

Summary
Engineering industrial microbes has been hampered by incomplete knowledge of cell biology. Thus an iterative engineering cycle of modeling, implementation, and analysis has been used to increase knowledge of the underlying biology while achieving engineering goals. Recent advances in systems biology technologies have drastically improved the amount of information that can be collected in each iteration. As well, synthetic biology tools are melding modeling and molecular implementation. These advances promise to move microbial engineering from the iterative approach to a design-oriented paradigm, similar to electrical circuits and architectural design. Genome scale metabolic models, new tools for controlling expression, and integrated –omics analysis are described as key contributors in moving the field toward design-based engineering.
Introduction
Industrial microbiology has benefitted greatly from the introduction of recombinant DNA technologies, and the myriad of options that have hereby been enabled. These options have allowed a generation of engineers to splice DNA in many different ways, but even though recombinant DNA technology was developed in the early 1970s, many biotech challenges remain unsolved. While we knew how to splice DNA, the past ~30 years have been spent trying to figure out what DNA to splice. In other engineering disciplines, such as electrical, civil, and chemical engineering, design-based engineering has enabled the construction of engineering devices (bridges, electric circuits, etc.) directly from plans, because of a largely complete mathematical understanding of the physics that govern these devices. Microbial engineering is qualitatively different, in that the objective to date has been to (a) retrofit existing organisms to produce a product-of-interest using (b) knowledge and models that can be limited in their predictive capabilities.
As such, microbial engineering has traditionally followed a cyclic pattern of modeling, implementation, and analysis (Figure 1a). Typically, integrating available knowledge, e.g., the biochemical reactions known in an organism, into a mathematical model and use this for simulations can be used to develop a design strategy. Implementation of the strategies, such as extending a metabolic pathway to a new compound by introducing new enzymes, or knocking out competing pathways, is accomplished using recombinant DNA techniques consisting of plasmids or genomic alterations. Finally, the strain would be analyzed, sometimes only characterizing physiological properties and product yields. As models are limited and do not encompass all the activities of the cell, this process is inevitably iterative. Therefore, the knowledge gained from the previous analysis is used to refine the model, and the cycle begins again. Advances in Systems Biology leverage analysis and modeling to better define how a cell behaves in particular circumstances. Likewise, advances in Synthetic biology are allowing implementation of novel gene circuits that are guided by predictive models of each molecular ‘part.’ Combined, these recent advances in Systems Biology and Synthetic Biology are changing the efficiency of this engineering cycle toward the goal of transitioning from iterative engineering (Figure 1a) to a linear design-based engineering (Figure 1b) where modeling and implementation are adequately sophisticated to achieve the design goal without iteration. In this review we will highlight techniques and tools that are helping to move us toward design-based engineering of microbes for industrial purposes.
Figure 1. Transitioning from iterative to linear design-based engineering of industrial microbes.

(a) Iterative engineering - Genome scale models describe the cellular processes under desired condition. These models guide implementation of engineering strategies. These implementations are characterized by –omics and integrated analysis tools, which are used to revise the model and improve predictive capability. (b) Design-based Engineering – Models and implementation tools are reliable enough that the expected outcome is usually achieved, as in civil and electrical engineering. Transitioning from iterative cycle to a linear design-based engineering can be achieved by advance by better modeling and implementation aided by enhanced analysis.
Modeling
Models are a key asset to leverage in the design of microbes for a desired phenotype. Predictive models that describe relevant cellular processes can be used by engineers to rewire existing cellular processes toward a desired function. Genome-scale metabolic models (GSMMs) have been used extensively to predict changes in enzyme reaction rates (fluxes) in response to nutrient or genetic perturbations [1]. These models rely on (1) conservation of mass through the network including electrons and (2) cellular requirements for biomass to help predict how the cell will regulate metabolic pathways. Because these models predict changes in the metabolic network, they are of great use in predicting cellular alterations that will increase the production of a desired product [2]. These models can be used in combination with metabolic optimization algorithms, such as OptKnock or OptGene, to scan through all possible gene knock outs in the cell, and identify knockouts that are predicted to increase production [3,4]. Improvement in production titers for a large number of products, including: lactic acid, L-valine, L-threonine, other amino acids, hydrogen, vanillin (See ref [2] for details), lycopene[5], ethanol[6], and sequiterpenes [7] have been achieved using such models.
Stoichiometric models have been created for many industrially-relevant microbes, through curating enzyme biochemistry literature and using comparative genomics approaches for organisms that have little biochemical evidence. Genome-scale metabolic models exist for a range of industrial microbes. Table 1 gives information for published industrial microbes. Because of the rapid drop in sequencing costs, models for new organisms are being added often. Currently models are being developed for Penicillium chrysogenum and Pichia pastoris, which will be useful for antibiotic and protein production. Many of the published models can be found at the Biochemical, Genetic and Genomic (BiGG) database (http://bigg.ucsd.edu/) or the BioMet Toolbox (http://sysbio.se/BioMet/).
Table 1.
