for studies of C. neoformans pathogenesis (26, 27). All mice were bred and maintained in the Institute for Animal Studies of the AECOM and given unrestricted access to food and water. All mouse experiments were conducted with prior approval from the Animal Care and Use Committee of AECOM, following established guidelines.
Cryptococcal strain
A serotype D strain (52D) of C. neoformans, ATCC 24067 (American Type Culture Collection, Manassas, VA), was used. This strain has been used extensively in studies of cryptococcal pathogenesis in mice (7, 21, 22, 27, 28). C. neoformans was grown for 52–56 h at 37°C with shaking in Difco Sabouraud Dextrose Broth (Becton Dickinson, Franklin Lakes, NJ), washed twice in sterile, endotoxin-free PBS pH 7.4 (Mediatech, Herndon, VA), counted in a hemocytometer using trypan blue for viability, and diluted to the desired concentration in PBS. For the i.n. infection, mice were anesthetized by isoflurane (Halocarbon, River Edge, NJ) inhalation and placed in a vertical position, and 1–5 × 105 CFUs of ATCC 24067 in a 20 ml volume was administered via the nares. For survival studies, infected mice were observed at least once daily.
Measurement of blood and tissue fungal burden
Using separate groups of mice than those used for the survival studies, sIgM−/− and control mice were bled retro-orbitally to collect blood samples, and then the mice were euthanized either 24 h or 7 or 28 d after i.n. infection, after which their lungs and brains were removed and homogenized in 1 ml HBSS (Lonza, Walkersville, MD). CFUs were determined by making 10-fold serial dilutions of each tissue, and plating each dilution on Sabouraud Dextrose agar plates (BBL, Sparks, MD). Each sample was processed in duplicate. Plates were incubated at 37°C for 48 h, after which, colonies were visually counted.
Characterization of the cellular composition of the lungs
The number of macrophages, B, and T cells in the lungs before and after i.n. infection with C. neoformans was determined by flow cytometry. Briefly, mice were infected as described previously, and euthanized 24 h, or 7, 28, 72, or 99 d postinfection. For isolation of cells from lung tissue, lungs were removed by dissection, washed in HBSS, and placed in tissue digestion buffer (1 mg/ml collagenase A [Roche Diagnostics, Mannheim, Germany], 30 μg/ml DNase I [Roche Diagnostics], 10% FBS [Atlanta Biologicals, Lawrenceville, GA], and 90% RPMI 1640 [Mediatech]) as described (29). Single-cell suspensions were produced by lacerating the lungs with sterile razor blades while in the digestion buffer and placing them into a 37°C incubator supplemented with 5% CO2 for 30 min. After incubation, lung pieces were pipetted into a 70-μm cell strainer (BD-Falcon, Bedford, MA) and the tissue was teased apart using a 3-ml syringe plunger (BD, Franklin Lakes, NJ). The cell suspension was pelleted at 1200 rpm at 4°C for 7 min and the erythrocytes were lysed in 5 ml ACK buffer (0.15 M NH4Cl, 10 mM KHCO3, 0.1 mM EDTA, adjusted to a pH of 7.2–7.4 with 1 N HCl [all reagents from Sigma-Aldrich, St. Louis, MO]) at room temperature (RT). The cells were then washed once and 5 ml 1% BSA-PBS was added, and passed through a 40-μm cell strainer (BD-Falcon). Finally, the cells were resuspended in 1% BSA-PBS and counted in a hemocytometer; an aliquot was appropriately diluted to a final concentration of 107 total cells per ml. The 100 μl of the cell suspension (106 cells) was removed for immunostaining. The following reagents were used: FITC conjugated anti-mouse CD19 (BD Biosciences, San Jose, CA), PE-Cy7 conjugated anti-mouse CD5, Cy5 conjugated anti-mouse IgM, Fc5μ fragment-specific (Jackson ImmunoResearch, West Grove, PA), PE-Cy5 conjugated anti-mouse CD4, allophycocyanin conjugated anti-mouse CD8, and FITC conjugated anti-mouse F4-80, a macrophage marker (all from e-Biosciences, San Diego, CA). These reagents have been used extensively for the detection of B cells, T cells, and macrophages in mice (7, 27, 29). Cells were incubated with the Abs for 1 h at 4°C, washed and fixed with 2% paraformaldehyde. Single-color controls and fluorescence-minus one controls were also included to ensure proper gating. Data was collected on a FACSCalibur (Becton Dickinson, Sunnyvale, CA) interfaced to Cell-Quest software (Macintosh version, Becton Dickinson). A total of 50,000 events per sample were analyzed. All analyses were performed using FlowJo v.7.2.2 for Windows software (TreeStar, Ashland, OR).
Measurement of cytokine levels
Separate groups of sIgM−/− and control mice than those that were used for the survival experiments were euthanized in the naive state, and 24 h, 7 d, or 28 d postinfection, and brain and lung homogenates were prepared and stored at −80°C prior to use. The concentration of cytokines in these tissues was determined using the Beadlyte Mouse 21-plex Cytokine Detection System. Levels of IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-9, IL-10, IL-12(p40), IL-12(p70), IL-13, IL-17, GM-CSF, IFN-γ, KC, MCP-1, MIP-1β, RANTES, TNF-α, and VEGF (Millipore, St. Charles, MO) were determined according to the manufacturer’s protocol, using a Luminex HT200 machine (Luminex, Austin, TX). Sera and lung and brain homogenates were stored at −80°C. Prior to use the samples were centrifuged at 4°C for 15 min at 13,000 × g (to separate lipids, following manufacturer’s recommendations) and used at a dilution of 1:2.
Determination of IgM in lung and IgM and IgG serum
Total and GXM-specific IgM, and IgG, were determined on serum samples obtained from the mice in the naive state and on days 7 and 28 postinfection using an IgM ELISA and a GXM-IgM ELISA, as described previously (22, 28). Levels of total IgM were similarly measured in serial dilutions of lung homogenates from control mice.
Measurement of C5a in the lungs
The level of C5a in the lungs of sIgM−/− and control mice in the naive state and 24 h, 7 d, and 28 d postinfection was determined by ELISA using the mouse complement component C5a DuoSet kit (R&D Systems, Minneapolis, MN). Briefly, 96-well microtiter plates were coated with 4 μg/ml rat anti-mouse C5a (R&D Systems) in PBS. Plates were incubated overnight at RT. Prior to use, the plates were washed and blocked with 1% BSA (Sigma-Aldrich) in PBS for 1 h at RT. Samples of lung homogenates from naive and C. neoformans-infected mice from each strain were diluted 1:100 in PBS, applied to the plate in duplicate, and serially diluted 1:2 in 1% BSA-PBS. A recombinant mouse C5a standard (R&D Systems) was included on the plates. The plates were incubated, washed, and detected with 200 ng/ml of biotinylated anti-mouse C5a (R&D Systems) for 2 h at RT. After incubation, the plates were washed and the biotinylated complex was detected with 1:200 dilution of streptavidin conjugated to HRP (R&D Systems) and incubated for 20 min at RT. A total of 100 μl SureBlue TMB (KPL, Gaithersburg, MD) was added and the reaction was stopped with 2 N H2SO4 (Fisher Scientific, Kalamazoo, MI). Absorbances were measured by an ELISA reader (Tecan, Männedorf, Switzerland) at a wavelength of 450 nm corrected to 540 nm. Wells with no sera were included as a negative control to determine the background absorbance of the reagents.
Ex vivo measurement of phagocytosis by alveolar macrophages
Using separate groups of mice than those used for the survival studies, sIgM−/− and control mice were euthanized either 24 h or 7 d postinfection with 5 × 105 C. neoformans to isolate alveolar macrophages for phagocytosis experiments. These times were chosen to evaluate the early response to C. neoformans. Alveolar macrophages were isolated from sIgM−/− and control mice as described previously (27, 30). Briefly, mice were euthanized and their tracheas were exposed by a skin incision. After the incision, a 20-gauge angiocath (BD) was advanced into the trachea and the lungs were lavaged 10 times with 0.8 ml of 1 mM EGTA (Sigma-Aldrich) in HBSS. The lavage fluids were pooled, and cells were collected by centrifugation. The cells were then resuspended in DMEM (Mediatech) containing 10% NCTC-109 (Invitrogen, Carlsbad, CA), 10% FCS (Atlanta Biologicals), and 1% MEM nonessential amino acids (Mediatech), and the cell suspension was allowed to attach to 96-well plates for 2 h, fixed with methanol, and then stained with Giemsa stain as described previously (27, 30). The phagocytic index (PI) was determined by examination of the wells with an inverted microscope at a magnification of ×400. The PI was defined as the number of internalized C. neoformans cells per 100 macrophages counted.
IgM reconstitution
To determine the role that IgM plays in phagocytosis of C. neoformans in the lungs, sIgM−/− mice were reconstituted with polyclonal IgM.
IgM preparation
Polyclonal IgM was prepared according to published methods (26, 31) with some modifications, as follows: C57BL/6 serum (Sigma-Aldrich) was thawed at RT and the Ig was precipitated with saturated (NH4)2SO4 and agitated at 4°C for 2 h, after which it was spun at 3000 rpm for 30 min and the supernatant was discarded. The precipitate was washed in 50% (NH4)2SO4 for 30 min at 3000 rpm, the supernatant was discarded, and then the precipitate was resuspended and washed extensively (4–5 times) and diluted in PBS. The precipitated Ig was run over a 2-ml anti-IgG agarose column (Sigma-Aldrich) and then over a 2-ml anti-IgM column (Sigma-Aldrich). The IgM was eluted with glycerin pH 2.7 and neutralized with pH 8.0 Tris-HCl. The IgM was concentrated, washed, tested for IgM and IgG reactivity, and quantified by ELISA. The IgM/IgG ratio of the solution that was administered was 7.5:1; in comparison, the ratio of IgM/IgG ratio in normal mouse serum is 1:20.
