

Abstract
Although malfunction of spinal cord water channels (aquaporins, AQP) likely contributes to severe disturbances in ion/water homeostasis after spinal cord injury (SCI), their roles are still poorly understood. Here we report and discuss the potential significance of changes in the AQP4 expression in human SCI that generates GFAP-labeled astrocytes devoid of AQP4, and GFAP-labeled astroglia that overexpress AQP4.
We used a rat model of contusion SCI to study observed changes in human SCI. AQP4-negative astrocytes are likely generated during the process of SCI-induced replacement of lost astrocytes, but their origin and role in SCI remains to be investigated. We found that AQP4-overexpression is likely triggered by hypoxia. Our transcriptional profiling of injured rat cords suggests that elevated AQP4-mediated water influx accompanies increased uptake of chloride and potassium ions which represents a protective astrocytic reaction to hypoxia. However, unbalanced water intake also results in astrocytic swelling that can contribute to motor impairment, but likely only in milder injuries. In severe rat SCI, a low abundance of AQP4-overexpressing astrocytes was found during the motor recovery phase. Our results suggest that severe rat contusion SCI is a better model to analyze AQP4 functions after SCI. We found that AQP4 increases in the chronic post-injury phase are associated with the development of pain-like behavior in SCI rats, while possible mechanisms underlying pain development may involve astrocytic swelling-induced glutamate release. In contrast, the formation and size of fluid-filled cavities occurring later after SCI does not appear to be affected by the extent of increased AQP4 levels. Therefore, the effect of therapeutic interventions targeting AQP4 will depend not only on the time interval after SCI or animal models, but also on the balance between protective role of increased AQP4 in hypoxia and deleterious effects of ongoing astrocytic swelling.
Keywords: Human Spinal Cord Injury, AQP4-negative astrocytes, Interleukin 1 receptor antagonist, AQP4-overexpressing astrocytes, Cytotoxic edema, Hypoxia, Bumetanide, Pain, Fluid-filled cavity, Motor recovery
I. Why study Aquaporins (AQPs) in spinal cord injury (SCI)?
It is estimated that there are 2.5 million people worldwide living with a SCI, and that 130,000 new injuries occur each year (Thuret et al., 2006). Devastating losses of motor, sensory and autonomic functions cannot be reversed, principally because of the lack of any effective therapeutic treatments.
Methylprednisolone is the only pharmacological agent that has been shown to produce a clinical improvement after SCI (Cayli et al., 2004) in a randomized blinded research design, but it is not approved by the FDA for SCI treatment (Rozet, 2008), and the actual benefits are debated (Miller, 2008; Rozet, 2008). Therefore, effective therapeutic interventions after SCI are greatly needed.
A dramatic SCI-induced deregulation of ion/water homeostasis significantly contributes to the final functional impairments (LoPachin et al., 1999). Therefore, a better understanding of the roles that AQPs play in the regulation of ion/water homeostasis in spinal cords may give important insights into some important outcomes resulting from SCI, including edema, the formation of fluid-filled progressively enlarging syringes, and final motor or sensory impairments. Detailed descriptions of the mechanisms underlying control of the ion water/water transport by different AQPs in the CNS is presented in other articles in this special issue of Neuroscience, so this study is primarily focused on describing current findings that implicate AQPs in the subsequent pathophysiology of SCI.
II. AQPs in spinal cords
Rodent spinal cords express transcripts for several AQPs: AQP4, AQP5, AQP8 and AQP9 (Oshio et al, 2004). With the exception of AQP5, expression of all other AQPs in rodent spinal cords has also been confirmed at the protein level. AQP4 protein is exclusively expressed in spinal astrocytes (Oshio et al., 2004; Nesic et al., 2006), while AQP9 proteins are found both in astrocytes and in neurons (Baudaut, 2009). AQP8 is found in ependymal cells around the central canal (Oshio et al., 2004), but detailed characterization of AQP8 and AQP9 in rodent spinal cords is lacking. Expression of AQP1 protein in sensory axons within dorsal horns has been confirmed in several studies on rodent spinal cords (Oshio et al., 2004; Shields et al., 2007; and Nesic et al., 2008), but the role of AQP1 in spinal cords remains unknown. AQP1 mRNA is not expected within spinal cords, since the axonal AQP1 protein in spinal cords originate in dorsal root ganglia neurons (Shields et al., 2007). With the exception of AQP4, none of the other AQPs found in rodent spinal cords has been confirmed in human spinal cords, so the challenging and intriguing study of AQPs' roles in different spinal pathologies has only just begun.