Genome-scale metabolic models of industrial microbes.
| Organism | Reactions | Metabolites | Genes | Regulation | Thermodynamics | Reference(s) |
|---|---|---|---|---|---|---|
| Aspergillus nidulans | 1,095 | 732 | 666 | [55] | ||
| A. niger | 2,168 | 1,045 | 871 | [56] | ||
| A. oryzae | 1,846 | 1,073 | 1,314 | [53] | ||
| Bacillus subtilis | 754 | 637 | 614 | [57] | ||
| Corynebacterium glutamicum | 495 | 408 | 411 | [58] | ||
| Escherichia coli | 2,077 | 1,039 | 1,260 | ✓ | ✓ | [12] |
| Lactococcus lactis | 621 | 422 | 358 | [59] | ||
| Mannheimia succinicproducens | 373 | 352 | 335 | [60] | ||
| Saccharomyces cerevisiae | 1,446 | 1,013 | 800 | [61] | ||
| 1,489 | 972 | 805 | ✓ | [10] | ||
| Streptomyces coelicolor | 700 | 500 | 971 | [62] |
While there is an increasing availability of stoichiometric models for a range of organisms, for E. coli and S. cerevisiae, additional model features are being added to improve the models predictive capabilities. The regulatory structure that controls the expression of different metabolic enzymes is not explicitly captured in stoichiometric models. In E. coli, Boolean logic representations of transcriptional regulation have been able to predict binary expression changes in response to various stimuli [8]. Such models contain at least some regulatory information for approximately half the metabolic reaction and can be trained using high throughput phenotyping data and transcriptome measurements [9]. Similar efforts have been carried out in yeast, accounting for 55 transcription factors and nutrient conditions [10].
Another improvement has been identifying the reversibility/irreversibility of enzymatic reactions to constrain the model from predicting thermodynamically infeasible reactions. This is achieved by calculating the thermodynamic free energy of metabolites [11,12]. Reactions that have a significant drop in free energy in the forward direction are irreversible, while reactions without significant changes in free energy will be reversible, and subject to mass action.
Currently, these models are limited to quantify metabolic fluxes, which surely are of significant value for biochemical production. However, many processes that may be of interest to engineers are not described by such models. In protein production, for example, various sorting/quality control processes are controlled by interactions with other proteins, not transcriptionally controlled, nor can these processes be predicted by mass action. Other processes, such as DNA repair, cell division, signaling pathways, stress responses, and trafficking between compartments have inputs and outputs that may be partially transcriptional in nature, but have many other interactions that cannot be described by current FBA-type models (a notable exception is the description of the JAK-STAT signaling pathway by stoichiometric models [13]).
Protein-protein interaction networks provide a scaffold for models that describe non-metabolic processes in the cell and are currently a useful resource for data analysis (described below). Such networks do not have the quantitative predictive power as GSMMs, but serve as scaffolds to identify which proteins may be relevant without specific knowledge of the mechanism. Protein interaction networks are available for several industrial organisms, such as B. subtilis, E. coli, S. cerevisiae, and S. pombe, and are stored at BioGrid, a central repository for these and other types of interactions (www.thebiogrid.org) [14]. These databases are being significantly upgraded by various efforts and this will lead to a more complete mapping of many cellular processes and move toward the usefulness of protein-protein interaction data in predicting cellular processes.
Implementation
Predictive models can offer hypotheses for improving the desired phenotype. In general, implementation has consisted of (a) strong plasmid-based over-expression, (b) knockout, or (c) introduction of a heterologous gene. As we move forward with more detailed models, it will be desirable to move from the binary options of strong over-expression and deletion to fine-tuned alteration of constitutive expression levels, as well as sophisticated responsive promoters to adjust protein expression through the course of the fermentation (Figure 2).
Figure 2. Biological ‘parts’ for controlling synthesis / degradation rates in the central dogma paradigm.