IgM administration
A group of sIgM−/− mice was given 50 μl IgM i.n. totaling 250 μg (reconstituted group, sIgM−/− and IgM), and a group of sIgM−/− and a group of control mice received PBS; then, all of the mice were infected i.n. with C. neoformans 1 h later. Mice were anesthetized with isoflourane, euthanized, and then bronchoalveolar lavage was performed as described previously, after which lavage fluid was plated and the cells were processed and scored for phagocytosis as described previously.
Histopathological examination
Using separate groups of mice than those used for the survival studies, sIgM−/− and control mice were euthanized either 7 (lungs) or 28 (lungs and brains) days postinfection to obtain samples for histological examination. The tissues were fixed in 1:10 buffered formalin (Fisher Scientific) and processed at the Histopathology Core of AECOM. Paraffin-embedded lung and brain tissues were stained with H&E and examined under a Zeiss AxioScope II microscope (Carl Zeiss, Thornwood, NY), using the ×2.5, ×10, and ×40 objectives, giving final magnifications of ×25, ×100, and ×400, respectively. Photomicrographs were captured using a camera mounted on the microscope. For the purpose of the illustration, the original photographs were processed in entirety by Adobe Photoshop CS3 for Windows (Adobe Systems, San Jose, CA) for balancing the contrast and color levels using the Auto Contrast and Auto Levels function of the software; resized maintaining their size ratios intact, and assembled into a single multipanel image in the prescribed format.
Statistics
Mouse survival data were evaluated by comparing Kaplan-Meier survival curves with a log-rank (Mantel-Cox) test. The unpaired Student t test or the Mann-Whitney U test was used for between-group comparisons depending on the normality of data distributions and sample size as appropriate; logarithmic transformations were applied on data involving very large numbers (such as cell numbers and CFU counts) prior to statistical comparison, to ensure normality and homoscedasticity of the variables. Data analyzed by Student t test were represented by bar graphs of the group means and SEs, and those analyzed by the Mann-Whitney U test were represented by scatterplots of the data points with a line indicating group medians. For all statistical evaluations, a two-tailed p value of ≤0.05 was considered significant, and p values of 0.05–0.1 were considered a trend toward significance; all p values were reported as numerical values. All statistical tests were performed using Prism v.5.03 for Windows (GraphPad Software, San Diego, CA).
Results
Survival and fungal burden
Using death as an end point, control mice survived significantly longer than sIgM−/− mice (log-rank test, p = 0.025) (Fig. 1). We evaluated survival for 100 d and terminated the experiment.
FIGURE 1.

Survival curves for sIgM−/− and C57×129Sv (control) mice challenged i.n. with C. neoformans ATCC 24067. The 1–5 × 105 CFUs was delivered i.n. in a 20-μl volume. Survival was evaluated up to 100 d. n = 12–13 mice per group. Asterisk (*) p = 0.025, comparing sIgM−/− versus control mice, log-rank test. The solid circles represent sIgM−/− mice; the open squares represent control mice.
Cellular composition of the lungs
Total lung lymphocytes, B, and T cells
We determined the levels of total lymphocytes, B, and T cells in the lungs of sIgM−/− and control mice in the naive state and 7 and 28 d postinfection to capture times that represent the early, innate (day 7), and mature, acquired (day 28) immune response (Fig. 2).
FIGURE 2.

Cellular composition of naive and post-C. neoformans infection lungs of control and sIgM−/− mice. A, Total lung lymphocytes. B–D, B cells and subsets. E and F, T cell subsets. G, Macrophages. White bars represent sIgM−/−; black bars, control mice. Asterisk (*), significant comparisons between strains; octothorpe (#), comparisons to sIgM−/− day 7 values; ampersand (&), comparisons to control mice day 7 values; p value range for significant comparisons, 0.0414 – <0.0001, unpaired Student t test. n = 3–5 mice per group.
Lung lymphocytes
There was no difference in total lung lymphocytes (Fig. 2A) between the mouse strains in the naive state or 7 d postinfection. However, both mouse strains had a significantly higher number of total lung lymphocytes on day 7 postinfection than in the naive state. Within strains, sIgM−/− mice had significantly fewer lymphocytes on day 28 than day 7 postinfection. Control mice had significantly more lymphocytes on day 28 postinfection than sIgM−/− mice.
Lung B cells
We evaluated total B cells and B cell subsets in the lungs of sIgM−/− and control mice. Compared with control mice, sIgM−/− mice had significantly higher total (CD19+) B cells 7 d postinfection (p < 0.0001) (Fig. 2B), higher B-1a B cells in the naive state (p = 0.0006) and 28 d postinfection (p = 0.0002) (Fig. 2C), and higher B-1b B cells 7 d postinfection (p < 0.0001) (Fig. 2D). Within strains, sIgM−/− had higher total B cells and B-1b B cells on 7 d and 28 d postinfection than in the naive state, and higher B-1a B cells on day 28 than on day 7 postinfection (Fig. 2B–D), whereas the control mice had lower total B cells and B-1b B cells on 7 d postinfection than in the naive state and on 28 d postinfection, and higher B-1a B cells on day 7 postinfection than in the naive state (Fig. 2B–D).
Lung T cells and macrophages
We evaluated T cell subsets (CD4+ and CD8+) and macrophages because of their importance in immunity to C. neoformans (6, 30, 32). Compared with sIgM−/− mice, control mice had more CD4+ T cells (Fig. 2E) on day 7 postinfection (p = 0.0003) and more macrophages (Fig. 2G) on days 7 (p = 0.0061) and 28 postinfection (p = 0.0027). Within strains, sIgM−/− mice had higher CD4+ and CD8+ T cells and macrophages on day 7 postinfection than in the naive state and higher CD8+ T cells and macrophages on day 28 than on day 7 postinfection. The control mice had higher macrophages on day 7 postinfection than in the naive state.
Fungal burden in the lungs, blood, and brain
We determined the fungal burden in the lungs, blood, and brains of sIgM−/− and control mice 24 h and 7, 28, and 72 d postinfection by enumerating CFUs in these tissues (Fig. 3A–C). We also determined blood CFUs on day 99 postinfection prior to terminating the survival experiment. sIgM−/− mice had lower but comparable (difference of means ~0.5 log) CFUs in the lungs compared with the control mice 24 h and on day 28 postinfection; however, lung CFUs in sIgM−/− were much lower than those in control mice on days 7 (difference of means, 1.33 log; p = 0.0506) and 72 postinfection (difference of means, 1.22 log; p = 0.21) (Fig. 3A). In contrast, sIgM−/− mice had higher but statistically comparable blood and brain CFUs than control mice at all times, with significantly higher blood CFUs on day 72 postinfection (difference of means, 1.14 log; p = 0.0032), with a comparably concomitant higher level in the brain (difference of means, 1.84 log; p = 0.11) (Fig. 3B, 3C).
FIGURE 3.
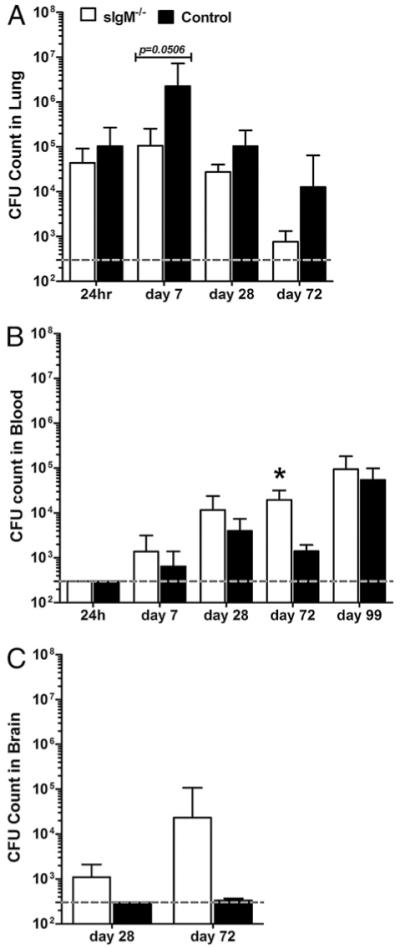
Organ fungal burden (CFUs) of C. neoformans-infected control and sIgM−/− mice. A, B, and C represent, respectively, CFUs enumerated in lungs, blood, and brain of separate groups of mice at the times postinfection indicated. Dashed grey line represents lower limit of detection of the assay. White bars represent sIgM−/−; black bars, control mice; asterisk (*), significant comparison between strains; blood CFU, day 72 postinfection, p = 0.0032. All comparisons by the Student t test; n =3–18 for lung and blood CFUs, 2–5 for brain CFUs.
The inflammatory response
A number of groups have demonstrated the importance of cytokines and chemokines in host resistance to CD (30, 33-37). We evaluated lung cytokines and chemokines in the naive state, 24 h, and on days 7 and 28 postinfection, because these times marked the early, innate (24 h, 7 d), and mature acquired (28 d) immune response, with day 28 marking the time of a difference in survival between the mouse strains. Brain cytokines were evaluated on days 7 and 28 postinfection.