III. AQP4
Widely distributed AQP4 expression in white and gray matter spinal cord astrocytes is found in both rodents (mice, Oshio et al., 2004; and rats, Nesic et al., 2006, Fig. 2A) and in humans (Misu et al., 2007; Fig. 1A). There is little doubt that AQP4's normal function in astrocytes is to enable fast water influx or efflux, driven by osmotic or hydrostatic pressures (Solenov et al., 1997; Yang et al., 2008). It has been reported that rat spinal cord tissue has the highest expression levels of AQP4 among all AQP4-expressing organs, including several different brain regions as well as various muscles including heart tissue (Shibuya et al., 2008). Widespread and high expression levels of AQP4 in mammalian spinal cords suggest the importance of potentially altered AQP4 functions in different pathological spinal conditions. AQP4 function has been implicated in spinal cord trauma (Nesic et al., 2006; Saddaoun et al., 2008), spinal cord ischemia (Xu et al., 2008); neuromyelitis optica (NMO) and multiple sclerosis (MS; Hinson et al., 2009; Misu et al., 2007), and amyotrophic lateral sclerosis (ALS; Nicaise et al., 2009). AQP4 expression changes are found in all those pathological conditions, including decreases in AQP4 levels (NMO), increases (ALS), and time-dependent combinations of both elevation and reduction (spinal cord trauma and ischemia). Fig. 1A shows typical AQP4 immunolabeling (brown) in white and gray matter astrocytes in uninjured human spinal cords (astrocytes are labeled for GFAP; pink). The same AQP4 expression pattern was found in cervical, thoracic and lumbar regions (not shown).
Fig. 2.
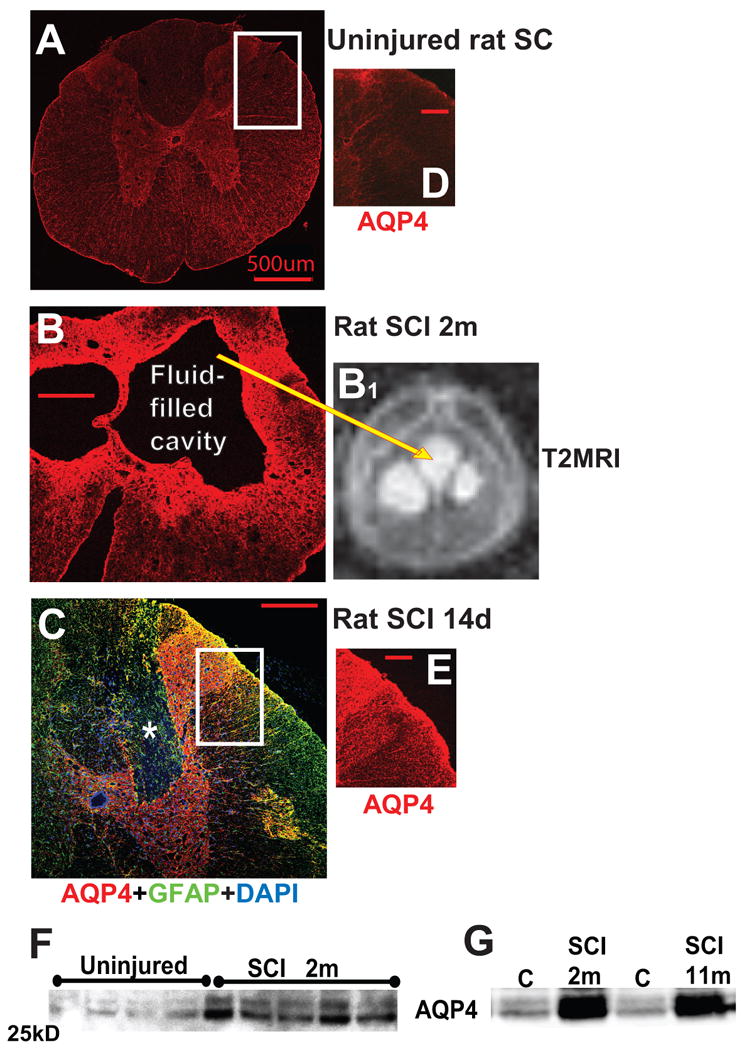
A) Immunofluorescent AQP4 labeling in the thoracic region of an uninjured rat spinal cord. Calibration line: 500 μm. B) AQP4 labeling at the lesion site (T10) of a chronically injured rat spinal cord (2 months after SCI) with large fluid-filled cavity. Calibration line: 200 μm. B1) Fluid accumulated within cavity is visible in T2 MRI image as white, hyperintense signal (see explanation in Fig. 4) in the same injured rat cord used for AQP4 immunolabeling in B. C) AQP4 (red) and GFAP (green) labeling of astrocytes in injured rat spinal cord segment (lesion site; T10) 14d after SCI, showing injury site (dorsal horns and dorsal columns) where GFAP-labeled astrocytes devoid of AQP4 were found (marked with white star). D, E) Comparison of AQP4 in equivalent imaged regions of uninjured (A) and injured (C) spinal cords, marked with white rectangles showing substantially more AQP4 labeling in injured cords where GFAP-labeled astrocytes were also