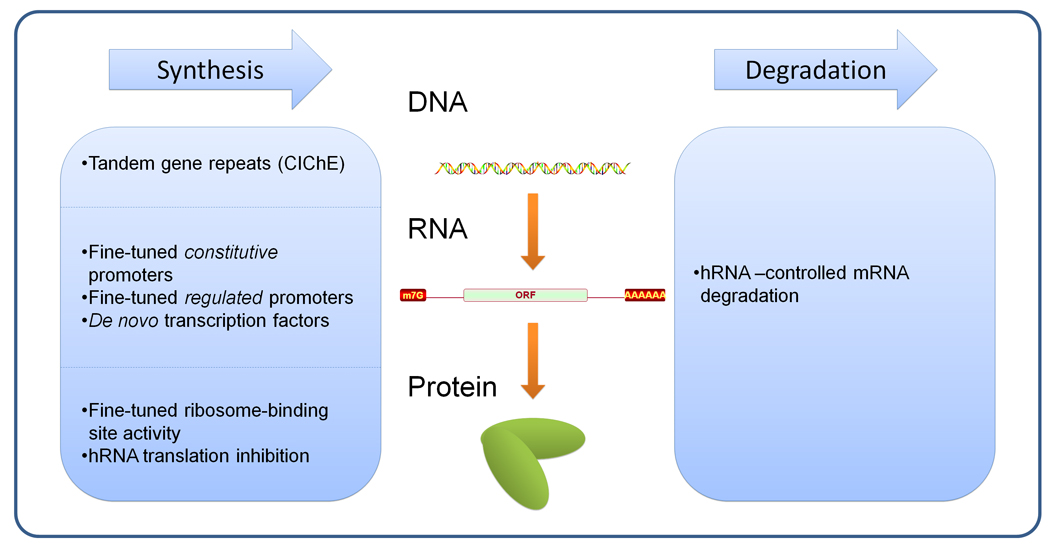
Well-characterized biological components are necessary to control DNA, mRNA, and proteins in the cell. These biological components should be fine-tunable, independent of other implementations of the same control scheme, and robust in a variety of circumstances. The left column contains technologies that affect the synthesis rates, and the right column explains biological components that affect the degradation rates. Technologies are discussed in detail in the text. hRNA – Hammerhead RNA.
Well-characterized, robust expression systems will aid in this development. The Standard Biological Parts initiative (http://partsregistry.org/) is an open-source repository of biological “parts” (promoters, ribosome-binding sites, reporters, etc.) with input/output data compiled for convenient use of the part [15]. Abstracted parameters, such as polymerases per second (PoPs), are more useful in design than gfp fluorescence or β-galactosidase units that are commonly reported in research literature. This catalog relies on a cloning strategy called Biobricks which follows an ordered design experience that is less prone to user error than ad hoc cloning [16]. Biobricks has been been used by engineers with no cloning experience to design and implement cellular engineering strategies as can be seen by the student designs in the International Genetically Engineered Machine Competition (iGEM) [17].
New expression systems are being developed that expand the precision of expression levels and allow novel regulation. These approaches allow (a) fine tuned expression rather than just strong over-expression and (b) scalability, because the control systems can be used orthogonally, allowing extensive multiplexing rather than only a few options.
Constitutive expression over a range of expression levels has been achieved through mutagenesis of existing promoters, followed by characterization of the derived promoter library. This has been achieved in E. coli [18] and yeast [19] and the concept should be extendible to most relevant microorganisms. Expression strength can also be controlled by changing the copy number of tandem repeats on the chromosome using an approach called Chemically Induced Chromosomal Evolution (CIChE) [20]. CIChE has the added advantage of stable copy numbers, whereas plasmid copy number can fluctuate significantly.
Beyond constitutive expression, regulatable promoters allow engineers to dynamically control expression levels in responses to environmental changes, making new bioprocessing strategies possible. Similar to the mutation-based diversification used to create the constitutive promoters, Ellis, et. al. has created libraries of tetracycline and lactose responsive promoters in E. coli with altered response characteristics to their respective inducers [21]. The researchers then design a gene circuit and implement it with components of the library, a simple example of design-based engineering. Nevoigt et. al. has adjusted the oxygen sensitivity of the DAN1 promoter, allowing similar design possibilities [22].
De novo transcriptional regulatory networks can also be introduced into the cell, to co-regulate different groups of genes in parallel. This has been achieved by evolving zinc-finger transcription factors (TF) to recognize non-native promoter sequences [23]. Conceptually, an unlimited number of transcription factor/promoter combinations could be used independently to regulate genes. (for a more extensive review of these possibilities, see [24]). Combined with the mutation-based diversification of promoter characteristics, a range of promoter strengths could be made for a heterologous zinc-finger TF in both prokaryotes and eukaryotes.
Besides mRNA synthesis, other processes can affect expression strength. Hammerhead RNA (hRNA) detectors/regulators can control mRNA stability and translation efficiency (see [25] for an overview of RNA-based tools for controlling protein expression). These hRNAs can be evolved to detect a wide range of small molecule substrates, allowing different chemical inputs to affect protein synthesis. hRNAs are also scalable because different input/output hRNAs can operate without unintended cross-talk. Protein synthesis can also be controlled by altering the RBS and a recent model can be used to design specific translation activities [26].
Other means of controlling cellular function have moved beyond controlling RNA/protein abundance or are based on inheritance. To improve flux through a metabolic pathway, Dueber et. al. has developed a method to tether sequential heterologous enzymes in series [27]. By this, the pathways mimic the biological process of channeling providing increased local concentrations of intermediates and decreased average concentrations which (a) increases flux, (b) decreases toxicity of intermediates, and (c) avoids undesired side reactions. Light-controlled conditional protein interactions can allow direct control over the activity of enzymes, independent of transcription/translation [28,29]. Finally, genetic stability can be problematic where intermediates or products of a heterologous pathway are toxic to the cell. By switching from plasmids to tandem gene repeats on the chromosome, the CIChE method can extend genetic stability by tenfold [20].