Lungs
Between the mouse strains, there were comparable levels of chemokines or cytokines (IL-1α, IL-1β, IL-6, IL-17, IFN-γ, TNF-α, IL-10, and IL-13) in the lungs in the naive state (Fig. 4, cytokine panels as indicated), and only a few statistically significant differences and trends at other times. Compared with sIgM−/− mice, control mice had more IL-1α, IL-1β (p = 0.0499), IL-12 (p = 0.0298), IL-17, IL-4, and IL-10 24 h postinfection; and more IL-1β, IL-6 (p = 0.0267), IFN-γ, and IL-4 on day 28 postinfection (Fig. 4, cytokine panels as indicated). In contrast, sIgM−/− mice had more IL-17 (p = 0.0335) and IL-13 (p = 0.018) than control mice on day 28 postinfection (Fig. 4, cytokine panels as indicated).
FIGURE 4.

Cytokine and chemokine levels in lung homogenates of naive and C. neoformans-infected control and sIgM−/− mice. Cytokine and chemokine panels as indicated on the figure. Open circles represent sIgM−/−; closed circles, control mice. Asterisk (*), significant comparisons between strains. The p values for comparisons at postinfection times, IL-1β 24 h, 0.0499; IL-4 24 h, 0.0092; IL-4 d 28, 0.0027; IL-6 d 28, 0.0267; IL-12p40 24 h, 0.0298; IL-13 d 28, 0.018; IL-17 d 28, 0.0335. All comparisons by the Mann-Whitney U test, p values as indicated; n = 4–16 mice per group.
Brain
In the naive state, both mouse strains had similar and comparable median levels of brain cytokines and chemokines, with the exception of IL-10, which was higher in the brains of sIgM−/− than control mice (Fig. 5). On day 28 postinfection, sIgM−/− mice had lower levels of IL-1β and IL-10 (not statistically significant), and higher levels of RANTES (p = 0.0176), MCP-1 (not statistically significant), and IL-17 (p = 0.0101) than control mice. Within strains, between the naive and postinfection state, the sIgM−/− mice exhibited a decrease in IL-1β and IL-10, whereas levels of these mediators were unchanged over this period in the control mice; both strains showed significantly higher median levels of RANTES postinfection than in the naive state (Fig. 5). The lack of statistical significance in various comparisons were determined to be partially due to computed cytokine values lower than the limit of assay detection for some mice, but these mice were not excluded from analyses because the biological significance of the lack of cytokine response is unclear.
FIGURE 5.
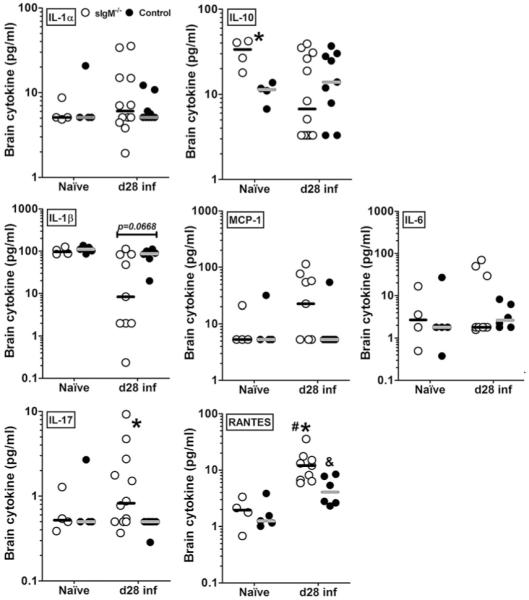
Cytokine and chemokine levels in brain homogenates from naive and C. neoformans-infected control and sIgM−/− mice. Cytokine and chemokine panels as indicated on the figure. Open circles represent sIgM−/−; closed circles, control mice. Asterisk (*), significant comparisons between strains; IL-10, naive, p = 0.0286; IL-17, day 28, p = 0.0101; IL-1β, day 28, p = 0.0245; RANTES, day 28, p = 0.0176; octothorpe (#), comparison within sIgM−/− values; RANTES, naive versus d 28, p = 0.0028; ampersand (&), comparison within control mice, RANTES, naive versus d 28, p = 0.0303. All comparisons were achieved using the Mann-Whitney U test, p values as indicated; n = 4–12 mice per group.
Serum total and specific Ab response
Total and GXM-specific IgM were determined in the serum of control mice, and total and GXM-specific IgG, in the serum of sIgM−/− and control mice in the naive state, and at 24 h and on day 7 and day 28 postinfection (Fig. 6A–D). Total serum IgM in the control mice in the naive state was higher than at 24 h postinfection and levels were higher postinfection: on day 7 and day 28 IgM and GXM-specific IgM were significantly higher than the 24 h postinfection levels. Total serum IgG levels were similar and comparable between the two mouse strains at all times, except on day 7 postinfection when the control mice had nominally higher IgG than sIgM−/− mice. Control mice had significantly higher GXM-specific IgG than sIgM−/− mice in the naive state (p = 0.0401) and on day 7 postinfection (p < 0.0001). Within strains, sIgM−/− mice in the naive state had higher serum IgG than at 24 h postinfection and levels were higher postinfection: day 28 IgG, as well as day 7 and day 28 GXM-specific IgG, were significantly higher than the 24 h postinfection levels. Compared with 24 h postinfection, total serum IgG in control mice was higher in the naive state, and at days 7 and 28 postinfection. In contrast, GXM-specific IgG in the control mice was similar and comparable across all times, except at day 7 postinfection, when it was significantly lower than the 24 h postinfection levels.
FIGURE 6.
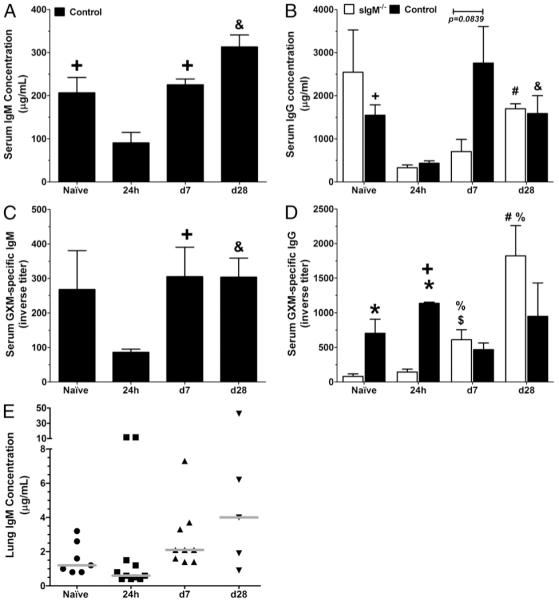
Serum IgM and IgG and lung IgM levels in naive and of C. neoformans-infected control and sIgM−/− mice. Asterisk (*), comparison between strains; ampersand (&), control mice; day 28 versus other times; plus (+), control mice; 24 h versus naive and day 7; octothorpe (#), sIgM−/−; day 28 versus other times; percent (%), sIgM−/−;24h versus naive and day 7; dollar ($), sIgM−/−; naive versus day 7. A and C, Total serum IgM and GXM-specific IgM, respectively, in control mice (black bars). Naive IgM > 24 h (p = 0.0205), day 7 and day 28 postinfection (p =0.0001 and <0.0001, respectively), and day 7 and 28 GXM-specific IgM > 24h(p =0.0315 and 0.0079, respectively). B and D, Total IgG and GXM-specific IgG, respectively, in both mouse strains (sIgM−/−, white bars). Control GXM-specific IgG > sIgM−/− naive (p = 0.0401) and day 7 (p < 0.0001) postinfection. sIgM−/− mice, total IgG day 28 > 24h (p < 0.0001) postinfection; GXM-specific IgG day 7 (p = 0.0278) and day 28 (p = 0.0087) > 24 h postinfection. Control mice, total IgG naive (p = 0.0029), day 7 (p = 0.0527) and day 28 (p = 0.0344) > 24 h postinfection; and GXM-specific IgG day 7 < (p = 0.0025) 24 h postinfection. E, Lung total IgM concentration control mice. IgM on day 7 (p = 0.0269) < 24 h and day 28 (p = 0.0531) postinfection. Serum comparisons were performed by Student t test; lung comparisons, by the Mann-Whitney U test. n = 5–8 mice per group for serum; 5–11 mice per group for lungs.
IgM levels in the lungs
To determine whether there is IgM in the lungs of control mice at the time of infection and postinfection, we measured total IgM in lung homogenates in the naive state, and at 24 h and on day 7 and day 28 postinfection. There was detectable IgM at each of these times, with a higher level than at 24 h postinfection, on day 7 (p = 0.0269) and day 28 (p = 0.0531) (Fig. 6E); the pattern of response followed that of the total serum IgM in these mice (Fig. 6A). There was no detectable IgM in homogenates from sIgM−/− mice (not shown).
Levels of C5a in the lungs
C5a anaphylatoxin can negatively regulate the Th1 response (38), enhance Th17 (39), and promote T cell recruitment (40) as well as the inflammatory response (41). Hence, we determined lung C5a levels in both strains of mice in the naive state, 24 h, 7, and 28 d postinfection (Fig. 7). The sIgM−/− mice had nominally higher mean levels of C5a than the control mice in the naive state, 24 h, and on day 28 postinfection, with the difference of means being significant at 24 h postinfection (p = 0.0172). For both strains of mice, 24 h lung C5a levels were significantly lower than the naive levels, as well as the levels on day 28 postinfection.
