

Abstract
MicroRNAs (miRNAs) are small noncoding RNAs that act as post-transcriptional repressors of gene expression in organisms ranging from plants to humans. A widespread role for miRNAs in diverse molecular processes driving initiation and progression of various tumor types has recently been described. In this review, we discuss the etiology of aberrant expression of miRNAs in human cancers and their role in tumor metastasis, which may define miRNAs as oncogenes or tumor suppressors. Moreover, we highlight the genomic/epigenetic alterations and transcriptional/post-transcriptional mechanisms associated with misexpression of miRNAs in cancer. A better understanding of miRNA biology may ultimately yield further insight into the molecular mechanisms of tumorigenesis and new therapeutic strategies against cancer.
microRNAs
Over the past decade, in different species and in a range of tissues several classes of small regulatory RNAs have been identified including miRNAs, small (18-25 nucleotides (nt) in length), noncoding, single-stranded RNAs. MiRNAs negatively regulate gene expression, either by translational inhibition or exonucleolytic mRNA decay, targeted through imperfect complementarity between the miRNA and the 3′ untranslated region (3′UTR) of the mRNA [1]. Depending on the targeted mRNAs, both mechanisms ultimately result in reduced protein levels and profound consequences on cellular homeostasis. Recent bioinformatics and experimental reports suggest that over 30% of human genes are direct targets of miRNAs [2], further indicating a role for miRNAs in almost every biological process, including cell cycle regulation, cell growth, apoptosis, cell differentiation and stress response. Recent genome-wide analyses have identified dysregulated miRNA expression in human malignancies (Table 1) [3], and a potential dual role in tumor formation: miRNAs can modulate oncogenic or tumor-suppressor pathways, including p53, c-MYC, RAS and BCR-ABL, while expression of miRNAs themselves can be regulated by oncogenes or tumor suppressors.
Table 1. miRNA signatures in human cancers.
Representative examples of miRNA expression signatures in the most common human cancers.
| Tumor | Results | References | |
|---|---|---|---|
| Chronic lymphocytic leukemia | CLL cells show a unique signaturethat associate with the presence or absence of disease progression and cytogenetic abnormalities. | 18, 53, 92 | |
| Acute myeloid leukemia | CD34(+) cells and AML cases differentially express miRNAs that associate with several cytogenetic groups (11q23 translocations and isolated trisomy 8). | 19 | |
| Acute promyelocytic leukemia (APL) | Identified 12 miRNAs in APL blasts compared to normal promyelocytes; miR-342 and let-7c are modulated by retinoic acid. | 100 | |
| Multiple Myeloma (MM) | First identification of miRNA signature in myeloma cell lines and patients. | 23 | |
| Acute lymphoblastic leukemia (ALL) | Identification of miRNA signature in ALL patients. | 101 | |
| B-cell lymphomas (DLBCL) and follicolar lymphomas (FL) | DLBCL- and FL-specific miRNA signatures. 98% of all 111 cases were correctly identified based on the expression of 4 miRNA (miR-210/106a/17-5p/330). | 103 | |
| Barrett's esophagus and esophageal adenocarcinoma | Identification of several miRNAs differentially expressed in the progression from low grade-dysplasia Barrett's esophagus to adenocarcinoma. | 105 | |
| Breast cancer | Reported miRNomes of normal and breast cancer samples. Dysregulated miRNAs associate with invasive breast cancer pathologic features, such as ER status. | 22 | |
| Glioblastoma | Reported miRNA profiles from case-matching pairs of tumor and control samples. 9 miRNAs are overexpr- essed and 4 are underexpressed in tumors compared to normal. | 21 | |
| Hepatocellular carcinoma (HCC) | Reported miRNA profiles in HCC compared to adjacent normal tissue and additional chronic hepatitis specimens. Identified a new diagnostic tool for HCC. | 17 | |
| Ovarian cancer | Identification of miRNAs whose expression correlates with specific ovarian cancer pathologies, such as histotype, lymphovascular and organ invasion and involvement of ovarian surface. | 93 | |
| Lung adenocarcinoma | Reported molecular signatures that differ across tumor histology; miR-155 and let-7 correlate with survival. | 94 | |
| Papillary thyroid carcinoma | Reported upregulation of miRNA-221/222 in tumor cells and adjacent normal cells as compared to normal thyroid | 95 | |
| Endocrine pancreatic cancer | Reported miRNA signatures from endocrine and acinar tumors; miR-21 associates with proliferation index and liver metastasis | 96 | |
| Colon cancer | Reported differential expression levels for 39 miRNAs between colon tumors normal tissue. Colon tumors differentially express miRNAs according to their mismatch repair status. | 20 | |
| Gastric cancer | Identified 22 miRNAs upregulated and 13 downregulated in gastric cancer by comparing non-tumour mucosa to cancerous tissue,. | 97 | |
| Cervical cancer | Identification of miRNA signature in 102 cervical cancer by PCR-based miRNA assay. miR-200a and miR-9 predict patient survival. | 98 | |
| Clear-cell Kidney cancer (ccRCC) | Identified 26 miRNA downregulated and 9 upregulated in 28 ccRCC patient-matched specimens. Downregulated microRNAs are correlated with common chromosome deletion in ccRCC. | 99 | |
| Sarcoma | microRNA expression is able to classify different histological types of sarcoma reflecting differentiation status and apparent lineage of the tumors. | 102 | |
| Bladder cancer | Identification of miRNA differentially expressed between normal urothelium/cancer and different disease stages. miR-129 predicts disease progression. | 104 | |
We will briefly describe miRNA biogenesis, discuss recent controversies that have emerged over the role of miRNAs in tumor formation and progression, and examine molecular mechanisms involved in miRNA dysregulation in cancer.
MiRNA biogenesis: from nucleus to cytoplasm
Since their discovery (Box 1), hundreds of miRNAs have been identified and, at present, the human miRNA database contains 721 miRNAs or approximately 2-3% of the total number of genes in the human genome (Box 2). MiRNAs are produced through a multistep process, including two distinct biogenetic pathways (Figure 1) [extensively reviewed in 4]. During the miRNA maturation processes, transcriptional and post-transcriptional levels are strictly regulated, ensuring precise production. Disruptions in the maturation process can contribute to altered miRNA levels, a hallmark of many human diseases including cancer, and we describe the most recent advances in the field of miRNAs and cancer [3].
Figure 1.

miRNA biogenesis. Canonical miRNAs are transcribed by RNA polymerase II to generate the primary transcript (pri-miRNAs), a long capped and polyadenylated RNA with a hairpin-shaped structure (left). Cropping is the first step in the maturation mediated by the Microprocessor complex, essentially composed of the RNase III enzyme Drosha and the molecular anchor Di George syndrome critical region 8 (DGCR8), and produces a ∼65 nt hairpin RNA called precursor-miRNA (pre-miRNA). Pre-miRNA has a short stem with a 2-3 nt overhang, which is recognized by the Exportin-5-Ran-GTP complex. After export from the nucleus, the pre-miRNA is subjected to the dicing step operated by Dicer with its partner TRBP and Arogonaute proteins 1-4 (AGO). This processing step creates the final duplex and allows the formation of the RNA-Induced Silencing Complex (RISC), which mediates miRNA activity. This multistep process used for independent miRNA transcription units or for intronic miRNAs. In flies and mammals, some miRNAs called mirtrons (right and Box 2) are located in short introns and bypass the Microprocessor complex-dependent step. After the splicing and production of the mature mRNA, the excised intron is debranched and trimmed to produce the pre-miRNA, which follows the canonical pathway for miRNAs biogenesis beginning at the export step.