AQP4-positive, indicating AQP4 overexpression in those astroglia. Calibration line: 200 μm. F) AQP4 Western blot of four uninjured and five injured samples 2months after SCI. As expected, the blot showed two clearly identifiable AQP4 bands (M1 and M23), with the M23 isoform being more abundant. Quantitative analysis of both bands before and after SCI and at different time points consistently showed that SCI induced similar increases in both isoforms in SCI rats (n=17 for uninjured and n=28 fro SCI rats). G) A representative Western blot of two uninjured samples (C) 2 and 11 months after sham treatment, and two SCI samples (at the lesion site) 2 and 11 months after SCI showed significant increases in AQP4 levels persisting up to 11 months after SCI (the last time point measured) without decline in the intensity of either M23 or M1, suggesting permanent upregulation of AQP4 in chronically injured rat spinal cords at the lesion site.
Fig. 1.
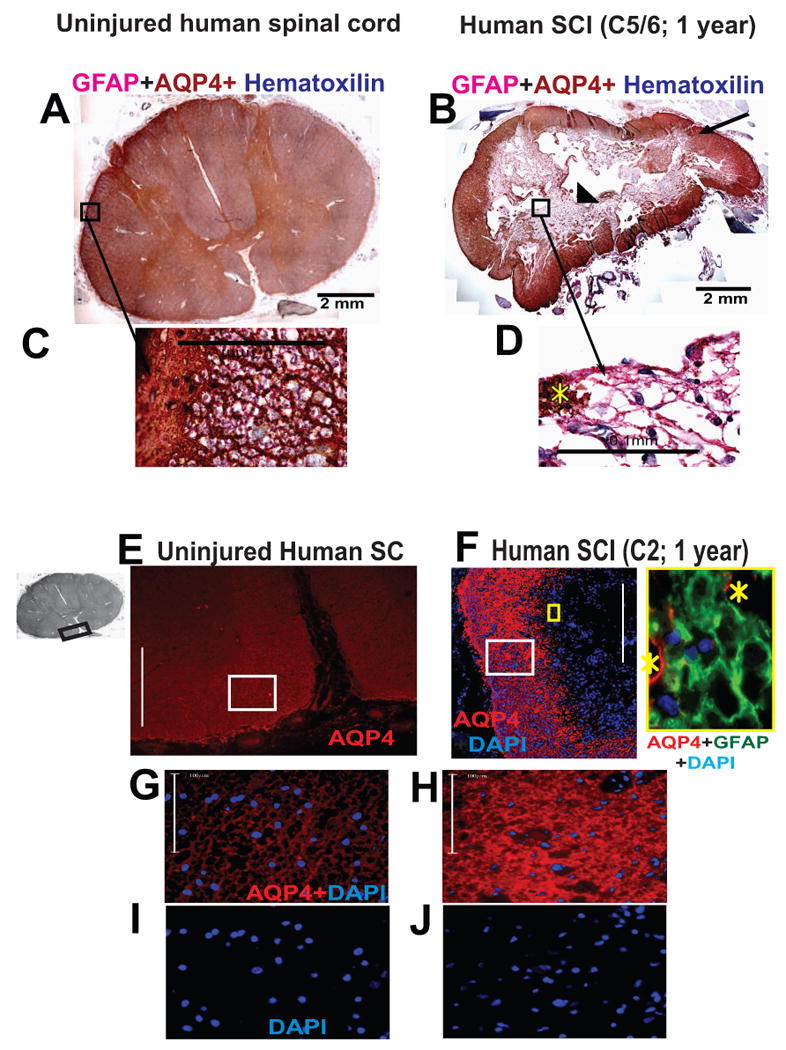
An immunohistochemical procedure was used to detect GFAP and AQP4 in human spinal cord sections. GFAP was detected with an alkaline phosphatase secondary detection system and a substrate that gave a pink precipitate from fast red chromogen. AQP4 was detected with LSAB2 system-horse radish peroxidase and DAB substrate that gave a brown end-product at the site of the target antigen. Counterstaining to detect nuclei was performed with Harris hematoxylin (dark blue). A) Uninjured human spinal cord section labeled with GFAP (red) and AQP4 (brown). Here we present cervical spinal cord section, but the same AQP4 expression pattern was found in thoracic and lumbar sections. B) Injured human spinal cord at the epicenter (C7), isolated from an SCI patient, one year after SCI. This case (No. 29) is described in Guest et al., 2005. C) Large magnification of astrocytes in the region of glia limitans externa (marked with white star) of uninjured cord clearly showing GFAP and AQP4 co-expression. Yellow star marks “islands” of AQP4 labeling among AQP4-negative astrocytes. Calibration line: 100 μm. D) GFAP-labeled astrocytes (pink) around the cyst were devoid of AQP4. Calibration line: 100 μm. E) Immunofluoresecent detection of AQP4 (red) in ventral white matter of the uninjured spinal cords; the region of the spinal cord used in this image is marked with a black rectangle in the gray image of the whole section (insert on the left). Calibration line: 400 μm. F) Equivalent AQP4 labeling in the white mater rim in C2, one year after SCI (Patient No. 28; Guest et al., 2005) showed both AQP4-devoid region at the center of the section and visibly