FIGURE 7.

C5a concentration in the lungs of naive and C. neoformans-infected control and sIgM−/− mice. Lung C5a levels were determined by ELISA as described in the text at the indicated times. Asterisk (*) represents comparisons between strains; ampersand (&), comparisons to C57 day 24 h; octothorpe (#), comparisons to sIgM 24 h < sIgM−/− mice > control mice 24 h postinfection (p = 0.0172). The 24 h < naive (sIgM−/−, p = 0.0408; control, p = 0.0327) and day 28 (sIgM−/−, p = 0.087; control, p = 0.0479) postinfection. All comparisons were done by Student t test. n = 6–10 mice per group.
Phagocytosis by alveolar macrophages
In vivo phagocytosis has been shown to be an important correlate of survival in pulmonary models of infection with the 24067 strain (30). We examined in vivo phagocytosis, ex vivo, 24 h, and on day 7 postinfection (Fig. 8). These times were selected to assess the response to C. neoformans in the lungs at the time that naturally occurring, innate B cell/IgM responses would be operative. Fig. 8A and 8B demonstrate the microscopic appearance of cells with internalized C. neoformans that were enumerated. Fig. 8C and 8D represent the cell counts 24 h and day 7 postinfection, respectively; at both times, compared with sIgM−/− mice, control mice had significantly more phagocytosing cells (p < 0.0025), more phagocytosing cells with one to two C. neoformans (p < 0.0025) and more phagocytosing cells with three or more C. neoformans (p = 0.0147 at 24 h; and p = 0.051 on day 28). These experiments revealed a significantly higher PI (p < 0.0025) for macrophages that were obtained from control than sIgM−/− mice (Fig. 8E) at both times.
FIGURE 8.
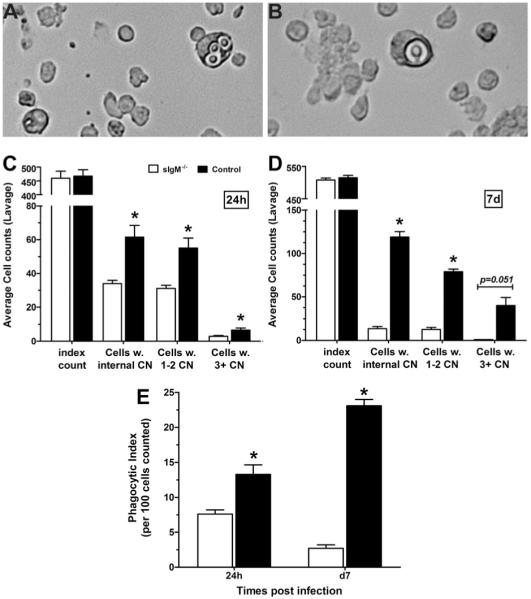
In vivo phagocytosis by alveolar lavage cells from C. neoformans-infected control and sIgM−/− mice. A and B, Microscopic appearance of internalized C. neoformans in enumerated cells. C and D, Cell counts 24 h postinfection and day 7 postinfection, respectively; at both times, phagocytosing cells control mice > sIgM−/− (p < 0.0025), and more cells with one to two C. neoformans (p < 0.0025) and three or more C. neoformans (p =0.0147 at 24 h; and 0.051 at day 7). E, PI control > sIgM−/−, both times (p < 0.0025). All comparisons were performed by Student t test; n = 11–12 mice per group at 24 h; three mice per group at day 28.
Reconstitution of sIgM−/− mice and measurement of phagocytosis
After administration of 250 μg polyclonal IgM, sIgM−/−/IgM (reconstituted) mice had 0.37, 1.05, 0.77, and 0.75 μg/ml IgM (n = 4) in lung homogenates; control mice had 2.75, 1.69, 0.52. 0.45, and 0.23 μg/ml (n = 5). The mean concentrations of IgG in the homogenates of the three experimental groups were not significantly different: sIgM−/−/IgM (reconstituted) 4.88 ± 2.09 μg/ml; sIgM−/−/PBS 3.51 ± 0.65 μg/ml; control 3.55 ± 4.06 μg/ml. The PI of alveolar lavage cells from sIgM−/−/IgM (reconstituted) mice was similar to that of control mice, and that of sIgM−/− (sIgM−/−/PBS, sIgM−/− control for reconstitution) was significantly less than that of control (p = 0.0024) and of reconstituted sIgM−/−/IgM mice (p = 0.0219) (Fig. 9). Hence, i.n. administration of IgM to sIgM−/− mice led to measurable levels of IgM in the lungs of sIgM−/− mice, no significant difference in IgG and an increase in the level of in vivo phagocytosis comparable to that of control mice.
FIGURE 9.

Phagocytosis by alveolar lavage cells from C. neoformans-infected sIgM−/−, control and IgM-reconstituted sIgM mice. The phagocytic index of alveolar lavage cells from sIgM−/− (white bar) < control (black bar; p = 0.0024) and reconstituted sIgM−/− mice (sIgM plus IgM, striped bar; p = 0.0219); no difference between reconstituted sIgM and control mice. All comparisons were performed with the Student t test; n = 11 sIgM−/−, 12 control and 4 sIgM plus IgM mice.
Histopathological examination of the lungs and brains
The histopathological appearance of lung tissue was evaluated on days 7 and 28 postinfection to compare the early, innate, and mature acquired response to C. neoformans in both mouse strains. Brain tissue was evaluated on day 28 postinfection. There were no findings in the lungs on day 7 postinfection. On day 28 postinfection, the lungs of control mice had evidence of necrosis, histiocytic infiltrates, and intralesional C. neoformans with the typical “soap bubble” appearance (Fig. 10A, 10B,). In contrast, the lungs of sIgM−/− mice had less exuberant cellular infiltrates, with perivascular inflammation and cellular infiltration around blood vessels and bronchioles without visible C. neoformans (Fig. 10C, 10D). The brains of sIgM−/− mice had evidence of meningitis with visible C. neoformans (Fig. 10E, 10F). No lesions were detected in the brains of control mice (Fig. 10G, 10H).
FIGURE 10.

Lung and brain pathology in sIgM−/− and control mice. The histopathological appearance of H&E stained lung and brain tissue on day 28 postinfection. Lungs of control mice had evidence of necrosis, histiocytic infiltrates, and intralesional C. neoformans with the typical “soap bubble” appearance (A, original magnification ×25; B, original magnification ×400). Lungs of sIgM−/− mice had less exuberant cellular infiltrates, with perivascular inflammation and cellular infiltration around blood vessels and bronchioles without visible C. neoformans (C, original magnification ×25; D, original magnification ×400). The brains of sIgM−/− mice had evidence of meningitis with visible C. neoformans (E, original magnification ×100; F, original magnification ×400). No lesions were detected in the brains of control mice (G, original magnification ×100; H, original magnification ×400).
Discussion
The role that Ab immunity plays in resistance to CD remains unresolved. Data from human studies reveals associations between HIV-associated CD and depletion of IgM memory B cells, and reduced expression of GXM-reactive IgM and solid organ transplant-associated CD (11-13). However, association is not causation and studies to establish causation cannot be performed in humans at this time. In recent years, mice that lack secreted IgM (sIgM−/− mice) have been used to establish the importance of naturally occurring (naive) IgM and B cells in resistance to numerous distinct pathogens (31, 42-47). In this study, we used sIgM−/− mice to evaluate the role of naturally occurring (naive) IgM in resistance to CD after i.n. infection with C. neoformans. The infection model reported herein uses a relatively low inoculum (1–5 × 105 CFUs), to which C57×129 (the control strain used in this study) were resistant. We note that this feature of our model recapitulates the resistance of immunologically normal humans to CD, despite ample evidence for chronic infection (latency) (see Ref. 3). Furthermore, although mouse susceptibility to CD depends on multiple factors, such as genetic background, C. neoformans strain, inoculum, and route of infection, we note that many groups use infection models in which mice that lack an immune subset or factor of interest are compared with mice that are resistant to death under the same experimental conditions (34, 37, 48).
The central result of this study is that sIgM−/− mice were more susceptible to death after i.n. challenge with C. neoformans than IgM-sufficient (control) mice. Compared with IgM-deficient mice, resistance in IgM-sufficient mice was associated with early Th1 polarization in the lungs and significantly more in vivo phagocytosis of C. neoformans. To test the hypothesis that IgM contributes to the phagocytic activity of control mice, we delivered purified serum IgM to sIgM−/− mice and evaluated its effect on phagocytosis in the lungs 24 h postinfection. IgM was delivered i.n. to recapitulate the presence of pleural B-1 B cell-derived IgM in the lungs, because IgM does not enter the lungs from the circulation. Remarkably, there was measureable IgM in the lungs of reconstituted sIgM−/− mice and the percent of phagocytosing cells in these mice increased significantly to a level that was comparable to that of control mice. Although determining the effect of administered IgM on survival or the inflammatory response was beyond the scope of this study, our findings echo results from many other models in which naturally occurring IgM enhanced pathogen clearance, prevented dissemination (15, 31, 42-44, 49) and/or reconstituted a resistant phenotype in IgM-deficient mice (31, 43, 50). To our knowledge, our data are the first to provide proof that naturally occurring (naive) IgM contributes to early host defense against C. neoformans in naive mice. However, we note that the role that IgM plays in resistance to CD is likely to be tissue-specific, given that sIgM−/− mice were more resistant to C. neoformans than control mice in an i.p. infection model (22).