MiRNA expression in cancer
Although initially identified in B-cell leukemia [5], alterations in the expression of miRNAs are now considered a common characteristic of all human tumors. Compared to normal tissue of the same type, most tumors display a distinct miRNA expression signature (Table 1). In 2006, Lu and collaborators demonstrated that expression profiles of miRNAs could accurately classify human cancers, on the basis of their embryonic lineage and differentiation states [6]: tumors of endodermal origin, such as colon, liver, pancreas and stomach, clustered together and were separate from hematopoietic malignancies clusters [6-7]. Furthermore, 129 of the 217 miRNAs analyzed were globally reduced in tumors compared to normal tissues [6]. Based on these data and on evidence showing that miRNAs can direct tissue-specific developmental functions [8], the global repression of miRNA levels might promote a less differentiated cellular state. Although an exciting possibility, miRNA microarray data alone is not able to establish a causative link between cancer and general miRNA knockdown; decreases in miRNA levels could simply be a byproduct of the neoplastic state. Experimental proof supporting an etiological role for miRNA repression in cancer came in 2007 from Kumar and collaborators [9], who demonstrated that the global knockdown of mature miRNAs by RNA interference (RNAi)-targeting of the processing enzymes Dicer, DGCR8 and Drosha enhanced tumorigenicity of cancer cell lines in vitro and accelerated xenograft tumor formation in vivo, independent of the tissue of origin. Indeed, following subcutaneous injection into nude mice cells with impaired miRNA processing diplayed increased invasion into the surrounding normal tissues, including skeletal muscle, adipose tissue and nerve sheaths [9]. Importantly, this effect on tumorigenicity and motility is only observed in cell lines that are already transformed. Defective miRNA biogenesis appears to only enhance the aggressiveness of malignant cells and is not sufficient to promote de novo transformation.
Two recent reports define one of the underlying genetic causes of impaired miRNA processing in human cancer [10-11]. Hemizygous deletion of the gene that encodes Dicer occurs in 27% of tumors of different tissue origins [10-11], whereas homozygous deletion has yet to be reported. Conditional Dicer mutation in different mouse models of cancer (e.g. K-Ras driven lung cancer [10] and Rb driven retinoblastoma [11]) downregulates miRNAs and accelerates the formation of early neoplastic lesions, which progress to aggressive and metastatic tumors only when Dicer is partially active. Therefore, Dicer is the first example of a gene that functions as a tumor suppressor in the context of haploinsufficiency but not in homozygous loss-of-function. Because Dicer haploinsufficency accounts for only 27% of the human tumors, this condition only partially explains the widespread loss of miRNAs in human tumors.
The above observations raise the question: are all miRNAs tumor suppressors? A miRNA microarray analyses of retinas from mice with Rb-driven retinoblastoma carrying Dicer haploinsufficency (monoallelic deletion of Dicer), identified only 11 downregulated miRNAs compared to mice with Rb-driven retinoblastoma. Among the downregulated miRNAs were two let-7 family members (let-7c and let-7i) [11]. Therefore, it seems likely that only a small subgroup of miRNAs has tumor-suppressor activity (e.g. the let-7 family, whose members are significantly downregulated in human lung cancer and correlate with a poor prognosis) [12]. Moreover, let-7 family members negatively regulate the expression of oncogenes, including RAS, MYC and HMGA2, indicating that the loss of let-7 might contribute to the pathogenesis of several types of human tumors [13-15].
A large-scale microarray analysis of 540 samples, including 363 solid tumors from the 6 most frequent malignancies (breast, prostate, lung, stomach, pancreas and thyroid) and 177 normal tissues demonstrated a more complex role for miRNAs in cancer by identifying a specific miRNA “miRNoma” expression signature. The miRNoma consists of 36-overexpressed and 21-downregulated miRNAs [16]. This study, together with genome-wide profiling analyses of different tumors (Table 1), support the view that miRNA alterations in cancer consist of both downregulated and overexpressed miRNAs with putative tumor-suppressive and oncogenic functions. One of the first oncogenic miRNAs identified was the cluster miR-17-92 (also known as oncomiR-1), which is processed from a polycistronic transcript into 7 miRNAs (mir-17-5p, miR-17-3p, miR-18a, miR19a, miR-20a, miR-19b-1 and miR-92-1). Cluster miR-17-92 is highly expressed in a range of hematopoietic malignancies, particularly B-cell lymphomas [24]. Although transgenic mice with moderate miR-17-92 cluster overexpression only develop lympho-proliferative phenotypes [25], forced high-level miR-17-92 cluster expression in the context of Myc-driven B-cell lymphoma leads to a dramatic increase in tumorigenicity in vivo; in this mouse model, tumors appear in ∼51 days compared to 3-6 months in Myc-driven B-cell lymphoma control mice [24]. In contrast, homozygous deletion of the miR-17-92 locus in mice results in premature death of B cells at the pro-B and pre-B stages, resulting in lymphopenia [26]. Functional dissection of the miR-17-92 cluster in the context of B-cell transformation in vivo reveales that miR-19a and b mediate the oncogenic activity of the entire cluster, at least in part, by targeting the tumor suppressor PTEN and thereby activating Akt-mTOR signaling to promote cell survival [27-28]. Another target of the miR-17-92 cluster in B cells appears to be the pro-apoptotic protein Bim, a critical regulator of B-cell survival and a tumor suppressor in the Eμ-Myc model of B-cell lymphoma [25-26]. The Bim 3′UTR contains multiple binding sites for the miR-17-92 cluster and Bim expression is increased in miR-17-92-null pre-B cells and reduced in B cells that overexpress miR-17-92, strongly indicating that Bim is a direct target of the miR-17-92 cluster. Overall, these data demonstrate that the miR-17-92 cluster controls normal B-cell differentiation and abnormal expression of this cluster represses several tumor-suppressive activities, resulting in potent oncogenic activity and contributing to the onset of B-cell lymphomas.
Another example of an oncogenic miRNA is miR-155, which is overexpressed in different types of B-cell malignancies, including pediatric Burkitt's lymphoma, Hodgkin's lymphoma, aggressive CLL and diffuse large B-cell lymphoma (DLBCL) [29-30]. In transgenic mice that specifically overexpress miR-155 in B cells [31], large pre-B cells undergo polyclonal expansion and after 9 months, the mice develop ALL/high-grade lymphoma. In addition, miR-155-mediated repression of SHIP and C/EBPβ protein expression is responsible for the accumulation of large pre-B cells and the frank leukemia phenotype in these transgenic mice [32]. However, since months elapse before the onset of the B-cell leukemia, miR-155 overexpression is probably an early event in the transformation process; an additional genetic alteration, such as Myc overexpression, might be necessary to induce the full malignant phenotype.
The notion that miRNAs can act as tumor suppressors or oncogenes, depending on the tissue context and target genes, is illustrated in the following examples. MiR-221 and miR-222 inhibit erythropoiesis by inhibiting the expression of the oncogene c-KIT and could be considered tumor suppressors [33]. However, upregulation of miR-221 and miR-222 is part of the miRNA progression signature in liver tumorigenesis, and both miRs stimulate colony formation of liver cancer cells in vitro and enhance tumor growth in a mouse model of liver cancer [34]. In an elegant study by Pineau [34], mouse p53 -/- liver progenitors were immortalized by c-MYC expression, and early immortalized progenitors were in turn transduced with a liver specific miR-221-expressing vector. Lower rates of tumor-free survival were observed for miR-221-transduced hepatic progenitors-injected mice when compared with empty vector control. The suppressive effect was due in part to miR-221 mediated direct down-regulation of DDIT4, a tumor-suppressor and an essential regulator of the mTOR kinase signaling, at both the mRNA and protein levels (35). Moreover, recent reports demonstrate that miR-221/222 directly suppress, in different cellular contexts, at least six other tumor suppressors p57 [36], PTEN [37], TIMP3 [37], p27 [38], BIM [39] and FOXO3 [40] by promoting proliferative and invasive phenotypes characteristic of frankly malignant cells. These studies further suggest another level of disruption exerted by miR-221/222 cluster, the PTEN-PI3K-AKT-mTOR axis, which is relevant to liver, lung and breast tumorigenesis, and show that a single miRNA, through its ability to modulate different genes involved in the same pathway, might act as a strong inhibitor of the entire cellular pathway. Collectively, these studies support the notion that targeting miRNAs could have greater therapeutic potential than single gene–directed drugs.