more AQP4 labeling compared to uninjured white or gray matter human spinal cords. Calibration line: 400 μm. The insert framed in yellow depicts high magnification image of astrocytes (GFAP labeled green) devoid of AQP4 (yellow square in the low magnification image). Yellow starts mark AQP4 labeling (red). G) High magnification images of the region marked by the white rectangle in E. Calibration line: 100 μm. H) High magnification images of the region marked by the white rectangle in F. Calibration line: 100 μm. I) Nuclear counterstain (DAPI) in the same image (shown in G). J) Nuclear counterstain (DAPI) in the same image (shown in H).
IV. Human SCI: AQP4-negative and AQP4-overexpressing astrocytes
AQP4 changes in human SCI have not been previously reported. We analyzed cords from three SCI patients suffering from trauma to the cervical segments at C5/6 (survival time 1 year), C2 (survival time 1 year) and C8 (survival time 2 years). Detailed histological analysis of these spinal cords is described in Guest et al., 2005; as patients No. 29, 28 and 59, respectively.
Immunohistochemical analysis of AQP4 labeling in all three injured cords at the lesion site showed the presence of GFAP-labeled astrocytes with and without AQP4. In contrast, GFAP-labeled astrocytes devoid of AQP4 have not been found in adult uninjured spinal cords. We and others have previously reported AQP4-labeled astrocytic processes that are not co-labeled with GFAP, reflecting extensive arborization of astrocytic processes not recognized by GFAP labeling (Nesic et al., 2006; Simard et al., 2003). However, GFAP-labeled processes without AQP4 have not been reported. We found AQP4-negative astrocytes at the lesion epicenter in all analyzed samples from three SCI patients, who differed in age, gender and the extent of SCI (complete vs. incomplete). We cannot rule out that causes other than SCI contributed to the AQP4 changes reported here; however, it is unlikely that three very different pathologies diagnosed as cause of death (e.g. pneumonia in patient #28, heart attack in patient #29 and sepsis in patient #59) all affected spinal cord astrocytes at the lesion site in the same manner, i.e., by inhibiting AQP4 expression. Similarly, it is doubtful that all three SCI patients suffered from the same undiagnosed disease that affected their spinal cord AQP4 in the same way. Furthermore, co-morbidity or deterioration of human spinal cord samples before or after autopsy, as possible causes of AQP4 absence, are also very unlikely because AQP4-negative astrocytes were also found in injured rat spinal cords (as shown in Fig. 2C).