As reported in other studies (22, 26), sIgM−/− mice had higher levels of lung B-1a B cells than control mice in the naive state. B-1a B cells, which are distinguished by surface expression of CD5 (16), are the source of naturally occurring (naive) IgM in mice (15). As a central component of innate immunity, naturally occurring IgM is produced without prior Ag stimulation and has intrinsic carbohydrate and polysaccharide reactivity, including with T-independent 2 Ags (15), of which GXM is an example (51). For another encapsulated pathogen, Streptococcus pneumoniae, B-1a B cells produced Abs to phophorylcholine (PC), whereas B-1b B cells produced Abs to pneumococcal capsular polysaccharide (PPS), and PC-binding Abs were required for natural resistance to S. pneumoniae in naive mice, whereas PPS-specific Abs were required for protection in immunized mice (49). C. neoformans vesicles, which are involved in capsular assembly, are rich in PC and phosphatidylcholine (52), and the C. neoformans cell wall is amply decorated with carbohydrate determinants, including β glucans, which are recognized by protective mAbs to lamanarin (53). Of relevance to innate immunity to C. neoformans Ags in mice, Abs to PC and phosphatidylcholine are derived from B-1a B cells (54, 55), GXM Abs are likely to be derived from B-1b B cells (49) and the B-1 B cell compartment is held to constitute a general carbohydrate Ab repertoire (15). Hence, B-1 B cell-derived IgM could be a crucial component of the innate response to C. neoformans.
Our data show that lung homogenates from control mice in the naive state and postinfection with C. neoformans contained IgM. These mice also exhibited increased total and GXM-specific serum IgM postinfection, but lung IgM was most likely derived from pleural B-1 B cells (50), because IgM does not enter the lungs from the circulation. In other models, B-1 B cell-derived IgM induced early T cell recruitment (56) and viral aggregation/neutralization (31) via immune complex-mediated complement activation. Complement component 3 (C3) is required for natural resistance to CD in mice (57) and for the generation of C5a. Given that transfer of naive IgM to sIgM−/− mice induced C3 activation via the classical complement pathway in other models (31, 43), C3 activation could be impaired in naive C. neoformans-infected sIgM−/− mice, as described for S. pneumoniae-infected sIgM−/− mice (44). However, given that sIgM−/− and control mice had similar levels of total lung IgG, C3 activation and production of C5a could have been elicited by IgG-containing immune complexes in sIgM−/− mice. However, sIgM−/− mice had lower levels of serum GXM-specific IgG than control mice, which could have contributed to reduced opsonic activity against C. neoformans. sIgM−/− mice have been shown to mount normal IgG responses to TI-2 Ags (26), including after i.p. infection with C. neoformans (22), although human studies reveal associations between IgM memory B cell depletion, reduced PPS-binding IgM, reduced PPS vaccine responses and/or an increased risk for disease (58-61). Hence, the ability of human and mouse B cell subsets to produce GXM-specific IgG in different tissues and the role that IgM and IgG play in C3 activation could differ between species.
Apart from Ab production, B-1 B cells secrete cytokines and can exert direct antimicrobial effects (8, 9, 62). For example, B-1 B cell IL-10 production inhibited macrophage efficacy against some pathogens (63, 64). Although our data does not rule out the possibility that lung B-1 B cell derived IL-10 inhibited alveolar macrophage phagocytosis of C. neoformans, there was no statistically significant difference in lung IL-10 levels between sIgM−/− and control mice at any time that was examined in this study. In fact, control mice exhibited a trend toward higher levels of IL-10 24 h postinfection and more B-1a B cells on day 7 postinfection than in the naive state, suggesting a role for B-1a B cell-derived IL-10 cells in their response, perhaps in counterbalancing Th1-induced inflammation (7). B-1a B cells are a major source of IL-10 in mice (65). B-1 B cells have also been shown to differentiate into phagocytes (B cell-derived mononuclear phagocytes [BDMP]) in vitro (9). BDMPs had less activity against Paracoccidiodes and Coxiella than macrophages (62, 63), but they exhibited significantly more phagocytosis and killing of C. neoformans than macrophages (8). However, the latter required a Th1-polarized environment and was only demonstrated for IgM-producing B-1 B cells from normal mice. Hence, our findings support the hypothesis that in control mice, Th1 polarization, as evidenced by their higher levels of IL-12 24 h postinfection (see below), in concert with and/or induced by lung IgM, enhanced macrophage and/or BDMP phagocytosis of C. neoformans. This hypothesis requires further investigation as it is currently unknown whether BDMPs develop in vivo. Nonetheless, our data demonstrating IgM in lung homogenates of control mice and the ability of naive IgM to enhance phagocytosis in sIgM−/− mice strongly support the conclusion that the phenotype of C. neoformans-infected sIgM−/− mice in this study resulted from the lack of IgM, rather than an expanded B-1 B cell repertoire.
Our data show that control mice had higher levels of lung IL-12 and IL-1β 24 h postinfection and more CD4+ T cells and macrophages in their lungs on day 7, implicating IgM in Th1 polarization and macrophage recruitment. The role that macrophages play in host defense against C. neoformans in mice is complex and context/animal model dependent. In some studies using chlodronate-mediated macrophage depletion, macrophages exerted a detrimental effect (66-69), but in another they did not (70). In contrast, studies of in vivo macrophage phagocytosis revealed a beneficial effect (30, 71), with the caveat that the cytokine milieu is a key determinant of macrophage efficacy against C. neoformans (69, 71, 72). Th1 and Th17 cytokines, for example, IL-12 and IL-17, respectively, promoted C. neoformans uptake by macrophages and limited intracellular replication and extrusion in vitro (72). Our data show that control mice had a (>1 log) trend toward more IL-17 in their lungs than sIgM−/− mice 24 h postinfection, whereas sIgM−/− mice had a trend toward more IL-17 on day 7 and significantly more on day 28. Given that IL-17 was shown to enhance fungal clearance without reducing dissemination (48, 73), this might help explain the trend toward lower CFUs in the lungs of sIgM−/− mice at these times, despite trends toward more blood CFUs. Although the statistically significant difference in IL-17 between control and sIgM−/− mice on day 28 postinfection, and in other cytokines at other times, was numerically small, we note that the amount of cytokines needed to mediate a biological effect against C. neoformans in vivo is unknown. Hence, more studies, focusing on quantitative and qualitative T cell and/or macrophage polarization are needed to assemble a better understanding of the effect of IgM deficiency on the cytokine milieu.
CFU data show that sIgM−/− mice exhibited more late C. neoformans dissemination to the bloodstream and brain than control mice. C. neoformans dissemination in mice has been shown to occur by Trojan horse-dependent and Trojan horse-independent mechanisms (66, 74). In a model of the former, monocytes with more than two intracellular C. neoformans were less likely to enter the brain (66). Consistent with this result, our data show that control mice had more C. neoformans-phagocytosing macrophages with three or more C. neoformans than sIgM−/− mice and were more resistant to dissemination and death. It is well documented that CD4+ T cells prevent C. neoformans dissemination from the lungs (75) and that C. neoformans-laden macrophages are more likely to disseminate in T cell-deficient mice (76). Hence, it is interesting to note that sIgM−/− mice had fewer CD4+ T cells in their lungs on day 7 postinfection. Given that control mice exhibited more Th1 polarization (IL-12, CD4+ T cell recruitment), more C. neoformans phagocytosing cells with more intracellular yeast, and more C. neoformans containment in the lungs histopathologically; our data suggest that the IgM sufficient state enhances the early cellular response to C. neoformans in the lungs, thereby reducing the likelihood of dissemination. We note that this scenario recapitulates the course of HIV-associated CD, which occurs almost exclusively in the setting of CD4+ T cell deficiency, commonly presents as disseminated disease (77) and is associated with reduced levels of the human source of naturally occurring IgM (IgM memory B cells) (11).
Another factor that could have contributed to the pathogenesis of CD in our study is C5a. B-1 B cell-derived IgM triggered C5a via immune complex formation activation of the classical complement pathway (40). C3-independent and alternative complement pathway generation of C5a have also been described (78, 79). We found that both strains of mice had higher levels of C5a in the naive state than in postinfection states. We cannot explain this finding. Given that each data point in our study was obtained from a different group of mice, we focused on between-strain, rather than across-time comparisons. IgM−/− mice had higher C5a levels than control mice 24 h postinfection, although it was statistically significant, this difference was modest in amount. C5a-deficient mice are more susceptible to C. neoformans (80, 81), but C5a also mediates effects that could increase the inflammatory response to C. neoformans. For example, C5a negatively regulated Th1 responses by inhibiting IL-12 (38), enhanced the Th17 response (39), and decreased the integrity of the blood brain barrier (BBB) (82). IL-17 can also perturb tight junctions and disrupt the BBB (83). Hence, compared with control mice, higher levels of C5a in sIgM−/− mice could have inhibited phagocytosis, compounding the effect of IgM deficiency, and enhanced inflammatory damage in the brain, favoring C. neoformans dissemination and BBB disruption. Consistent with the latter, sIgM−/− mice had histopathological evidence of meningitis, more brain CFUs and higher levels of brain IL-17 and RANTES than control mice. RANTES was increased in C. neoformans-infected brains in another model (84). C5a has also been implicated in airway hyper-responsiveness (85) and induction of IL-13 in rats with CD (33), perhaps explaining the higher levels of IL-13 on day 28 postinfection in sIgM−/− mice.