Metastasis is responsible for more than 90% of cancer-related mortality and arises through a multistep process, starting with cancer cell migration away from the primary tumor and invasion through the basement membrane [41]. The epithelial-to-mesenchymal transition (EMT), characterized by the conversion of polarized immotile epithelial cells to motile mesenchymal cells, is now recognized as an important component of this multistep process [42]. Several transcription factors are important for EMT. The two zinc-finger E-box-binding homeobox (ZEB) factors (ZEB1 and ZEB2), induce EMT by repressing E-cadherin transcription and promoting vimentin transcription [43]. Recent reports [44-46] linking EMT to miRNAs provide compelling evidence that miR-200 family (miR-200a, miR-200b, miR-200c, miR-141 and miR-429) and miR-205 control EMT. Ectopic expression of the miR-200 family or miR-205 downregulates ZEB1 and ZEB2 and upregulates E-cadherin, followed by mesenchymal–epithelial transition (MET) in cells that had previously undergone EMT [44-46]. More intriguingly and by contrast, ZEB1 and ZEB2 can also directly suppress transcription of the miR-200 family by binding to the E-box or Z-box in their putative common promoter, thereby generating a miR-200 family-ZEB1/ZEB2 loop to maintain EMT and MET equilibrium [47-48]. Loss of these miRNAs in tumors, as reported in invasive breast cancer cell lines with mesenchymal phenotypes and in regions of metaplastic breast cancer specimens lacking E-cadherin, activates the EMT, whereas in normal tissue homeostasis these miRNAs act as a control switch between EMT and MET.
The studies described above highlight the magnitude of miRNA genes in the pathogenesis and progression of human cancer and provide insight into new opportunities for cancer treatment by modulating miRNA pathways and activities.
Causes of abnormal miRNA expression in cancer
As aberrant miRNA levels strongly associate with disease, precise control of miRNAs levels is essential for maintaining normal cellular homeostasis. In the following section, we review the major mechanisms involved in miRNA dysregulation in cancer (Figure 2).
Figure 2.

Mechanisms of miRNA dysregulation. MiRNA gene expression is a multistep tightly regulated process and any alteration might contribute to miRNA dysregulation and neoplastic transformation. These mechanisms of miRNA regulation can be differentiated into 1) mechanisms targeting the miRNA genes, including genomic alterations (deletions, amplifications or translocations), epigenetics changes (methylation and histone modification), polymorphisms or mutation, and transcriptional alteration; and 2) mechanisms modulating the activity of the multistep processing enzymes (Drosha, DGCR8, Exportin 5, Dicer and TBRP).
Genetic abnormalities and miRNAs
Genomic abnormalities, such as chromosomal rearrangements, genomic amplifications, deletions or mutations, alter miRNA genes much like they affect protein coding genes. The first example of a genomic alteration affecting miRNA expression was reported in 1989; a chromosomal rearrangement involving the translocation of c-MYC into the regulatory elements of an elusive genomic entity, called BCL3, on chromosome 17 was identified in human leukemia cells [49]. Fifteen years after the initial discovery, the BCL3 gene was found to produce the small noncoding RNA, miR-142, and overexpression of the translocated MYC gene is driven by the upstream miRNA 142 promoter [49].
In 2004, an in silico study showed that more than half of miRNAs map to genomic regions that are frequently altered in cancer [50]: 65 miRNAs map to loss of heterozygosity regions (LOH) (e.g. miR-15a/16-1), where the majority of tumor-suppressor genes are located [50]; 15 miRNAs are in amplified regions (e.g. miR-17-92 cluster, miR-155), which harbor most oncogenes [50]; 61 miRNAs are in breakpoint regions and fragile sites (FRA) (e.g. let-7 family members), common sites for sister-chromatid exchange, translocation, deletion and tumor-associated viral integration [51]. An extensive study of high-resolution array-based comparative genomic hybridization on 227 human ovarian cancers, breast cancers and melanoma specimens shows concordance between the expression of miRNAs and DNA copy number (75% concordance for amplified/deleted and upregulated/downregulated miRNAs) [52], providing evidence that genomic alterations at miRNA loci have an etiologic role in cancer.
MiRNAs 15a and miR-16-1 are deleted in 68% of CLL patients [5], as shown by cell-hybrid studies using mouse thymidine kinase-deficient-fibroblast and human CLL cells carrying a 13q14.3 translocation, the most frequently deleted region in human CLL that contains a tumor-suppressor gene. Furthermore, a sequencing-based screen for miRNAs dysregulated in some CLL patients identified a germ-line mutation in the primary precursor of miR-15a/16-1 that impairs their processing [53], and in a model mouse for human CLL (NBZ mouse), a point mutation occurring in the 3′ flanking region of miR-16-1 correlates with reduced miR-16-1 expression [54]. Finally, Dalla-Favera and colleagues generated transgenic mice carrying conditional alleles that mimic the minimal deletion region 13q14.3 or that specifically delete the miR-15a/16-1 cluster without affecting the expression of the host gene DLEU2 [55]. These mice exhibit a full spectrum of CLL-associated phenotypes, consistent with the DLEU2/miR-15a/16-1 locus having a tumor-suppressor role in the B-cell lineage. In these studies, miR-15a/16-1 modulate cell proliferation by decreasing the expression of CCND2, CCND3, CCNDE, CDK4 and CDK6. While these findings are in contrast with previous studies suggesting that miR-15a/16-1 influence cell survival by regulating BCL2 [56-57], the difference between these alternative mechanisms is not straightforward. MiRNA genes are commonly duplicated, and there is a duplicate copy of the miR-15a/16-1 gene located on a different chromosome. Redundancy could provide a safeguard against loss, and in this specific case it could explain why Bcl2 expression does not increase in vivo in DLEU2/miR-15a/16-1-deleted mice.
Amplification of miRNAs also occurs in cancer, exemplified by the human oncogenic cluster miR-17-92, which is located at chromosome 13q31 [58]. Overexpression of miR-17-92 increases MYC-induced lymphomagenesis, and this region is preferentially amplified in cancers such as DLBCL, follicular lymphoma, mantle cell lymphoma and primary cutaneous B-cell lymphoma. It is now recognized that cancer is mainly a genetic disease, however, genetic lesions alone cannot explain the complexity of the aberrations that arise in cancer cells.
Epigenetics and miRNAs
Epigenetics, defined as heritable changes in gene activity that are independent of DNA sequence, play a prominent role in the initiation and progression of cancer. Three main epigenetic events regulate tumor-associated genes: aberrant hypermethylation of tumor-suppressor genes, global DNA hypomethylation and post-translational modifications of histones [59]. MiRNAs can be targets of epigenetic events that, in some instances, can explain the perturbation of miRNA expression in cancer (Figure 3) [59]. Saito and colleagues have shown that DNA methylation status and chromatin structure around miRNA genes differ between bladder cancer cells and normal human fibroblasts [60]. They further demonstrate that inhibitors of DNA methylation and histone deacetylation induce the expression of miR-127 only in cancer cells [60]. In addition, the epigenetic activation of miR-127, which is silenced in cancer cells and downregulated in 75% of primary prostate and bladder tumors, downregulates expression of the BCL6 proto-oncogene [60], suggesting that miR-127 is a tumor suppressor. Another study, which employed the same experimental approach, reported that miR-124a is unmethylated in the normal human colon but hypermethylated in the majority of colon tumors analyzed [61]. Similarly, epigenetic silencing of miR-124a upregulates the oncogene CDK6 kinase [61]. Therefore, methylation of miR-124 and miR-127 genes influences the expression of two oncogenic proteins, which are not normally regulated by methylation.