A representative example of AQP4 labeling in human SCI (Fig. 1B, Patient No. 29; Guest et al., 2005) showed typical lesion morphology with a cavity at the epicenter – the cell/tissue-free center of the cord (cells were labeled with hematoxylin, blue). Most of the GFAP-labeled astrocytes (pink, Fig. 1B; marked with arrow head; Fig. 1D) surrounding the cyst were devoid of AQP4 (AQP4 labeling was brown), while astrocytes in the spared white matter rim had intense AQP4 labeling (Fig. 1B; marked with arrow). A similar finding was obtained using immunofluorescent labeling (Fig. 1F); GFAP-labeled astrocytes (green; high magnification insert) were not co-labeled with AQP4 (red) around the lesion epicenter (marked by the nuclear DAPI staining, blue), which was devoid of AQP4 immunolabeling (low magnification image).
AQP4 expression was markedly increased in the spared white matter in a chronically injured human spinal cord (Fig. 1F, SCI patient No. 28; Guest et al., 2005; the injury was at C2, and the survival time was 1 year), compared to the white matter AQP4 in uninjured human spinal cords (Fig. 1E; black rectangle in the image of the whole cord shows ventral white matter region presented here). Large-scale magnification of (Fig. 1H) of the area marked with the white rectangle in Fig. 1F showed dense and intense AQP4 labeling that was significantly higher than in the equivalent white matter region of an uninjured cord (Fig. 1G). AQP4 upregulation likely did not result from the increased density and number of AQP4-expressing astrocytes in the spared white matter because of the similar number of nuclei per area in the peripheral white matter in both uninjured and injured cords (Figs. 1I, J).
Therefore, it appears that chronically injured human spinal cords possess at least two types of GFAP-labeled astroglia: AQP4-negative and AQP4-overexpressing cells. These astrocytes are perhaps interacting, given that we found AQP4-positive and negative GFAP-immunolabeled processes in close proximity (see yellow stars in Fig 1D and 1F, high magnification insert). Alternatively, AQP4-negative astrocytes may have AQP4 proteins localized in some membrane compartments. The heterogeneity of the astrocytic population has been well established, especially in injured cords (elegantly reviewed in White and Jakeman, 2008), but differential AQP4 expression in different subsets of astrocytes has not been reported.
V. AQP4-overexpressing astrocytes
V. A) AQP4-overexpressing and AQP4-negative astrocytes in a rat model of SCI
To shed light on the mechanisms and consequences of AQP4 changes found in human SCI, we investigated AQP4 changes in a rat model of contusion SCI. Contusion injuries are the most prevalent in clinical settings, and a rat model of contusion SCI mimics some of the basic histopathological outcomes found in human SCI (Beattie and Bresnahan, 2000). We employed the widely used and well-characterized Infinite Horizon device (Scheff et al., 2003) to produce contusion injury to the thoracic regions (T10) of male Sprague Dawley rats, as described in detail in Nesic et al., 2008.
Uninjured rat spinal cords have an AQP4 expression pattern (Fig. 2A) similar to that in uninjured human spinal cords, with dense astrocytic expression of AQP4 in both gray and white matter. In contrast to the human SCI we assessed, almost all astrocytes in chronically injured rat cords (1, 2, 3 or 5 months after SCI; n=3/group) were overexpressing AQP4 (Fig. 2B). However, at earlier time points after SCI in rats, there were more AQP4-negative astrocytes, similar to chronically injured human cords (Fig. 2C, marked with white star; see Fig. 7A). Immunofluorescent analysis also showed that next to GFAP-labeled, AQP4-negative astrocytes (Fig. 2C, marked with white star), AQP4-positive astrocytes expressed higher levels of AQP4 14d after SCI (Fig. 2C, E; n=3) than did astrocytes in uninjured cords (Fig. 2A, D; n=3). Therefore, rat SCI in the subacute phase post-SCI resembled chronically injured human spinal cords, since they both contained two identifiable populations of astroglia defined via AQP4 expression. This result suggests that recovery processes after SCI including the transition of astrocytes from AQP4-negative (see Part VII) to AQP4-positive astrocytes is more expeditious in rat than in human injured spinal cords.
Fig. 7.
Described in the text.
V. B) AQP4-overexpressing astrocytes: cytotoxic edema
As a result of a large abundance of AQP4-overexpressing astrocytes in chronically injured rat SCI, overall AQP4 levels were significantly and persistently increased by several-fold, not only at the mRNA (Nesic et al., 2005), but also at the protein level (Fig. 2F; Nesic et al., 2006). Considering that AQP4 increases at 11 months were comparable to those at 2 months after SCI (Fig. 2G), we predict that AQP4 upregulation is a life-long astrocytic alteration after rodent SCI.