In summary, this article reports a previously unrecognized role for naturally occurring (naive) IgM in resistance to CD in mice. IgM-deficient mice were more susceptible to death after i.n. challenge with C. neoformans than control mice. Mechanistically, transfer of purified, naive IgM to sIgM−/− mice increased their level of in vivo phagocytosis of C. neoformans, providing proof of the principle that IgM can enhance the cellular response to C. neoformans. More work is needed to identify the C. neoformans specificity of naturally occurring (naive) IgM and to establish a causal link between IgM-mediated enhancement of phagocytosis and acquired immunity to C. neoformans. Nonetheless, the model presented in this study recapitulates important aspects of human CD, including that immunologically normal individuals are largely resistant to disease. Despite having more B-1a B cells, sIgM−/− mice do not produce IgM. This enabled us to evaluate the effect of IgM on the pathogenesis of CD independently of a decrease in B cells, such as that reported in HIV-associated CD (11). The data presented in this study show that the absence of IgM is the most likely cause of the susceptibility phenotype of sIgM−/− mice, providing a plausible explanation for the relationship between lower levels of GXM-reactive IgM and solid organ transplant-associated CD (13), and the risk for CD in HIV-infected individuals (12, 23). However, there are important caveats to our model, including that the survival and immunological phenotypes of both mouse strains could differ with a higher or lower inoculum or a different C. neoformans strain, which underscore the importance of continued basic scientific studies in mice and translational studies in humans. Along these lines, we believe that further studies to establish the feasibility of using IgM memory B cell level/amount and secreted IgM repertoires as predictive biomarkers for CD and/or as possible targets for immunotherapeutic agents for HIV-associated CD are warranted.
Acknowledgments
This work was supported by National Institutes of Health Grants R01 AI 35370, 45459, and 44347 (to L.P.) and Molecular Pathogenesis of Infectious Diseases Training Grant T32 AI 007506-10 (to K.S.).
Abbreviations used in this paper
- AECOM
Albert Einstein College of Medicine
- BBB
blood brain barrier
- BDMP
B cell-derived mononuclear phagocytes
- C3
complement component 3
- CD
cryptococcal disease
- GXM
glucuronoxylomannan
- i.n.
intranasal
- PC
phophorylcholine
- PI
phagocytic index
- PPS
pneumococcal capsular polysaccharide
- RT
room temperature
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Goldman DL, Khine H, Abadi J, Lindenberg DL, Pirofski L, Niang R, Casadevall A. Serologic Evidence for Cryptococcus neoformans Infection in Early Childhood. Pediatrics. 2001;107:e66. doi: 10.1542/peds.107.5.e66. [DOI] [PubMed] [Google Scholar]
- 2.Abadi J, Pirofski L. Antibodies reactive with the cryptococcal capsular polysaccharide glucuronoxylomannan are present in sera from children with and without HIV infection. J. Infect. Dis. 1999;180:915–919. doi: 10.1086/314953. [DOI] [PubMed] [Google Scholar]
- 3.Datta K, Pirofski L. Towards a vaccine for Cryptococcus neoformans: principles and caveats. FEMS. Yeast Res. 2006;6:525–536. doi: 10.1111/j.1567-1364.2006.00073.x. [DOI] [PubMed] [Google Scholar]
- 4.Casadevall A, Perfect JR. Cryptococcus neoformans. 1st Ed. American Society of Microbiology; Washington, D.C.: 1998. Human cryptococcosis; pp. 407–457. [Google Scholar]
- 5.Garcia-Hermoso D, Janbon G, Dromer F. Epidemiological evidence for dormant Cryptococcus neoformans infection. J. Clin. Microbiol. 1999;37:3204–3209. doi: 10.1128/jcm.37.10.3204-3209.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huffnagle GB, Lipscomb MF, Lovchik JA, Hoag KA, Street NE. The role of CD4+ and CD8+ T cells in the protective inflammatory response to a pulmonary cryptococcal infection. J. Leukoc. Biol. 1994;55:35–42. doi: 10.1002/jlb.55.1.35. [DOI] [PubMed] [Google Scholar]
- 7.Rivera J, Zaragoza O, Casadevall A. Antibody-mediated protection against Cryptococcus neoformans is dependent on B cells. Infect. Immun. 2005;73:1141–1150. doi: 10.1128/IAI.73.2.1141-1150.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ghosn EE, Russo M, Almeida SR. Nitric oxide-dependent killing of Cryptococcus neoformans by B-1-derived mononuclear phagocyte. J. Leukoc. Biol. 2006;80:36–44. doi: 10.1189/jlb.1005603. [DOI] [PubMed] [Google Scholar]
- 9.Almeida SR, Aroeira LS, Frymuller E, Dias MA, Bogsan CS, Lopes JD, Mariano M. Mouse B-1 cell-derived mononuclear phagocyte, a novel cellular component of acute non-specific inflammatory exudate. Int. Immunol. 2001;13:1193–1201. doi: 10.1093/intimm/13.9.1193. [DOI] [PubMed] [Google Scholar]
- 10.Casadevall A, Pirofski LA. Antibody-mediated regulation of cellular immunity and the inflammatory response. Trends Immunol. 2003;24:474–478. doi: 10.1016/s1471-4906(03)00228-x. [DOI] [PubMed] [Google Scholar]
- 11.Subramaniam K, Metzger B, Hanau LH, Guh A, Rucker L, Badri S, Pirofski LA. IgM(+) memory B cell expression predicts HIV-associated cryptococcosis status. J. Infect. Dis. 2009;200:244–251. doi: 10.1086/599318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Subramaniam K, French N, Pirofski LA. Cryptococcus neoformans-reactive and total immunoglobulin profiles of human immunodeficiency virus-infected and uninfected Ugandans. Clin. Diagn. Lab. Immunol. 2005;12:1168–1176. doi: 10.1128/CDLI.12.10.1168-1176.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jalali Z, Ng L, Singh N, Pirofski LA. Antibody response to Cryptococcus neoformans capsular polysaccharide glucuronoxylomannan in patients after solid-organ transplantation. Clin. Vaccine Immunol. 2006;13:740–746. doi: 10.1128/CVI.00139-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carsetti R, Rosado MM, Wardmann H. Peripheral development of B cells in mouse and man. Immunol. Rev. 2004;197:179–191. doi: 10.1111/j.0105-2896.2004.0109.x. [DOI] [PubMed] [Google Scholar]
- 15.Baumgarth N, Tung JW, Herzenberg LA. Inherent specificities in natural antibodies: a key to immune defense against pathogen invasion. Springer Semin. Immunopathol. 2005;26:347–362. doi: 10.1007/s00281-004-0182-2. [DOI] [PubMed] [Google Scholar]
- 16.Hardy RR. B-1 B cell development. J. Immunol. 2006;177:2749–2754. doi: 10.4049/jimmunol.177.5.2749. [DOI] [PubMed] [Google Scholar]
- 17.Feldmesser M, Mednick A, Casadevall A. Antibody-mediated protection in murine Cryptococcus neoformans infection is associated with pleotrophic effects on cytokine and leukocyte responses. Infect. Immun. 2002;70:1571–1580. doi: 10.1128/IAI.70.3.1571-1580.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mukherjee J, Scharff MD, Casadevall A. Protective murine monoclonal antibodies to Cryptococcus neoformans. Infect. Immun. 1992;60:4534–4541. doi: 10.1128/iai.60.11.4534-4541.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakouzi A, Valadon P, Nosanchuk JD, Green N, Casadevall A. Molecular basis for immunoglobulin M specificity to epitopes in Cryptococcus neoformans polysaccharide that elicit protective and nonprotective antibodies. Infect. Immun. 2001;69:3398–3409. doi: 10.1128/IAI.69.5.3398-3409.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fleuridor R, Zhong Z, Pirofski L. A human IgM monoclonal antibody prolongs survival of mice with lethal cryptococcosis. J. Infect. Dis. 1998;178:1213–1216. doi: 10.1086/515688. [DOI] [PubMed] [Google Scholar]
- 21.Maitta R, Datta K, Chang Q, Luo R, Subramaniam K, Witover B, Pirofski L. Protective and non-protective human IgM monoclonal antibodies to Cryptococcus neoformans glucuronoxylomannan manifest different specificity and gene use. Infect. Immun. 2004;72:4810–4818. doi: 10.1128/IAI.72.8.4810-4818.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Subramaniam KS, Datta K, Marks MS, Pirofski LA. Improved survival of mice deficient in secretory IgM following systemic infection with Cryptococcus neoformans. Infect. Immun. 2009;78:441–452. doi: 10.1128/IAI.00506-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Houpt DC, Pfrommer GS, Young BJ, Larson TA, Kozel TR. Occurrences, immunoglobulin classes, and biological activities of antibodies in normal human serum that are reactive with Cryptococcus neoformans glucuronoxylomannan. Infect. Immun. 1994;62:2857–2864. doi: 10.1128/iai.62.7.2857-2864.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fleuridor R, Lyles RH, Pirofski L. Quantitative and qualitative differences in the serum antibody profiles of HIV-infected persons with and without Cryptococcus neoformans meningitis. J. Infect. Dis. 1999;180:1526–1536. doi: 10.1086/315102. [DOI] [PubMed] [Google Scholar]