Figure 3.
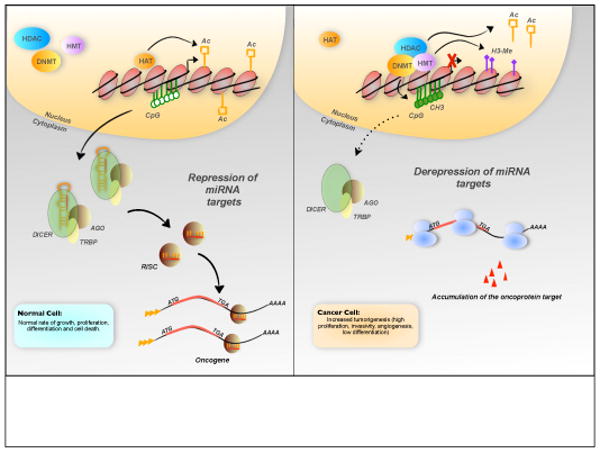
Epigenetic control of miRNA expression. Downregulation of miRNAs that function as tumor suppressors could lead to tumor formation and aggressive phenotypes through the loss of translational repression of several oncoproteins. Different mechanisms could be responsible for these events, and here, we illustrate the effect of epigenetic silencing mediated by methylation and loss of acetylation on the miRNA gene. Gain of repressive histone marks, such as histone tri-methylation (for examples: tri-methylation of lysine 27 on histone H3, H3K27me3), could prevent transcriptional activation of miRNA genes. (Abbreviations: CH3, methyl-cytosine; HMT, histone methyl-transferase; HDAC, histone deacetylase; DNMT, DNA methyl-transferase; HAT, histone acetyl-transferase; CpG, CpG islands; Ac, acetyl group; H3-Me, trimethylation of the histone H3 at the K4 residue; ATG, translation start codon; TGA, translation stop codon)
In several hematopoietic tumors, a combination of genetic and epigenetic mechanisms coordinately inactivate miR-203 [62]. Expression of miR-203 seems to be restricted to specific cell types [63], and its inhibition induces increased cell proliferation [64], suggesting an antiproliferative function. In fact, miR-203 is silenced by the loss of one allele and by promoter CpG hypermethylation in the other allele. Because ABL1 (a nonreceptor tyrosine kinase involved in the development of diverse hematopoietic malignancies) is a miR-203 target, silencing miR-203 in hematopoietic tumor cells could result in a proliferative advantage. An alternative to the interpretation that hypermethylation-mediated repression of tumor suppressors gives tumor cells a proliferative advantage is that hypermethylation-mediated silencing of miRNA could protect normal tissues and miRNA activation could promote neoplastic transformation [65]. In fact, let-7a-3 is heavily methylated in normal human tissues but hypomethylated in some lung adenocarcinomas [65]. Interestingly, hypomethylation facilitates let-7a-3 reactivation, enhances tumor phenotypes and induces oncogenic changes in transcription profiles [65]. However, let-7a-3 is methylated in a large cohort of human ovarian cancer samples and this methylation correlates with better survival rates for patients [66]. Further studies are necessary to reconcile the tumor-suppressor function of let-7 and epigenetic regulation.
DNA methylation is closely linked to histone modifications and miRNAs genes are also targets for histone alteration in cancer. Interestingly, DNA methylation and silencing of the miR-124 gene associates with the presence of repressor proteins MeCP2 and MBD2 and the absence of active histone marks, such as acetylation of histones H3 and H4 or trimethylation of histone H3 at lysine-4 (K4) [61].
The scenario is further complicated by the fact that miRNAs are not only regulated by epigentic mechanisms, but miRNAs can also regulate enzymes that are involved in the methylation of the CpG islands of tumor suppressor genes. In fact, the miR-29 family targets the de novo DNA methyltransferases DNMT3A and DNMT3B, and enforced expression of miR-29 family members in lung [67] and AML cancer cells [68] causes demethylation of the CpG islands in the promoter regions of tumor-suppressor genes, allowing their reactivation and further loss of tumorigenicity. Recently [69], two other miRNAs, miR-148a and miR-152, were shown to target DNMT1 in cholangiocarcinoma cells with concomitant activation of the methylation-sensitive tumor-suppressor genes RASSF1A and p16INK4A [69].
In conclusion, epigenetic changes complemented by genetic inactivation due to mutation or deletion can shed light on the mechanisms that partially account for the miRNA dysregulation in cancer.
Transcriptional control of miRNAs
Transcription is the major point of regulation in miRNA biogenesis and numerous factors control transcription from miRNA genes (Figure 4). The miR-17-92 cluster is frequently amplified in lymphoma and functions cooperatively as an oncogene, possibly by targeting apoptotic factors activated in response to MYC overexpression [24, 70]. O'Donnell and collaborators have shown that MYC induces the expression of the miR-17/92 cluster, and chromatin immunoprecipitation experiments reveal that MYC binds the first intron of the host gene of miR-17-92 cluster, which produces the long noncoding RNA C13orf25 [58]. Indeed, the authors identified the E2F1 mRNA, which creates a reciprocal positive-feedback loop with MYC, as a direct target of the miR-17-5p and miR-20a. Based on these results, the miR-17-92 cluster should limit the activity of E2F1, thereby reducing MYC-induced cell proliferation. In this context, the miR-17-92 cluster would function as tumor suppressor; however, taking into consideration that excessive E2F1 induces apoptosis [71], this cluster might be activated by MYC to counter the apoptotic activity of E2F1, allowing MYC mediated-proliferation. Human homologues of this miRNA gene cluster are also located on chromosome 7 in the MCM7 gene (miR-106b/93/25) and on chromosome X (miR-106a/18b/20b/19b-2/92a/363). The miR-17-92 cluster and the homologues miR-106a/363 are also upregulated after estradiol treatment in breast cancer cells, and miR-17-92 cluster expression is highly correlated with the level of estrogen receptor-α (ERα) in breast cancers [72]. Interestingly, miR-18a/19b/20b downregulate ERα [72], whereas miR-20a/17–5p/106a/20b downregulate the ERα transcriptional p160 coactivator, AIB1 [73], implicating both clusters in the regulation of ERα transcriptional activity upon estrogenic stimulation. Petrocca and colleagues have demonstrated that E2F1 activates the cluster on chromosome 7 and in parallel the host gene, MCM7 [74]. Indeed, miR-106b and miR-93 target E2F1, creating a negative feedback loop of transcriptional regulation [74].
Figure 4.
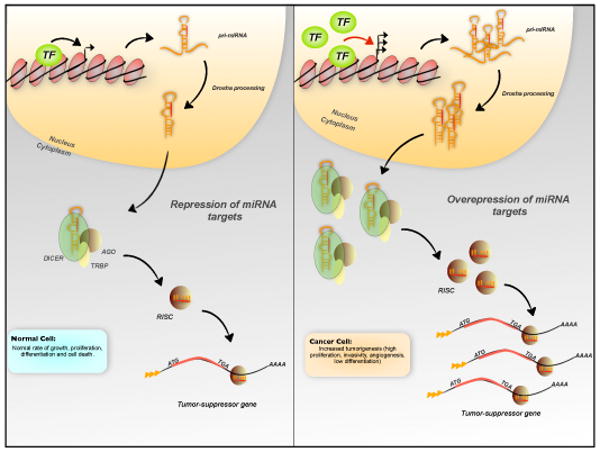
Transcriptional control of miRNA expression. Overexpression of miRNAs might induce tumor formation through the repression of several tumor-suppressor genes. Overexpression and/or activation of a transcription factor at inappropriate times or in the wrong tissues could explain the increased level of miRNAs. (Abbreviations: TF, transcription factor; ATG, translation start codon; TGA, translation stop codon)
Elevated miR-221 and miR-222 expression has been causally linked to proliferation [38], apoptosis [75] and migration [76] in several cancer cell lines. However, our knowledge of the molecular mechanisms mediating miR-221/222 function in cancer is limited. Two recent reports explore this issue. The activation of miR-221/222 is regulated specifically in lung and liver tumors, at least in part, by the MET oncogene through activation of the c-JUN transcription factor, which binds directly to the promoter of miR-221/222 cluster [37]. In breast cancer, the miR-221/222 cluster is negatively regulated by ERα, through the recruitment of its corepressor proteins, silencing mediator of retinoic acid and thyroid hormone receptor (SMRT) and nuclear receptor corepressor (NCoR) [40].