Analysis
As models are not 100% predictive and molecular implementation may not perform as expected, the engineered cells must be analyzed after implementation. Over the past decade, technological advancements have enabled –omics measurement, the characterization of all instances of a class of molecules in a cell. In the wake of this massive amount of data that is generated, model-based analytic tools are allowing researchers to compress high dimensionality data to simpler, biologically-based insights, which can ultimately improve the model.
Transcriptomics examines the expression level of mRNAs at a specific physical condition. Various technologies have been developed to quantify the transcriptome, including hybridization (DNA microarray) and sequence-based approaches. The development of novel high-throughput sequencing methods has provided a new tool for both mapping and quantifying transcriptomes. This method, termed RNA-Seq (RNA sequencing), is being developed to profile transcriptomes and allows high throughput sequencing of cDNA in order to quantify mRNA in the cell [30]. Willenbrock et al. has shown that RNA-Seq correlates well with traditional microarray hybridization using synthetic RNA samples [31]. As well, the sensitivity in terms of reproducibility and relative quantification were equivalent. As microarray analysis requires an available genome sequence, custom chips for each organism, and complex statistical tools, RNA-seq, though a platform still under active development, overcomes these hybridization limitations, as well as enables analysis of sequence variants (for example SNPs [32]), and provides quantitative estimates of RNA abundance in the cell [33].
Proteomics attempts to quantify the level of all proteins in the cell. In the past, mRNA levels were used as a (sometimes poor) proxy for protein levels. Nowadays, methods combining mass spectrometry and liquid chromatography, either gel-based or non gel-based protein separation, are used to improve complex proteome identification. Since proteomics methods were developed in the middle of 1990’s, interactomics [34,35] and phosphoproteomics [36] have also become interesting for many research applications, characterizing the state of a protein. Now, various methods are developed for proteomics such as MALDI-TOF for peptide mass finger printing, and electrospray ionization (ESI) Fourier transform ion cyclotron resonance (FT-ICR) coupling with tandem mass spectrometry (MS/MS) for peptide identification. However, quantitative proteomic data must be validated by independent methods in order to overcome the heterogeneity of the biological samples [37]. Proteome analysis of Saccharomyces cerevisiae response to carbon and nitrogen limitation was done by using shotgun approach, multidimensional protein identification technology (MudPIT), in combination with the metabolic or in vitro labeling of proteins. The comparison of transcript and protein levels clearly shows that up-regulation in response to glucose limitation is transcriptionally controlled while up-regulation in response to nitrogen limitation is controlled at the post transcriptional level [38]. In a similar study the combination of transcriptome and proteome analysis by MudPIT allowed for global mapping of the regulatory network associated with the key protein kinase Snf1p in yeast [39].
Similar to proteomics, sophisticated mass spectrometry permits metabolomic studies. Metabolomics is achieved by quantifying cellular metabolites and systemically studying the unique chemical fingerprints that specific cellular processes leave behind. The methods coupling electrospray ionization with mass spectrometry is well developed and used for detecting the metabolomes in S. cerevisiae. With ESI-MS, up to 84% of the metabolites in S. cerevisiae can be identified [40]. A complete quantitative metabolomics workflow in a microplate format has recently been published [41]. Although the protocol is developed and validated for S. cerevisiae, this protocol can be adapted and applied for high throughput metabolomes studies in different organisms.
With this massive flow of complex data from mRNA, protein, and metabolite levels, integrative analysis is crucial and needed to prevent bottlenecks during the analytical step [42,43]. Integration comes from leveraging existing information about the cell through models (as described above) to interpret –omics data. At present, genotype-to-phetotype databases (G2P) are currently being improved using various integrative tools. The core modules for construction and improving genotype-to-phenotype databases include object model, exchange (data) format, ontology, globally unique identifier (GUID) and web service [44]. According to the availability of databases, a great number of mathematical models and integrative strategies have been developed and established as systems biology tools to integrate and visualize multidimensional data for examples PromoT [45], Metatool [46], PathSys and BiologicalNetworks [35] and YANAsquare [47]. Subnetwork analysis allows complex system with multistationarity such as cellular networks to be analyzed separately [48]. An algorithm to identify key metabolites and subnetwork structures in a biological response has been successfully developed by integrating transcriptome with topological information from genome-scale metabolic model without direct measurement of metabolite concentrations [49,50].
Many models are significantly revised after incorporating new biological information from the –omics analysis [51]. For example, a metabolic model of A. niger was significantly improved by combining metabolome and metabolic flux data with existing genome and literature data [52,53]. By integrating metabolic flux and transcriptome data with both interaction networks and stoichiometric models, a new approach to correlate mRNA and metabolic flux data was developed [34]. Through integrated data analysis, a better understanding of the biological system is possible, allowing more realistic models to be developed for bio-based industries.
Conclusions & Future Directions
As synthetic genomes are quickly becoming a reality [54], we are then faced with an analogous question as 30 years ago. We know how to synthesize DNA, but what DNA should we synthesize? These research efforts mentioned here are qualitatively changing the nature of the cyclic engineering paradigm for industrial microbes. Thanks to increased analysis capability, much more information is gathered in each iteration. As well, with better models and molecular tools, better hypotheses and more controlled implementation is now possible.