- 25.Deshaw M, Pirofski LA. Antibodies to the Cryptococcus neoformans capsular glucuronoxylomannan are ubiquitous in serum from HIV+ and HIV− individuals. Clin. Exp. Immunol. 1995;99:425–432. doi: 10.1111/j.1365-2249.1995.tb05568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boes M, Esau C, Fischer MB, Schmidt T, Carroll M, Chen J. Enhanced B-1 cell development, but impaired IgG antibody responses in mice deficient in secreted IgM. J. Immunol. 1998;160:4776–4787. [PubMed] [Google Scholar]
- 27.Rivera J, Mukherjee J, Weiss LM, Casadevall A. Antibody efficacy in murine pulmonary Cryptococcus neoformans infection: a role for nitric oxide. J. Immunol. 2002;168:3419–3427. doi: 10.4049/jimmunol.168.7.3419. [DOI] [PubMed] [Google Scholar]
- 28.Datta K, Lees A, Pirofski LA. Therapeutic efficacy of a conjugate vaccine containing a peptide mimotope of cryptococcal capsular polysaccharide glucuronoxylomannan. Clin. Vaccine Immunol. 2008;15:1176–1187. doi: 10.1128/CVI.00130-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marks M, Burns T, Abadi M, Seyoum B, Thornton J, Tuomanen E, Pirofski LA. Influence of neutropenia on the course of serotype 8 pneumococcal pneumonia in mice. Infect. Immun. 2007;75:1586–1597. doi: 10.1128/IAI.01579-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zaragoza O, Alvarez M, Telzak A, Rivera J, Casadevall A. The relative susceptibility of mouse strains to pulmonary Cryptococcus neoformans infection is associated with pleiotropic differences in the immune response. Infect. Immun. 2007;75:2729–2739. doi: 10.1128/IAI.00094-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jayasekera JP, Moseman EA, Carroll MC. Natural antibody and complement mediate neutralization of influenza virus in the absence of prior immunity. J. Virol. 2007;81:3487–3494. doi: 10.1128/JVI.02128-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hill JO, Dunn PL. A T cell-independent protective host response against Cryptococcus neoformans expressed at the primary site of infection in the lung. Infect. Immun. 1993;61:5302–5308. doi: 10.1128/iai.61.12.5302-5308.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goldman DL, Davis J, Bommarito F, Shao X, Casadevall A. Enhanced allergic inflammation and airway responsiveness in rats with chronic Cryptococcus neoformans infection: potential role for fungal pulmonary infection in the pathogenesis of asthma. J. Infect. Dis. 2006;193:1178–1186. doi: 10.1086/501363. [DOI] [PubMed] [Google Scholar]
- 34.Müller U, Stenzel W, Köhler G, Werner C, Polte T, Hansen G, Schütze N, Straubinger RK, Blessing M, McKenzie AN, et al. IL-13 induces disease-promoting type 2 cytokines, alternatively activated macrophages and allergic inflammation during pulmonary infection of mice with Cryptococcus neoformans. J. Immunol. 2007;179:5367–5377. doi: 10.4049/jimmunol.179.8.5367. [DOI] [PubMed] [Google Scholar]
- 35.Herring AC, Lee J, McDonald RA, Toews GB, Huffnagle GB. Induction of interleukin-12 and gamma interferon requires tumor necrosis factor alpha for protective T1-cell-mediated immunity to pulmonary Cryptococcus neoformans infection. Infect. Immun. 2002;70:2959–2964. doi: 10.1128/IAI.70.6.2959-2964.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoag KA, Street NE, Huffnagle GB, Lipscomb MF. Early cytokine production in pulmonary Cryptococcus neoformans infections distinguishes susceptible and resistant mice. Am. J. Respir. Cell Mol. Biol. 1995;13:487–495. doi: 10.1165/ajrcmb.13.4.7546779. [DOI] [PubMed] [Google Scholar]
- 37.Guillot L, Carroll SF, Homer R, Qureshi ST. Enhanced innate immune responsiveness to pulmonary Cryptococcus neoformans infection is associated with resistance to progressive infection. Infect. Immun. 2008;76:4745–4756. doi: 10.1128/IAI.00341-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hawlisch H, Belkaid Y, Baelder R, Hildeman D, Gerard C, Köhl J. C5a negatively regulates toll-like receptor 4-induced immune responses. Immunity. 2005;22:415–426. doi: 10.1016/j.immuni.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 39.Fang C, Zhang X, Miwa T, Song WC. Complement promotes the development of inflammatory T-helper 17 cells through synergistic interaction with Toll-like receptor signaling and interleukin-6 production. Blood. 2009;114:1005–1015. doi: 10.1182/blood-2009-01-198283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsuji RF, Kawikova I, Ramabhadran R, Akahira-Azuma M, Taub D, Hugli TE, Gerard C, Askenase PW. Early local generation of C5a initiates the elicitation of contact sensitivity by leading to early T cell recruitment. J. Immunol. 2000;165:1588–1598. doi: 10.4049/jimmunol.165.3.1588. [DOI] [PubMed] [Google Scholar]
- 41.Guo RF, Ward PA. Role of C5a in inflammatory responses. Annu. Rev. Immunol. 2005;23:821–852. doi: 10.1146/annurev.immunol.23.021704.115835. [DOI] [PubMed] [Google Scholar]
- 42.Alugupalli KR, Gerstein RM, Chen J, Szomolanyi-Tsuda E, Woodland RT, Leong JM. The resolution of relapsing fever borreliosis requires IgM and is concurrent with expansion of B1b lymphocytes. J. Immunol. 2003;170:3819–3827. doi: 10.4049/jimmunol.170.7.3819. [DOI] [PubMed] [Google Scholar]
- 43.Boes M, Prodeus AP, Schmidt T, Carroll MC, Chen J. A critical role of natural immunoglobulin M in immediate defense against systemic bacterial infection. J. Exp. Med. 1998;188:2381–2386. doi: 10.1084/jem.188.12.2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brown JS, Hussell T, Gilliland SM, Holden DW, Paton JC, Ehrenstein MR, Walport MJ, Botto M. The classical pathway is the dominant complement pathway required for innate immunity to Streptococcus pneumoniae. Proc. Natl. Acad. Sci. USA. 2002;99:16969–16974. doi: 10.1073/pnas.012669199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Diamond MS, Shrestha B, Marri A, Mahan D, Engle M. B cells and antibody play critical roles in the immediate defense of disseminated infection by West Nile encephalitis virus. J. Virol. 2003;77:2578–2586. doi: 10.1128/JVI.77.4.2578-2586.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Choi YS, Baumgarth N. Dual role for B-1a cells in immunity to influenza virus infection. J. Exp. Med. 2008;205:3053–3064. doi: 10.1084/jem.20080979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rajan B, Ramalingam T, Rajan TV. Critical role for IgM in host protection in experimental filarial infection. J. Immunol. 2005;175:1827–1833. doi: 10.4049/jimmunol.175.3.1827. [DOI] [PubMed] [Google Scholar]
- 48.Kleinschek MA, Muller U, Brodie SJ, Stenzel W, Kohler G, Blumenschein WM, Straubinger RK, McClanahan T, Kastelein RA, Alber G. IL-23 enhances the inflammatory cell response in Cryptococcus neoformans infection and induces a cytokine pattern distinct from IL-12. J. Immunol. 2006;176:1098–1106. doi: 10.4049/jimmunol.176.2.1098. [DOI] [PubMed] [Google Scholar]
- 49.Haas KM, Poe JC, Steeber DA, Tedder TF. B-1a and B-1b cells exhibit distinct developmental requirements and have unique functional roles in innate and adaptive immunity to S. pneumoniae. Immunity. 2005;23:7–18. doi: 10.1016/j.immuni.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 50.Baumgarth N, Herman OC, Jager GC, Brown LE, Herzenberg LA, Chen J. B-1 and B-2 cell-derived immunoglobulin M antibodies are nonredundant components of the protective response to influenza virus infection. J. Exp. Med. 2000;192:271–280. doi: 10.1084/jem.192.2.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sundstrom JB, Cherniak R. The glucuronoxylomannan of Cryptococcus neoformans serotype A is a type 2 T-cell-independent antigen. Infect. Immun. 1992;60:4080–4087. doi: 10.1128/iai.60.10.4080-4087.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rodrigues ML, Nimrichter L, Oliveira DL, Frases S, Miranda K, Zaragoza O, Alvarez M, Nakouzi A, Feldmesser M, Casadevall A. Vesicular polysaccharide export in Cryptococcus neoformans is a eukaryotic solution to the problem of fungal trans-cell wall transport. Eukaryot. Cell. 2007;6:48–59. doi: 10.1128/EC.00318-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rachini A, Pietrella D, Lupo P, Torosantucci A, Chiani P, Bromuro C, Proietti C, Bistoni F, Cassone A, Vecchiarelli A. An anti-betaglucan monoclonal antibody inhibits growth and capsule formation of Cryptococcus neoformans in vitro and exerts therapeutic, anticryptococcal activity in vivo. Infect. Immun. 2007;75:5085–5094. doi: 10.1128/IAI.00278-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang H, Clarke SH. Positive selection focuses the VH12 B-cell repertoire towards a single B1 specificity with survival function. Immunol. Rev. 2004;197:51–59. doi: 10.1111/j.0105-2896.2004.0098.x. [DOI] [PubMed] [Google Scholar]