Recently, miR-34a and miR-34b/c were reported as direct, conserved p53 target genes that presumably mediate p53 activities including apoptosis, cell cycle arrest and senescence [77]. However, miR-34a is also regulated independently of p53 during oncogene-induced senescence [78]. A number of different oncogenes induce senescence, including B-RAF in human benign tumors and N-Ras, K-Ras, H-Ras, B-Ras and E2f3 in mouse [79]. A strong upregulation of miR-34a is observed during B-RAF-induced senescence in human fibroblasts that is directly dependent on the ETS family transcription factor ELK1 and independent of p53. Indeed, miR-34a targets several important oncogenes, including MYC, CCND1, CDK6, MET and the antiapoptotic BCL2, inducing an irreversible growth arrest that acts as a barrier against tumorigenesis [78]. However, in this cellular system, inhibition of miR-34a alone did not prevent the appearance of phenotypic hallmarks of senescence, indicating that miR-34a is important but is not the sole player in oncogene-induced senescence. Other mediators remain to be identified.
Post-transcriptional control of miRNAs
Emerging evidence in various developmental and disease contexts supports extensive post-transcriptional control of miRNA expression; however, for most miRNAs, mechanisms of post-transcriptional regulation remain poorly understood. Processing by Drosha is an important point of regulation. Exposure of the p53 wild-type HCT116 human colon cancer cell line to the DNA-damaging agent doxorubicin, which also induces p53, increases the expression of several mature miRNAs that have growth-suppressive functions and are downregulated in cancer (such as miR-15, miR-16 and miR-145) [80]. These authors also demonstrated that p53 associates with Drosha in doxorubicin-treated cells and promotes the Drosha-mediated processing of certain tumor-suppressor miRNAs in response to DNA damage [80]. Therefore, the loss of p53 in cancer reduces the levels of miRNAs that are either transcriptionally regulated by p53 or regulated by p53-induced Drosha processing.
MiR-18a, a member of the oncogenic cluster miR-17-92, is also regulated by Drosha-mediated processing by hnRNP A1, which binds to the loop of pri-miR-18a, rearranges the RNA secondary structure, and creates a more favorable cleavage site for Drosha and DGCR8 complex [81]. Another interesting example of post-transcriptional regulation at the RNA-processing level comes from Yamagata and colleagues [82], who reported that estrogen suppresses the levels of a set of miRNAs, including miR-195 and miR-125a, in mice and human cultured cells. By in vitro assays, estrogen-bound ERα was shown to inhibit the maturation of these miRNAs by associating with the Drosha complex and preventing the conversion of pri-miRNAs into pre-miRNAs. The negative regulation of miRNAs mediated by ERα stabilizes estrogen target genes, such as human VEGF mRNA [83] by downregulating miRNA maturation, (e.g. miR-125a and miR-195).
The primary transcript of let-7 is expressed in both normal and cancer cells, whereas mature let-7 is detected only in normal cells, indicating that let-7 might be post-transcriptionally regulated [84]. In embryonic stem cells and embryonic carcinoma cells, an RNA binding protein, Lin28, binds to pre-let-7 in the cytoplasm and recruits the terminal uridyl transferase (TUTase) Zcchc11 to induce terminal uridylation of pre-let-7 [85-86]. The U tail (∼14 nt) that is added to the 3′ end of pre-let-7 blocks Dicer processing and facilitates the decay of pre-let-7 [86]. Given the proto-oncogenic activity of Lin28 [87], this pathway could restrict the expression of the tumor suppressor let-7 and contribute to its dysregulation in cancer.
Another example of post-transcriptional regulation is the presence of inactivating mutations in the gene encoding TAR RNA-binding protein 2 (TARBP2 or TRBP), an essential functional partner of DICER1, in human cancer cell lines and primary tumors with microsatellite instability [88]. Loss of TRBP destabilizes DICER1 following impairment of miRNA processing and enhancement of cellular transformation. More studies are clearly needed to understand how miRNA processing contributes to both somatic cell reprogramming and oncogenesis.
Concluding remarks
The rapidly progressing field of small regulatory RNAs continues to reveal the diversity and complexity of the RNA world. However, despite remarkable recent progress, the connection between cancer and miRNAs remains incompletely understood and many important questions remain. First, genome-wide analyses for alterations in miRNA genes or for copy number alterations in various human tumors are needed to identify all of the putative tumor-suppressor and oncogenic miRNAs. At the same time, more sophisticated in vivo models are needed to identify and define miRNA functions. Because miRNAs can have widespread effects on the transcriptome, their full biological properties are unlikely to be explained by the suppression of a single or few proteins. In fact, the major challenge is to define all of the mRNA targets and cancer-related pathways that are controlled by the dysregulated miRNAs in neoplasia. Towards this aim, new and more sophisticated computational target-prediction methods will be helpful. Further studies are also necessary to better understand silencing by an endogenous miRNA in the context of normal cellular homeostasis, to identify the factors that influence miRNA and target co-localization, and to understand how miRNA turnover is regulated. Although miRNAs only moderately suppress their targets, miRNAs could exert both strong and broad effects, largely because they suppress many genes and because they are implicated in multiple feedback loops with other regulators of gene expression.
Exploring the roles of miRNAs in these intimate crosstalks will help us to understand the causes of cancer and other diseases and in creating new alternative therapeutics. In this regard, Mendell and colleagues have cleared the path to the clinic for a miRNA-based therapy [89]: the investigators have shown that restoration in vitro of miR-26a strongly downregulated in MYC-induced liver tumors in mice [89], regulates the expression of the cyclins D2 and E2, and induces a G1 arrest of human liver cancer cells. Based on these striking effects, an adeno-associated virus (AAV) vector, highly efficient at transducing hepatocytes in vivo and enginereed to express miR-26a, was intravenously injected into a mouse model of liver cancer: 8 out of 10 animals treated with the miR-26a construct did not develop disease, and the 2 animals with disease showed lower transduction efficiency of the vector [89]. More recently, the Weinberg group explored another in vivo approach to modulate miRNA function with possible therapeutic implication. Their approach involved the use of the sponge vector [90], a vector expressing miRNA target sites used to saturate an endogenous miRNA and prevent it from regulating its natural targets [91]. In this case, miR-9 was identified as a pro-metastatic miRNA in breast cancer [90], and miRNA-sponge-mediated suppression of miR-9 in the highly metastatic 4T1 mouse mammary tumor cells reduced lung metastasis by 50%, although no effect was observed in the onset of the primary tumor. Future studies should improve our ability to identify new miRNAs with therapeutic potential and to use miRNA-based therapies to treat cancers.
Box1. The discovery of miRNAs
During a loss-of-function study in Caenorhabditis elegans in 1981, mutations in lin-4 gene led to continued synthesis of larval-specific cuticle 1. At that time, together with lin-14, lin-29 and lin-28, these genes were classified as heterochronic genes, capable of controlling the timing of specific post-embryonic developmental events in C. elegans.