Further developments in models that capture the change in metabolite concentrations will be useful in predicting flux distribution as well as potentially toxic levels of a particular metabolite. To improve molecular implementation, a larger catalog of well characterized parts will need to be available to the research community. The standard biological parts initiative has, thus far, been an open source initiative. With suitable intellectual property security, private biological “parts” companies could do rigorous characterization and sell “components” to microbial engineers, significantly accelerating the accumulation of available parts. And finally, the use of –omics data to create phenotype-predictive models of protein-protein interactions would significantly extend the biological processes that could be engineered. In the end, the far, far end, experimental analysis may become an academic exercise for undergraduate, as is the state in electrical circuit design. At this point, we will have reached a paradigm of design-based engineering of industrial microbes.
Figure 3. The conceptual work flow of integrated analysis of –omics data to formalize genotype-to-phenotype database.

Advances in -omics measurement lead to the accumulation of high throughput -omics data. With systematic and integrative analysis tools, genotype-to-phenotype database are currently being developed under the core modules outlined in this figure. Standardized object models and data formats facilitate the exchange of information between different data systems and are essential for unambiguous transmission of data between computers. A well defined ontology enables the representation of domain-specific knowledge, as well as, the relationship between those domains and leads to powerful database searching. GUIDs solve data integration problems that result from ambiguity in name, or identity, of biological concepts and objects. Standard protocols for web service afford machine-to-machine interaction over the internet and simplify the task of exploring distributed data, forming the basis of the service-oriented architecture (SOA) [44].
Acknowledgements
We thank NIH F32 Kirschstein NRSA fellowship, Thailand Science and Technology Ministry, The Knut and Alice Wallenberg Foundation and the Chalmers Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Patil KR, Akesson M, Nielsen J. Use of genome-scale microbial models for metabolic engineering. Curr Opin Biotechnol. 2004;15:64–69. doi: 10.1016/j.copbio.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 2.Feist AM, Palsson BO. The growing scope of applications of genome-scale metabolic reconstructions using Escherichia coli. Nat Biotechnol. 2008;26:659–667. doi: 10.1038/nbt1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Patil KR, Rocha I, Forster J, Nielsen J. Evolutionary programming as a platform for in silico metabolic engineering. BMC Bioinformatics. 2005;6:308. doi: 10.1186/1471-2105-6-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pharkya P, Burgard AP, Maranas CD. OptStrain: a computational framework for redesign of microbial production systems. Genome Res. 2004;14:2367–2376. doi: 10.1101/gr.2872004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alper H, Miyaoku K, Stephanopoulos G. Construction of lycopene-overproducing E. coli strains by combining systematic and combinatorial gene knockout targets. Nat Biotechnol. 2005;23:612–616. doi: 10.1038/nbt1083. [DOI] [PubMed] [Google Scholar]
- 6.Bro C, Regenberg B, Forster J, Nielsen J. In silico aided metabolic engineering of Saccharomyces cerevisiae for improved bioethanol production. Metab Eng. 2006;8:102–111. doi: 10.1016/j.ymben.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 7. Asadollahi MA, Maury J, Patil KR, Schalk M, Clark A, Nielsen J. Enhancing sesquiterpene production in Saccharomyces cerevisiae through in silico driven metabolic engineering. Metab Eng. 2009;11:328–334. doi: 10.1016/j.ymben.2009.07.001. This example uses a genome scale model of S. cerevisiae to predict genomic alterations that will increase flux to isoprenoid precursors.
- 8.Covert MW, Palsson BO. Transcriptional regulation in constraints-based metabolic models of Escherichia coli. J Biol Chem. 2002;277:28058–28064. doi: 10.1074/jbc.M201691200. [DOI] [PubMed] [Google Scholar]
- 9.Covert MW, Knight EM, Reed JL, Herrgard MJ, Palsson BO. Integrating high-throughput and computational data elucidates bacterial networks. Nature. 2004;429:92–96. doi: 10.1038/nature02456. [DOI] [PubMed] [Google Scholar]
- 10.Herrgard MJ, Lee BS, Portnoy V, Palsson BO. Integrated analysis of regulatory and metabolic networks reveals novel regulatory mechanisms in Saccharomyces cerevisiae. Genome Res. 2006;16:627–635. doi: 10.1101/gr.4083206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Henry CS, Broadbelt LJ, Hatzimanikatis V. Thermodynamics-based metabolic flux analysis. Biophys J. 2007;92:1792–1805. doi: 10.1529/biophysj.106.093138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Feist AM, Henry CS, Reed JL, Krummenacker M, Joyce AR, Karp PD, Broadbelt LJ, Hatzimanikatis V, Palsson BO. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol Syst Biol. 2007;3:121. doi: 10.1038/msb4100155. One of the most advanced models to date. This E. coli model covers 28% of the genes in the genome and contains regulatory information and has been checked for thermodynamic consistency.