- 55.Baumgarth N, Herman OC, Jager GC, Brown L, Herzenberg LA, Herzenberg LA. Innate and acquired humoral immunities to influenza virus are mediated by distinct arms of the immune system. Proc. Natl. Acad. Sci. USA. 1999;96:2250–2255. doi: 10.1073/pnas.96.5.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tsuji RF, Szczepanik M, Kawikova I, Paliwal V, Campos RA, Itakura A, Akahira-Azuma M, Baumgarth N, Herzenberg LA, Askenase PW. B cell-dependent T cell responses: IgM antibodies are required to elicit contact sensitivity. J. Exp. Med. 2002;196:1277–1290. doi: 10.1084/jem.20020649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shapiro S, Beenhouwer DO, Feldmesser M, Taborda C, Carroll MC, Casadevall A, Scharff MD. Immunoglobulin G monoclonal antibodies to Cryptococcus neoformans protect mice deficient in complement component C3. Infect. Immun. 2002;70:2598–2604. doi: 10.1128/IAI.70.5.2598-2604.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shi Y, Yamazaki T, Okubo Y, Uehara Y, Sugane K, Agematsu K. Regulation of aged humoral immune defense against pneumococcal bacteria by IgM memory B cell. J. Immunol. 2005;175:3262–3267. doi: 10.4049/jimmunol.175.5.3262. [DOI] [PubMed] [Google Scholar]
- 59.Hart M, Steel A, Clark SA, Moyle G, Nelson M, Henderson DC, Wilson R, Gotch F, Gazzard B, Kelleher P. Loss of discrete memory B cell subsets is associated with impaired immunization responses in HIV-1 infection and may be a risk factor for invasive pneumococcal disease. J. Immunol. 2007;178:8212–8220. doi: 10.4049/jimmunol.178.12.8212. [DOI] [PubMed] [Google Scholar]
- 60.Clutterbuck EA, Oh S, Hamaluba M, Westcar S, Beverley PC, Pollard AJ. Serotype-specific and age-dependent generation of pneumococcal polysaccharide-specific memory B-cell and antibody responses to immunization with a pneumococcal conjugate vaccine. Clin. Vaccine Immunol. 2008;15:182–193. doi: 10.1128/CVI.00336-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kruetzmann S, Rosado MM, Weber H, Germing U, Tournilhac O, Peter HH, Berner R, Peters A, Boehm T, Plebani A, et al. Human immunoglobulin M memory B cells controlling Streptococcus pneumoniae infections are generated in the spleen. J. Exp. Med. 2003;197:939–945. doi: 10.1084/jem.20022020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Popi AF, Zamboni DS, Mortara RA, Mariano M. Microbicidal property of B1 cell derived mononuclear phagocyte. Immunobiology. 2009;214:664–673. doi: 10.1016/j.imbio.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 63.Popi AF, Lopes JD, Mariano M. Interleukin-10 secreted by B-1 cells modulates the phagocytic activity of murine macrophages in vitro. Immunology. 2004;113:348–354. doi: 10.1111/j.1365-2567.2004.01969.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.O’Garra A, Howard M. IL-10 production by CD5 B cells. Ann. N. Y. Acad. Sci. 1992;651:182–199. doi: 10.1111/j.1749-6632.1992.tb24615.x. [DOI] [PubMed] [Google Scholar]
- 65.O’Garra A, Chang R, Go N, Hastings R, Haughton G, Howard M. Ly-1 B (B-1) cells are the main source of B cell-derived interleukin 10. Eur. J. Immunol. 1992;22:711–717. doi: 10.1002/eji.1830220314. [DOI] [PubMed] [Google Scholar]
- 66.Charlier C, Nielsen K, Daou S, Brigitte M, Chretien F, Dromer F. Evidence of a role for monocytes in dissemination and brain invasion by Cryptococcus neoformans. Infect. Immun. 2009;77:120–127. doi: 10.1128/IAI.01065-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shao X, Mednick A, Alvarez M, van Rooijen N, Casadevall A, Goldman DL. An innate immune system cell is a major determinant of species-related susceptibility differences to fungal pneumonia. J. Immunol. 2005;175:3244–3251. doi: 10.4049/jimmunol.175.5.3244. [DOI] [PubMed] [Google Scholar]
- 68.Kechichian TB, Shea J, Del Poeta M. Depletion of alveolar macrophages decreases the dissemination of a glucosylceramide-deficient mutant of Cryptococcus neoformans in immunodeficient mice. Infect. Immun. 2007;75:4792–4798. doi: 10.1128/IAI.00587-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Guerrero A, Jain N, Wang X, Fries BC. Cryptococcus neoformans variants generated by phenotypic switching differ in virulence through effects on macrophage activation. Infect. Immun. 2010;78:1049–1057. doi: 10.1128/IAI.01049-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shea JM, Kechichian TB, Luberto C, Del Poeta M. The cryptococcal enzyme inositol phosphosphingolipid-phospholipase C confers resistance to the antifungal effects of macrophages and promotes fungal dissemination to the central nervous system. Infect. Immun. 2006;74:5977–5988. doi: 10.1128/IAI.00768-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hardison SE, Ravi S, Wozniak KL, Young ML, Olszewski MA, Wormley FL., Jr. Pulmonary infection with an interferon-{gamma}-producing Cryptococcus neoformans strain results in classical macrophage activation and protection. Am. J. Pathol. 2010;176:774–785. doi: 10.2353/ajpath.2010.090634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Voelz K, Lammas DA, May RC. Cytokine signaling regulates the outcome of intracellular macrophage parasitism by Cryptococcus neoformans. Infect. Immun. 2009;77:3450–3457. doi: 10.1128/IAI.00297-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang Y, Wang F, Tompkins KC, McNamara A, Jain AV, Moore BB, Toews GB, Huffnagle GB, Olszewski MA. Robust Th1 and Th17 immunity supports pulmonary clearance but cannot prevent systemic dissemination of highly virulent Cryptococcus neoformans H99. Am. J. Pathol. 2009;175:2489–2500. doi: 10.2353/ajpath.2009.090530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chang YC, Stins MF, McCaffery MJ, Miller GF, Pare DR, Dam T, Paul-Satyaseela M, Kim KS, Kwon-Chung KJ, Paul-Satyasee M. Cryptococcal yeast cells invade the central nervous system via transcellular penetration of the blood-brain barrier. Infect. Immun. 2004;72:4985–4995. doi: 10.1128/IAI.72.9.4985-4995.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hill JO. CD4+ T cells cause multinucleated giant cells to form around Cryptococcus neoformans and confine the yeast within the primary site of infection in the respiratory tract. J. Exp. Med. 1992;175:1685–1695. doi: 10.1084/jem.175.6.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Luberto C, Martinez-Mariño B, Taraskiewicz D, Bolaños B, Chitano P, Toffaletti DL, Cox GM, Perfect JR, Hannun YA, Balish E, Del Poeta M. Identification of App1 as a regulator of phagocytosis and virulence of Cryptococcus neoformans. J. Clin. Invest. 2003;112:1080–1094. doi: 10.1172/JCI18309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dromer F, Bernede-Bauduin C, Guillemot D, Lortholary O, French Cryptococcosis Study Group Major role for amphotericin B-flucytosine combination in severe cryptococcosis. PLoS One. 2008;3:e2870. doi: 10.1371/journal.pone.0002870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR, Lambris JD, Warner RL, Flierl MA, Hoesel LM, et al. Generation of C5a in the absence of C3: a new complement activation pathway. Nat. Med. 2006;12:682–687. doi: 10.1038/nm1419. [DOI] [PubMed] [Google Scholar]
- 79.Banda NK, Levitt B, Wood AK, Takahashi K, Stahl GL, Holers VM, Arend WP. Complement activation pathways in murine immune complex-induced arthritis and in C3a and C5a generation in vitro. Clin. Exp. Immunol. 2010;159:100–108. doi: 10.1111/j.1365-2249.2009.04035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dromer F, Perronne C, Barge J, Vilde JL, Yeni P. Role of IgG and complement component C5 in the initial course of experimental cryptococcosis. Clin. Exp. Immunol. 1989;78:412–417. [PMC free article] [PubMed] [Google Scholar]
- 81.Rhodes JC. Contribution of complement component C5 to the pathogenesis of experimental murine cryptococcosis. Sabouraudia. 1985;23:225–234. doi: 10.1080/00362178585380331. [DOI] [PubMed] [Google Scholar]
- 82.Jacob A, Hack B, Chiang E, Garcia JG, Quigg RJ, Alexander JJ. C5a alters blood-brain barrier integrity in experimental lupus. FASEB J. 2010 doi: 10.1096/fj.09-138834. doi: 10.1096/fj.09-138834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kebir H, Kreymborg K, Ifergan I, Dodelet-Devillers A, Cayrol R, Bernard M, Giuliani F, Arbour N, Becher B, Prat A. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat. Med. 2007;13:1173–1175. doi: 10.1038/nm1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Uicker WC, Doyle HA, McCracken JP, Langlois M, Buchanan KL. Cytokine and chemokine expression in the central nervous system associated with protective cell-mediated immunity against Cryptococcus neoformans. Med. Mycol. 2005;43:27–38. doi: 10.1080/13693780410001731510. [DOI] [PubMed] [Google Scholar]
- 85.Köhl J, Baelder R, Lewkowich IP, Pandey MK, Hawlisch H, Wang L, Best J, Herman NS, Sproles AA, Zwirner J, et al. A regulatory role for the C5a anaphylatoxin in type 2 immunity in asthma. J. Clin. Invest. 2006;116:783–796. doi: 10.1172/JCI26582. [DOI] [PMC free article] [PubMed] [Google Scholar]