Ruvkun and collaborators found that deletions of the 3′UTR of lin-14 mRNA led to abnormal protein accumulation in the later larval stages, highlighting the existence of a regulatory element in the 3′UTR of lin-14 mRNA (reviewed in [106]). Since lin-4 temporally downregulates lin-14 protein, the authors hypothesized that lin-4 gene product was a trans-acting factor that bound to the 3′UTR of lin-14 and negatively regulated it. Finally in 1993, two independent studies demonstrated that lin-4 does not encode a protein but produces two small RNA transcripts of approximately 22 and 61 nts containing sequences that are complementary to the 3′UTR of lin-14. Accordingly, the authors suggested that the temporal regulation of lin-14 translation is controlled by lin-4 RNA via antisense RNA-RNA interaction involving the small RNA lin-4 and the 3′UTR of lin-14. Seven years later, the let-7 gene was identified as another heterochronic gene that produces a small 21 nt RNA with sequences complementary to the 3′UTRs of lin-14, lin-28, lin-41, lin-42 and daf-12.
At that time, these discoveries were considered merely new pieces in the gene expression regulation puzzle for the small temporal RNAs in worms. This idea completely changed when independent groups investigated whether similar RNAs played a general role in gene regulation. Working in different organisms and cellular systems, researchers from three laboratories isolated a group of RNAs with the same characteristics of lin-4 and let-7, providing evidence for the existence of a large class of small RNAs with potential regulatory roles. Because of their small size, the authors referred to these novel RNAs as microRNAs.
Box2. Genomics of miRNAs
Almost 50% of mammalian miRNAs are located in the introns of protein coding genes or long noncoding RNAs transcripts, whereas the remainder are independent transcription units with specific core promoter elements and polyadenylation signals [reviewed in 1, 4]. Among the intragenic miRNAs, 40% are in the introns of protein coding genes, whereas ∼10% are in introns of long noncoding RNA transcripts [1]. Approximately 50% of mammalian miRNA loci are in close proximity to other miRNAs. These clustered miRNAs are transcribed as polycistronic messages in single transcription units or are overlapped in the host transcripts, within exons or introns, depending on splicing patterns of the host gene. Additionally, many miRNAs overlap with two or more transcription units that are transcribed on opposite DNA strands. Although the transcription of most miRNAs is mediated by RNA polymerase II, a small set of miRNAs associated with Alu repeats are transcribed by RNA polymerase III [107]. A small group of miRNA-like RNAs located in short introns occur in flies and mammals [108]. These small RNAs, named mirtrons, bypass canonical Drosha processing and rely on the splicing machinery and lariat-debranching enzymes to form pre-miRNA-like hairpins (Figure 1). Once a hairpin is produced, the mirtron pathway merges with the canonical miRNA pathway; it is exported by Exportin-5 and processed further by Dicer.
Acknowledgments
We sincerely thank Dr Kenneth Nephew for the critical reading of the manuscript, and Michela Garofalo and Flavia Pichiorri for suggestions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kim VN, Nam JW. Genomics of microRNA. Trends Genet. 2006;22:165–173. doi: 10.1016/j.tig.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 2.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and fucntion. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 3.Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet. 2009;10:704–714. doi: 10.1038/nrg2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim NV, et al. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol. 2009;10:126–139. doi: 10.1038/nrm2632. [DOI] [PubMed] [Google Scholar]
- 5.Calin GA, et al. Frequent deletion and downregulation of microRNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2004;99:15524–15529. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu J, et al. MicroRNA expression profiles classify human cancer. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 7.Garzon R, et al. MicroRNA fingerprints during human megakaryocytopoiesis. Proc Natl Acad Sci USA. 2006;103:5078–5083. doi: 10.1073/pnas.0600587103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stefani G, Slack F. Small non-coding RNAs in animal development. Nat Rev Mol Cell Biol. 2008;9:219–230. doi: 10.1038/nrm2347. [DOI] [PubMed] [Google Scholar]
- 9.Kumar MS, et al. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat Genet. 2007;39:673–677. doi: 10.1038/ng2003. [DOI] [PubMed] [Google Scholar]
- 10.Kumar MS, et al. Dicer1 functions as a haploinsufficient tumor suppressor. Genes Dev. 2009;23:2700–2704. doi: 10.1101/gad.1848209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lambertz I. Monoallelic but not biallelic loss of Dicer1 promotes tumorigenesis in vivo. Cell Death Differ. 2009 doi: 10.1038/cdd.2009.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takamizawa J, et al. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. 2004;64:3753–3756. doi: 10.1158/0008-5472.CAN-04-0637. [DOI] [PubMed] [Google Scholar]
- 13.Johnson SM, et al. RAS is regulated by the let-7 microRNA family. Cell. 2005;8:635–647. doi: 10.1016/j.cell.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 14.He XY, et al. The let-7a microRNA protects from growth of lung carcinoma by suppression of k-Ras and c-Myc in nude mice. J Cancer Res Clin Oncol. 2009 doi: 10.1007/s00432-009-0747-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mayr C, et al. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science. 2007;315:1576–1579. doi: 10.1126/science.1137999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Volinia S, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006;103:2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murakami Y, et al. Comprehensive analysis of microRNA expression patterns in hepatocellular carcinoma and non-tumorous tissues. Oncogene. 2006;25:2537–2545. doi: 10.1038/sj.onc.1209283. [DOI] [PubMed] [Google Scholar]
- 18.Calin GA, et al. MicroRNA profiling reveals distinct signatures in B cell chronic lymphocytic leukemias. Proc Natl Acad Sci U S A. 2004;101:11755–11760. doi: 10.1073/pnas.0404432101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garzon R, et al. MicroRNA signatures associated with cytogenetics and prognosis in acute myeloid leukemia. Blood. 2008;111:3183–3189. doi: 10.1182/blood-2007-07-098749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sarver AL, et al. Human colon cancer profiles show differential microRNA expression depending on mismatch repair status and are characteristic of undifferentiated proliferative states. BMC Cancer. 2009;18:9–401. doi: 10.1186/1471-2407-9-401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ciafre S, et al. Extensive modulation of a set of microRNAs in primary glioblastoma. Biochem Biophys Res Commun. 2005;334:1351–1358. doi: 10.1016/j.bbrc.2005.07.030. [DOI] [PubMed] [Google Scholar]
- 22.Iorio MV, et al. microRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65:7065–7070. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- 23.Pichiorri F, et al. MicroRNAs regulate critical genes associated with multiple myeloma pathogenesis. Proc Natl Acad Sci U S A. 2008;105:12885–12890. doi: 10.1073/pnas.0806202105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He L, et al. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–833. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao C, et al. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat Immunol. 2008;9:405–414. doi: 10.1038/ni1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ventura A, et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell. 2008;132:875–886. doi: 10.1016/j.cell.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mu P, et al. Genetic dissection of the miR-17∼92 cluster of microRNAs in Myc-induced B-cell lymphomas. Genes Dev. 2009;23:2806–2811. doi: 10.1101/gad.1872909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olive V, et al. miR-19 is a key oncogenic component of mir-17-92. Genes Dev. 2009;23:2839–2849. doi: 10.1101/gad.1861409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Metzler M, et al. High expression of precursor microRNA-155/BIC RNA in children with Burkitt lymphoma. Genes Chromosomes Cancer. 2004;39:167–169. doi: 10.1002/gcc.10316. [DOI] [PubMed] [Google Scholar]