Overexpression of AQP4 is a rate-limiting factor for astrocytic swelling, as elegantly shown by Yang et al., (2008). Furthermore, deletion of AQP4 in AQP4-null mice demonstrated significantly alleviated cytotoxic swelling of astrocytes in different neuropathological conditions (Zador et al., 2008). Therefore, significantly increased AQP4 levels in AQP4-overexpressing astrocytes likely reflect cytotoxic swelling of astrocytes in injured rat or human spinal cords. Astrocytic swelling in injured spinal cords has been documented by electron microscopy (Sharma and Ollson, 1990), but the possible impact on functional recovery has not been investigated until recently (Saadoun et al., 2008; described in the next paragraph). The impact of cytotoxic astrocytic swelling (and AQP4 upregulation) on the functional recovery after SCI will thus depend on the abundance of AQP4-overexpressing astrocytes in the time window that is relevant to motor or sensory recovery after SCI.
V. C) Effect on motor recovery
Gross motor recovery in rat SCI takes place in the first two weeks after SCI and plateaus by 3 wks post-SCI (Fig. 3A). Motor recovery of SCI rats was assessed using the Basso, Beattie, Bresnahan (BBB) scoring system that measures recovery of hind limb function, which is completely lost immediately after moderate or severe thoracic contusion injury (Basso et al., 1995). Uninjured rats have a BBB score of 21, while SCI rats with completely paralyzed hind limbs have a BBB score of 0. As shown in Fig. 3A, SCI rats recover their gross hind limb locomotor function to some degree by 21d post-SCI, but the extent of recovery depends on the severity of the initial insult. To test whether the severity of injury affects AQP4 changes after SCI, we used SCI rats with different injury levels, classified in three groups by an arbitrary criterion: (a) severe injury with BBB score of 8±0.87; moderate injury, BBB score = 12±1.32; and mild injury, BBB score = 16.5±2.12 (n=3 per group). The recovery of gross hindlimb motor function had plateaued by 3 wks in severe and moderate SCI, but not in mild SCI, where recovery continues, and reaches a plateau by 7wks post- SCI (not shown). We measured AQP4 levels in all SCI rats via Western blots (Fig. 3C). Our findings suggest that AQP4 levels at 3wks post-SCI in moderate and mild SCI rats are similar to control levels, in contrast to the severe group, which showed significantly lower AQP4 levels (p<0.05). Therefore, better motor recovery after different rat SCI was NOT associated with lower AQP4 levels (Fig. 3C), that would be expected if AQP4-mediated cytotoxic swelling negatively affects neuronal function (see Sykova et al., 2004; Mulligan and MacVicar; 2006) underlying motor recovery after SCI. Although moderate and mild SCI differed significantly in terms of the restored hindlimb motor functions, AQP4 levels were indistinguishable both at 21 (Fig. 3) and at 56d after SCI (not shown).
Fig. 3.
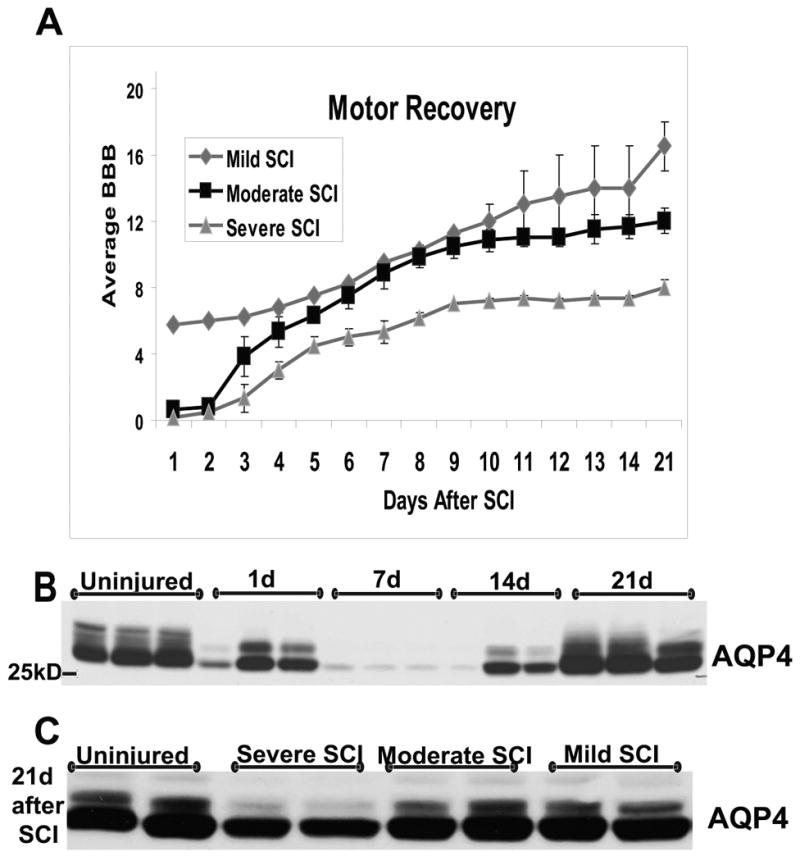
A) Motor recovery of SCI (n=9) with different severities of initial contusion injury to T10. Mild injuries did not produce immediate and complete paralysis of their hindlimbs (BBB scores were >0), in contrast to moderate or severe SCI. Motor recovery was assessed using the BBB scoring system up to 21d after SCI, when motor recovery is typically finalized in severe or moderate injury. At 21d, SCI rats were sacrificed and the lesion site used for Western blot analysis. Maintaining severely injured rats for longer than 21d poses a problem, because