- 13. Papin JA, Palsson BO. The JAK-STAT signaling network in the human B-cell: an extreme signaling pathway analysis. Biophys J. 2004;87:37–46. doi: 10.1529/biophysj.103.029884. This paper implements a stoichiometric model typically used to model metabolism in a signaling pathway. This may be a useful way to model other cellular processes.
- 14. Stark C, Breitkreutz BJ, Reguly T, Boucher L, Breitkreutz A, Tyers M. BioGRID: a general repository for interaction datasets. Nucleic Acids Research. 2006;34:D535–D539. doi: 10.1093/nar/gkj109. BioGRID is an online resource for biological interactions. The database contains interaction data for many industrially relevent organisms, and contains physical as well as genetic interactions.
- 15. Canton B, Labno A, Endy D. Refinement and standardization of synthetic biological parts and devices. Nat Biotechnol. 2008;26:787–793. doi: 10.1038/nbt1413. Standard Biological Parts will be essential to design-based engineering. This paper describes a strategy for cataloging parts in a way that is useful for cellular design.
- 16. Shetty RP, Endy D, Knight TF., Jr Engineering BioBrick vectors from BioBrick parts. J Biol Eng. 2008;2:5. doi: 10.1186/1754-1611-2-5. This paper describes the use of Biobricks as a platform for expressing proteins. The value of this approach is it systematizes the process, minimizing the opportunity for design errors that often plague cloning efforts.
- 17.Goodman C. Engineering ingenuity at iGEM. Nat Chem Biol. 2008;4:13. doi: 10.1038/nchembio0108-13. [DOI] [PubMed] [Google Scholar]
- 18.Alper H, Fischer C, Nevoigt E, Stephanopoulos G. Tuning genetic control through promoter engineering. Proc Natl Acad Sci U S A. 2005;102:12678–12683. doi: 10.1073/pnas.0504604102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nevoigt E, Kohnke J, Fischer CR, Alper H, Stahl U, Stephanopoulos G. Engineering of promoter replacement cassettes for fine-tuning of gene expression in Saccharomyces cerevisiae. Appl Environ Microbiol. 2006;72:5266–5273. doi: 10.1128/AEM.00530-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tyo KE, Ajikumar PK, Stephanopoulos G. Stabilized gene duplication enables long-term selection-free heterologous pathway expression. Nat Biotechnol. 2009;27:760–765. doi: 10.1038/nbt.1555. Tandem genes offer a preferrable approach to plasmids for multicopy expression of genes. CIChE allows high genetic stability, tunable expression, and does not require a selectable marker.
- 21. Ellis T, Wang X, Collins JJ. Diversity-based, model-guided construction of synthetic gene networks with predicted functions. Nat Biotechnol. 2009;27:465–471. doi: 10.1038/nbt.1536. This study generated a library of promoters with different response properties to their inducers. These characterized promoters were used to design a genetic circuit directly from computer simulation.
- 22.Nevoigt E, Fischer C, Mucha O, Matthaus F, Stahl U, Stephanopoulos G. Engineering promoter regulation. Biotechnol Bioeng. 2007;96:550–558. doi: 10.1002/bit.21129. [DOI] [PubMed] [Google Scholar]
- 23. Beerli RR, Dreier B, Barbas CF., 3rd Positive and negative regulation of endogenous genes by designed transcription factors. Proc Natl Acad Sci U S A. 2000;97:1495–1500. doi: 10.1073/pnas.040552697. Engineered zinc finger proteins are a useful way to impose new regulation on genes. This system can work in both bacteria and fungi.
- 24.Lu TK, Khalil AS, Collins JJ. Next-generation synthetic gene networks. Nat Biotechnol. 2009;27:1139–1150. doi: 10.1038/nbt.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Win MN, Liang JC, Smolke CD. Frameworks for programming biological function through RNA parts and devices. Chem Biol. 2009;16:298–310. doi: 10.1016/j.chembiol.2009.02.011. This paper describes the design strategies using RNA hammerheads. This strategy should have extensive use in a wide variety of organisms and in many applications.
- 26. Salis HM, Mirsky EA, Voigt CA. Automated design of synthetic ribosome binding sites to control protein expression. Nat Biotechnol. 2009;27:946–950. doi: 10.1038/nbt.1568. A ribosome-binding site model is able to accurately predict translational activity, based on DNA sequence. This allows predictive ways to alter protein synthesis properties in a genetic circuit.