- 30.Eis PS, et al. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci U S A. 2005;102:3627–3632. doi: 10.1073/pnas.0500613102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Costinean S, et al. Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in E(mu)-miR155 transgenic mice. Proc Natl Acad Sci U S A. 2006;103:7024–7029. doi: 10.1073/pnas.0602266103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Costinean S, et al. Src homology 2 domain-containing inositol-5-phosphatase and CCAAT enhancer-binding protein beta are targeted by miR-155 in B cells of Emicro-MiR-155 transgenic mice. Blood. 2009;114:1374–1382. doi: 10.1182/blood-2009-05-220814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Felli N, et al. MicroRNAs 221 and 222 inhibit normal erythropoiesis and erythroleukemic cell growth via kit receptor down-modulation. Proc Natl Acad Sci U S A. 2005;102:18081–18086. doi: 10.1073/pnas.0506216102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pineau P, et al. miR-221 overexpression contributes to liver tumorigenesis. Proc Natl Acad Sci U S A. 2010;107:264–269. doi: 10.1073/pnas.0907904107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.DeYoung MP, et al. Hypoxia regulates TSC1/2- mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev. 2008;22:239–251. doi: 10.1101/gad.1617608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Medina R, et al. MicroRNAs 221 and 222 bypass quiescence and compromise cell survival. Cancer Res. 2008;68:2773–2780. doi: 10.1158/0008-5472.CAN-07-6754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garofalo M, et al. miR-221&222 regulate TRAIL resistance and enhance tumorigenicity through PTEN and TIMP3 downregulation. Cancer Cell. 2009;16:498–509. doi: 10.1016/j.ccr.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 38.Le Sage, et al. Regulation of the p27(Kip1) tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. EMBO J. 2007;26:3699–3708. doi: 10.1038/sj.emboj.7601790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Terasawa K, et al. Sustained activation of ERK1/2 by NGF induces microRNA-221 and 222 in PC12 cells. FEBS J. 2009;276:3269–3276. doi: 10.1111/j.1742-4658.2009.07041.x. [DOI] [PubMed] [Google Scholar]
- 40.Di Leva G, et al. MicroRNA Cluster 221-222 and Estrogen Receptor α Interactions in Breast Cancer. J Natl Cancer Inst. 2010 doi: 10.1093/jnci/djq102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fidler IJ, et al. The pathogenesis of cancer metastasis: the “seed and the soil” hypothesis revisited. Nature Rev Cancer. 2003;3:453–458. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- 42.Thiery JP, Sleeman JP. Complex networks orchestrate epithelial–mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- 43.Peinado H, et al. Snail, ZEB and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415–428. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- 44.Gregory PA, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10:593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
- 45.Park SM, et al. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008;22:894–907. doi: 10.1101/gad.1640608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Korpal M, et al. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem. 2008;283:14910–14914. doi: 10.1074/jbc.C800074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bracken CP, et al. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. 2008;68:7846–7854. doi: 10.1158/0008-5472.CAN-08-1942. [DOI] [PubMed] [Google Scholar]
- 48.Burk U, et al. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008;9:582–589. doi: 10.1038/embor.2008.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Calin GA, Croce CM. MicroRNAs and chromosomal abnormalities in cancer cells. Oncogene. 2006;25:6202–6210. doi: 10.1038/sj.onc.1209910. [DOI] [PubMed] [Google Scholar]
- 50.Calin GA, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A. 2004;101:2999–3004. doi: 10.1073/pnas.0307323101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Richards RI. Fragile and unstable chromosomes in cancer: causes and consequences. Trends Genet. 2001;17:339–345. doi: 10.1016/s0168-9525(01)02303-4. [DOI] [PubMed] [Google Scholar]
- 52.Zhang L, et al. Genomic and epigenetic alterations deregulate microRNA expression in human epithelial ovarian cancer. Proc Natl Acad Sci U S A. 2008;105:7004–7009. doi: 10.1073/pnas.0801615105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Calin GA. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med. 2005;353:1793–1801. doi: 10.1056/NEJMoa050995. [DOI] [PubMed] [Google Scholar]
- 54.Salerno E, et al. Correcting miR-15a/16 genetic defect in New Zealand Black mouse model of CLL enhances drug sensitivity. Mol Cancer Ther. 2009;8:2684–2692. doi: 10.1158/1535-7163.MCT-09-0127. [DOI] [PubMed] [Google Scholar]
- 55.Klein U, et al. The DLEU2/miR-15a/16-1 Cluster Controls B Cell Proliferation and Its Deletion Leads to Chronic Lymphocytic Leukemia. Cancer Cell. 2010;17:28–40. doi: 10.1016/j.ccr.2009.11.019. [DOI] [PubMed] [Google Scholar]
- 56.Cimmino A, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A. 2005;102:13944–13949. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bonci D, et al. The miR-15a-miR-16-1 cluster controls prostate cancer by targeting multiple oncogenic activities. Nat Med. 2008;14:1271–1277. doi: 10.1038/nm.1880. [DOI] [PubMed] [Google Scholar]
- 58.Ota A, et al. Identification and characterization of a novel gene, C13orf25, as a target for 13q31-q32 amplification in malignant lymphoma. Cancer Res. 2004;64:3087–3095. doi: 10.1158/0008-5472.can-03-3773. [DOI] [PubMed] [Google Scholar]
- 59.Rouhi A, et al. MiRNAs, epigenetics, and cancer. Mamm Genome. 2008;19:517–525. doi: 10.1007/s00335-008-9133-x. [DOI] [PubMed] [Google Scholar]
- 60.Saito Y, et al. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell. 2006;9:435–443. doi: 10.1016/j.ccr.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 61.Lujambio A, et al. Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res. 2007;67:1424–1429. doi: 10.1158/0008-5472.CAN-06-4218. [DOI] [PubMed] [Google Scholar]
- 62.Bueno MJ, et al. Genetic and epigenetic silencing of microRNA-203 enhances ABL1 and BCR-ABL1 oncogene expression. Cancer Cell. 13:496–506. doi: 10.1016/j.ccr.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 63.Yi R, et al. Morphogenesis in skin is governed by discrete sets of differentially expressed microRNAs. Nat Genet. 2006;38:356–362. doi: 10.1038/ng1744. [DOI] [PubMed] [Google Scholar]
- 64.Cheng AM, et al. Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res. 2005;33:1290–1297. doi: 10.1093/nar/gki200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brueckner B, et al. The human let-7a-3 locus contains an epigenetically regulated microRNA gene with oncogenic function. Cancer Res. 2007;67:1419–1423. doi: 10.1158/0008-5472.CAN-06-4074. [DOI] [PubMed] [Google Scholar]
- 66.Lu L, et al. Hypermethylation of let-7a-3 in epithelial ovarian cancer is associated with low insulin-like growth factor-II expression and favorable prognosis. Cancer Res. 2007;67:10117–10122. doi: 10.1158/0008-5472.CAN-07-2544. [DOI] [PubMed] [Google Scholar]