those SCI rats develop severe pain-like behavior in chronically post-injury phase. B) Time course of AQP4 protein level changes in moderate SCI at the lesion site (T10). A representative AQP4 Western blot shows decreases in AQP4 levels at 1, 3 and 14d after SCI (n=3 per time point), and restoration of AQP4 to basal levels by 21d post-SCI. C) AQP4 Western blot (T10, 21d after SCI) in SCI rats with different levels of injury (A) showed that restoration of AQP4 levels happened in mild and moderate SCI, but not in severe, where AQP4 levels remained reduced.
The time-course study of AQP4 levels after moderate SCI (Fig. 3B) showed that AQP4 protein levels were also significantly reduced as observed in severe injury (at 21d) but only at earlier time points (1-14d), while they were not reduced in mild injury (not shown). As presented in Fig 3B, AQP4 levels were drastically decreased in the first week, and were restored by the 3rd week post-SCI in moderate SCI. However, AQP4 levels at 21d post-injury were still reduced in severe SCI (Fig. 3C), suggesting that reappearance of AQP4-expressing astrocytes after SCI depends on the severity of injury, and is delayed in severely injured rats. Therefore, possibly negative impact of AQP4-mediated astrocytic swelling on motor recovery will likely be fairly limited in severe injuries, considering the small number of AQP4-(over)expressing astrocytes in the relevant time window (and a smaller number of functional axons in contact with AQP4-overexpressing astrocytes; Fig. 3A, C). Even in moderate SCI, where motor recovery plateaus by day 14, it is unlikely that AQP4-driven astrocytic swelling may significantly impair processes underlying motor recovery. This result may explain why we did not find a correlation between motor recovery scores and AQP4 levels, as described above.
However, Saadoun et al. (2008) found that AQP4- null mice demonstrated significantly better motor recovery after SCI compared to wild type mice, due to reduced cytotoxic edema. They also documented that SCI in mice produced significant increases in AQP4 expression 2d after injury, when AQP4 levels were severely reduced in a rat contusion SCI (Fig. 3). Therefore, we can assume that the mouse model of compression SCI model used in Saadoun et al. (2008) study produces a milder insult compared to the moderate contusion SCI used in most of our studies, and much milder than the examples of human SCI analyzed here (that resembles severe rat SCI, see Fig. 7B). It is also possible that mouse astrocytes differ profoundly in their response to SCI, including AQP4 changes, compared to astrocytes in rats or humans. This is a plausible hypothesis given that inflammatory reactions in mouse SCI significantly differ from those in rat (Sroga et al., 2003).
Therefore, we propose that the effect of AQP4 deletion or inhibition on the motor recovery after SCI depends on the severity of initial injury and on the animal model or species chosen for the animal model in the study. Additionally, inhibition of AQP4 early after severe SCI may be even harmful, if a small number of surviving AQP4-expressing astrocytes exerts a protective role in reaction to hypoxia (described in Part VI; see Fig. 5), which is predictably dramatic after severe SCI. Unfortunately, the only viable approach for testing the hypotheses proposed here requires the use of specific AQP4 inhibitors that can be safely administered to different SCI models in various temporal paradigms – and these inhibitors have not yet been developed.