- 27.Dueber JE, Wu GC, Malmirchegini GR, Moon TS, Petzold CJ, Ullal AV, Prather KL, Keasling JD. Synthetic protein scaffolds provide modular control over metabolic flux. Nat Biotechnol. 2009;27:753–759. doi: 10.1038/nbt.1557. [DOI] [PubMed] [Google Scholar]
- 28.Levskaya A, Weiner OD, Lim WA, Voigt CA. Spatiotemporal control of cell signalling using a light-switchable protein interaction. Nature. 2009;461:997–1001. doi: 10.1038/nature08446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Levskaya A, Chevalier AA, Tabor JJ, Simpson ZB, Lavery LA, Levy M, Davidson EA, Scouras A, Ellington AD, Marcotte EM, et al. Synthetic biology: engineering Escherichia coli to see light. Nature. 2005;438:441–442. doi: 10.1038/nature04405. [DOI] [PubMed] [Google Scholar]
- 30.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Willenbrock H, Salomon J, Sokilde R, Barken KB, Hansen TN, Nielsen FC, Moller S, Litman T. Quantitative miRNA expression analysis: comparing microarrays with next-generation sequencing. RNA. 2009;15:2028–2034. doi: 10.1261/rna.1699809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barbazuk WB, Emrich SJ, Chen HD, Li L, Schnable PS. SNP discovery via 454 transcriptome sequencing. The Plant Journal. 2007;51:910–918. doi: 10.1111/j.1365-313X.2007.03193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, Wang X, Bodeau J, Tuch BB, Siddiqui A, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Meth. 2009;6:377–382. doi: 10.1038/nmeth.1315. Describe a method for single-cell gene expression profiling assay and compare mRNA-Seq with microarray techniques.
- 34. Moxley JF, Jewett MC, Antoniewicz MR, Villas-Boas SG, Alper H, Wheeler RT, Tong L, Hinnebusch AG, Ideker T, Nielsen J, et al. Linking high-resolution metabolic flux phenotypes and transcriptional regulation in yeast modulated by the global regulator Gcn4p. Proceedings of the National Academy of Sciences. 2009;106:6477–6482. doi: 10.1073/pnas.0811091106. An excellent example of model that correlate mRNA with metabolic data including interaction network and flux determination and the model is used to identify specific mechanisms which genetic regulation mediate metabolic flux phenotype.
- 35.Baitaluk M, Qian X, Godbole S, Raval A, Ray A, Gupta A. PathSys: integrating molecular interaction graphs for systems biology. BMC Bioinformatics. 2006;7:55. doi: 10.1186/1471-2105-7-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jacobs J, Monroe M, Qian W, Shen Y, Anderson G, Smith R. Ultra-sensitive, high throughput and quantitative proteomics measurements. International Journal of Mass Spectrometry. 2005;240:195–212. [Google Scholar]
- 37.Meyer HE, Stühler K. High-performance Proteomics as a Tool in Biomarker Discovery. PROTEOMICS. 2007;7:18–26. doi: 10.1002/pmic.200700183. [DOI] [PubMed] [Google Scholar]
- 38.Kolkman A, Daran-Lapujade P, Fullaondo A, Olsthoorn MMA, Pronk JT, Slijper M, Heck AJR. Proteome analysis of yeast response to various nutrient limitations. Mol Syst Biol. 2006;2 doi: 10.1038/msb4100069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Usaite R, Jewett MC, Oliveira AP, Yates JR, 3rd, Olsson L, Nielsen J. Reconstruction of the yeast Snf1 kinase regulatory network reveals its role as a global energy regulator. Mol Syst Biol. 2009;5:319. doi: 10.1038/msb.2009.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Højer-Pedersen J, Smedsgaard J, Nielsen J. The yeast metabolome addressed by electrospray ionization mass spectrometry: Initiation of a mass spectral library and its applications for metabolic footprinting by direct infusion mass spectrometry. Metabolomics. 2008;4:393–405. [Google Scholar]
- 41. Ewald JC, Heux Sp, Zamboni N. High-Throughput Quantitative Metabolomics: Workflow for Cultivation, Quenching, and Analysis of Yeast in a Multiwell Format. Analytical Chemistry. 2009;81:3623–3629. doi: 10.1021/ac900002u. Provide a recently developed protocol for high throughput metabolomes study.
- 42. Prokisch H, Scharfe C, Camp DG, II, Xiao W, David L, Andreoli C, Monroe ME, Moore RJ, Gritsenko MA, Kozany C, et al. Integrative Analysis of the Mitochondrial Proteome in Yeast. PLoS Biol. 2004;2:e160. doi: 10.1371/journal.pbio.0020160. Propose the systematic integrative analysis of the mitochondrial proteome in Saccharomyces cerevisiae which is done by perform a comparative analysis of their proteomic dataset with gene expression analysis and quantitative deletion phenotype screening dataset by another research group.
- 43.Shields DC, O'Halloran AM. Integrating genotypic data with transcriptomic and proteomic data. Comparative and Functional Genomics. 2002;3:22–27. doi: 10.1002/cfg.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Thorisson GA, Muilu J, Brookes AJ. Genotype-phenotype databases: challenges and solutions for the post-genomic era. Nat Rev Genet. 2009;10:9–18. doi: 10.1038/nrg2483. Review methods and relevant technologies which are required and currently improved for establishing genotype-to-phenotype databases.