- 67.Fabbri M, et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci USA. 2007;104:15805–15810. doi: 10.1073/pnas.0707628104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Garzon R, et al. MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene re-expression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood. 2009;113:6411–6418. doi: 10.1182/blood-2008-07-170589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Braconi C, et al. MicroRNA-dependent regulation of DNA methyltransferase-1 and tumor suppressor gene expression by interleukin-6 in human cholangiocytes. Hepatology. 2010;51:881–890. doi: 10.1002/hep.23381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.O'Donnell KA, et al. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- 71.Stanelle J, et al. E2F1-induced apoptosis: turning killers into therapeutics. Trends Mol Med. 2006;12:177–185. doi: 10.1016/j.molmed.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 72.Castellano L, et al. The estrogen receptor-α-induced microRNA signature regulates itself and its transcriptional response. Proc Natl Acad Sci U S A. 2009;106:15732–15737. doi: 10.1073/pnas.0906947106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hossain A, et al. Mir-17–5p regulates breast cancer cell proliferation by inhibiting translation of AIB1 mRNA. Mol Cell Biol. 2006;26:8191–8201. doi: 10.1128/MCB.00242-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Petrocca F, et al. E2F1-regulated microRNAs impair TGFbeta-dependent cell-cycle arrest and apoptosis in gastric cancer. Cancer Cell. 2008;13:272–286. doi: 10.1016/j.ccr.2008.02.013. [DOI] [PubMed] [Google Scholar]
- 75.Garofalo M, et al. MicroRNA signatures of TRAIL resistance in human non-small cell lung cancer. Oncogene. 2008a;27:3845–3855. doi: 10.1038/onc.2008.6. [DOI] [PubMed] [Google Scholar]
- 76.Felicetti F, et al. The promyelocytic leukemia zinc finger-microRNA-221/-222 pathway controls melanoma progression through multiple oncogenic mechanisms. Cancer Res. 2008;68:2745–2754. doi: 10.1158/0008-5472.CAN-07-2538. [DOI] [PubMed] [Google Scholar]
- 77.He L, et al. A microRNA component of the p53 tumor suppressor network. Nature. 2007;447:1130–1134. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Christoffersoen NR, et al. p53-independent upregulation of miR-34a during oncogene-induced senescence represses MYC. Cell Death Differ. 2010;17:236–245. doi: 10.1038/cdd.2009.109. [DOI] [PubMed] [Google Scholar]
- 79.Di Micco R, et al. Breaking news: high-speed race ends in arrest – how oncogenes induce senescence. Trends Cell Biol. 2007;17:529–536. doi: 10.1016/j.tcb.2007.07.012. [DOI] [PubMed] [Google Scholar]
- 80.Suzuki HI, et al. Modulation of microRNA processing by p53. Nature. 2009;460:529–533. doi: 10.1038/nature08199. [DOI] [PubMed] [Google Scholar]
- 81.Michlewski G, et al. Posttranscriptional regulation of miRNAs harboring conserved terminal loops. Mol Cell. 2008;32:383–393. doi: 10.1016/j.molcel.2008.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yamagata K, et al. Maturation of microRNA is hormonally regulated by a nuclear receptor. Mol Cell. 2009;36:340–347. doi: 10.1016/j.molcel.2009.08.017. [DOI] [PubMed] [Google Scholar]
- 83.Ruohola JK, et al. Vascular endothelial growth factors are differentially regulated by steroid hormones and antiestrogens in breast cancer cells. Mol Cell Endocrinol. 1999;149:29–40. doi: 10.1016/s0303-7207(99)00003-9. [DOI] [PubMed] [Google Scholar]
- 84.Pasquinelli AE, et al. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature. 2000;408:86–89. doi: 10.1038/35040556. [DOI] [PubMed] [Google Scholar]
- 85.Viswanathan SR, et al. Selective blockade of microRNA processing by Lin28. Science. 2008;320:97–100. doi: 10.1126/science.1154040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Heo I, et al. TUT4 in concert with Lin28 suppresses microRNA biogenesis through pre-microRNA uridylation. Cell. 2009;138:696–708. doi: 10.1016/j.cell.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 87.Gunaratne PH. Embryonic stem cell microRNAs: defining factors in induced pluripotent (iPS) and cancer (CSC) stem cells? Curr Stem Cell Res Ther. 2009;4:168–177. doi: 10.2174/157488809789057400. [DOI] [PubMed] [Google Scholar]
- 88.Melo SA, et al. A TARBP2 mutation in human cancer impairs microRNA processing and DICER1 function. Nat Genet. 2009;41:365–370. doi: 10.1038/ng.317. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 89.Kota J, et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell. 2009;137:1005–1017. doi: 10.1016/j.cell.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ma L, et al. miR-9, a MYC/MYC-N-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol. 2010;12:247–256. doi: 10.1038/ncb2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Brown BD, Naldini L. Exploiting and antagonizing microRNA regulation for therapeutic and experimental application. Nat Rev Cancer. 2009;10:578–585. doi: 10.1038/nrg2628. [DOI] [PubMed] [Google Scholar]
- 92.Visone R, et al. Karyotype-secific microRNA signature in chronic lymphocytic leukemia. Blood. 2009;114:3872–3879. doi: 10.1182/blood-2009-06-229211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Iorio MV, et al. MicroRNA signatures in human ovarian cancer. Cancer Res. 2007;67:8699–8707. doi: 10.1158/0008-5472.CAN-07-1936. [DOI] [PubMed] [Google Scholar]
- 94.Yanaihara N, et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell. 2006;9:189–198. doi: 10.1016/j.ccr.2006.01.025. [DOI] [PubMed] [Google Scholar]
- 95.Pallante P, et al. MicroRNA deregulation in human thyroid papillary carcinomas. Endocr Relat Cancer. 2006;13:497–508. doi: 10.1677/erc.1.01209. [DOI] [PubMed] [Google Scholar]
- 96.Roldo C, et al. MicroRNA expression abnormalities in pancreatic endocrine and acinar tumors are associated with distinctive pathologic features and clinical behavior. J Clin Oncol. 2006;24:4677–4684. doi: 10.1200/JCO.2005.05.5194. [DOI] [PubMed] [Google Scholar]
- 97.Ueda T, et al. Relation between microRNA expression and progression and prognosis of gastric cancer: a microRNA expression analysis. Lancet Oncol. 2010;11:136–146. doi: 10.1016/S1470-2045(09)70343-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hu X, et al. A microRNA expression signature for cervical cancer progression. Cancer Res. 2010;70:1441–1448. doi: 10.1158/0008-5472.CAN-09-3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Juan D, et al. Identification of a microRNA panel for clear-cell kidney cancer. Urology. 2009 doi: 10.1016/j.urology.2009.10.033. [DOI] [PubMed] [Google Scholar]
- 100.Careccia S, et al. A restricted signature of microRNAs distinguishes APL blasts from normal promyelocytes. Oncogene. 2009;28:4034–4040. doi: 10.1038/onc.2009.255. [DOI] [PubMed] [Google Scholar]
- 101.Zhang H, et al. Genome-wide analysis of small RNA and novel microRNA discovery in human acute lymphoblastic leukemia based on extensive sequencing approach. PloS One. 2009;4:e6849. doi: 10.1371/journal.pone.0006849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Subramanian S, et al. MicroRNA expression signature of human sarcomas. Oncogene. 2008;27:2015–2026. doi: 10.1038/sj.onc.1210836. [DOI] [PubMed] [Google Scholar]
- 103.Roehle A, et al. MicroRNA signatures characterize diffuse large B-cell lymphomas and follicular lymphomas. Br J Haematol. 2008;142:732–744. doi: 10.1111/j.1365-2141.2008.07237.x. [DOI] [PubMed] [Google Scholar]
- 104.Dyrskjot L, et al. Genomic profiling of microRNAs in bladder cancer: miR-129 is associated with poor outcome and promotes cell death in vitro. Cancer Res. 2009;69:4851–4860. doi: 10.1158/0008-5472.CAN-08-4043. [DOI] [PubMed] [Google Scholar]
- 105.Yang H, et al. MicroRNA expression signatures in Barrett's esophagus and esophageal adenocarcinoma. Clin Cancer Res. 2009;15:5744–5752. doi: 10.1158/1078-0432.CCR-09-0385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ruvkun G. The perfect storm of tiny RNAs. Nat Med. 2008;14:1041–1045. doi: 10.1038/nm1008-1041. [DOI] [PubMed] [Google Scholar]
- 107.Borchert GM, et al. RNA polymerase III transcribes human microRNAs. Nature Struct Mol Biol. 2006;13:1097–1101. doi: 10.1038/nsmb1167. [DOI] [PubMed] [Google Scholar]
- 108.Ruby JG, et al. Intronic microRNA precursors that bypass Drosha processing. Nature. 2007;448:83–86. doi: 10.1038/nature05983. [DOI] [PMC free article] [PubMed] [Google Scholar]