- 45.Mirschel S, Steinmetz K, Rempel M, Ginkel M, Gilles ED. PROMOT: modular modeling for systems biology. Bioinformatics. 2009;25:687–689. doi: 10.1093/bioinformatics/btp029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.von Kamp A, Schuster S. Metatool 5.0: fast and flexible elementary modes analysis. Bioinformatics. 2006;22:1930–1931. doi: 10.1093/bioinformatics/btl267. [DOI] [PubMed] [Google Scholar]
- 47.Schwarz R, Liang C, Kaleta C, Kuhnel M, Hoffmann E, Kuznetsov S, Hecker M, Griffiths G, Schuster S, Dandekar T. Integrated network reconstruction, visualization and analysis using YANAsquare. BMC Bioinformatics. 2007;8:313. doi: 10.1186/1471-2105-8-313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Conradi C, Flockerzi D, Raisch, Stelling Subnetwork analysis reveals dynamic features of complex (bio)chemical networks. Proceedings of the National Academy of Sciences. 2007;104:19175–19180. doi: 10.1073/pnas.0705731104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Patil KR, Nielsen J. Uncovering transcriptional regulation of metabolism by using metabolic network topology. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:2685–2689. doi: 10.1073/pnas.0406811102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oliveira A, Patil K, Nielsen J. Architecture of transcriptional regulatory circuits is knitted over the topology of bio-molecular interaction networks. BMC Systems Biology. 2008;2:17. doi: 10.1186/1752-0509-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucl. Acids Res. 2008 doi: 10.1093/nar/gkn923. gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Andersen M, Nielsen M, Nielsen J. Metabolic model integration of the bibliome, genome,metabolome and reactome of Aspergillus niger. Molecular Systems Biology. 2008;4:178. doi: 10.1038/msb.2008.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Vongsangnak W, Olsen P, Hansen K, Krogsgaard S, Nielsen J. Improved annotation through genome-scale metabolic modeling of Aspergillus oryzae. BMC Genomics. 2008;9:245. doi: 10.1186/1471-2164-9-245. Developed annotation strategy in this paper allows a genome scale model of Aspergillus oryzae to be revised and reconstructed. This improved annotation reduce the number of hypothetical proteins and newly predicted genes are assign to new putative functions in the model.
- 54.Gibson DG, Benders GA, Andrews-Pfannkoch C, Denisova EA, Baden-Tillson H, Zaveri J, Stockwell TB, Brownley A, Thomas DW, Algire MA, et al. Complete chemical synthesis, assembly, and cloning of a Mycoplasma genitalium genome. Science. 2008;319:1215–1220. doi: 10.1126/science.1151721. [DOI] [PubMed] [Google Scholar]
- 55.David H, Ozcelik IS, Hofmann G, Nielsen J. Analysis of Aspergillus nidulans metabolism at the genome-scale. BMC Genomics. 2008;9:163. doi: 10.1186/1471-2164-9-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Andersen MR, Nielsen ML, Nielsen J. Metabolic model integration of the bibliome, genome, metabolome and reactome of Aspergillus niger. Mol Syst Biol. 2008;4:178. doi: 10.1038/msb.2008.12. This model was built using a variety of data sources beyond genomic sequence and biochemical evidence. As built, this model demonstrates that sophisticated models can now be developed rapidly using Systems Biology approaches.
- 57.Park S, Schilling C, Palsson B. Compositions and methods for modeling Bacillus subtilis metabolism. US Patent. 2003
- 58.Kjeldsen KR, Nielsen J. In silico genome-scale reconstruction and validation of the Corynebacterium glutamicum metabolic network. Biotechnol Bioeng. 2009;102:583–597. doi: 10.1002/bit.22067. [DOI] [PubMed] [Google Scholar]
- 59.Oliveira AP, Nielsen J, Forster J. Modeling Lactococcus lactis using a genome-scale flux model. BMC Microbiol. 2005;5:39. doi: 10.1186/1471-2180-5-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hong SH, Kim JS, Lee SY, In YH, Choi SS, Rih JK, Kim CH, Jeong H, Hur CG, Kim JJ. The genome sequence of the capnophilic rumen bacterium Mannheimia succiniciproducens. Nat Biotechnol. 2004;22:1275–1281. doi: 10.1038/nbt1010. [DOI] [PubMed] [Google Scholar]
- 61. Nookaew I, Jewett MC, Meechai A, Thammarongtham C, Laoteng K, Cheevadhanarak S, Nielsen J, Bhumiratana S. The genome-scale metabolic model iIN800 of Saccharomyces cerevisiae and its validation: a scaffold to query lipid metabolism. BMC Syst Biol. 2008;2:71. doi: 10.1186/1752-0509-2-71. A recent genome-scale model of yeast. It specifically accounts for lipid metabolism and is useful in the study of lipid disease states.
- 62.Borodina I, Krabben P, Nielsen J. Genome-scale analysis of Streptomyces coelicolor A3(2) metabolism. Genome Res. 2005;15:820–829. doi: 10.1101/gr.3364705. [DOI] [PMC free article] [PubMed] [Google Scholar]