Fig. 5.
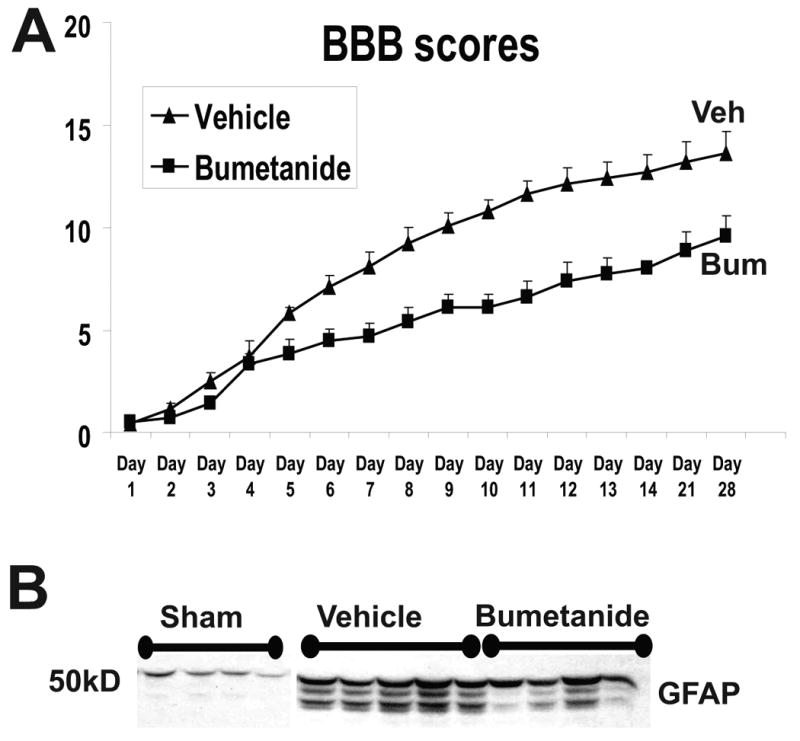
A) Motor recovery (BBB test) was assessed in two groups of SCI rats; a group that received vehicle (n=5) and the other bumetanide (0.1mM; n=5) for 4 weeks continuously via osmotic minipumps (Alzet minipump, 2.5 microl/h. Catheterization and intrathecal delivery is described in Nesic et al., 2008). Vehicle or bumetanide (SIGMA #B3023) were delivered intrathecally to avoid systemic effects of NKCC1 inhibition. Concentration of bumetanide was based on Willis et al., 2006. Values are presented as Mean+/-SE. The difference between BBB scores in vehicle-treated and bumetanide-treated SCI rats was significant from day 6 till day 28 post-SCI (p<0.05; ANOVA). B) GFAP Western blot was performed after the completion of the experiment described in A). GFAP levels were measured at the site of lesion (T10), where GFAP levels are significantly increased after SCI. However, bumetanide treatment reduced GFAP levels, consistent with its inhibitory effect on astrocytic swelling.
V. D) Effect on sensory recovery
In contrast to motor recovery, sensory impairments after SCI, such as autonomic dysreflexia (Krenz and Weaver, 1997) or neuropathic pain, appear later and persist into the chronic post-injury phase in rat SCI (reviewed in Hulsebosch et al., 2009), a time frame in which AQP4-overexpressing astrocytes are present in human SCI (Fig. 1), and predominate in rat SCI, (Fig. 2), irrespective of the severity of injury. Therefore, we hypothesize that robust AQP4 upregulation (“AQP4 overexpression”) and resultant cytotoxic edema may be more relevant to sensory recovery after SCI, regardless of the severity of SCI. We have already shown that increased AQP4 mRNA levels are correlated with pain-like behavior in SCI rats (Nesic et al., 2005). SCI rats that develop pain-like behavior (mechanical allodynia) have > 2-fold higher levels of AQP4 mRNA than do SCI rats that do not develop pain-like behavior (n=4; p<0.05). Therefore, AQP4 upregulation in AQP4-expressing astrocytes may play a role in the development of chronic pain. A large body of evidence suggests an important role that astrocytes play in development of neuropathic pain (for review see Milligan and Watkins, 2009), including SCI pain (Gwak and Hulsebosch; 2009; Carlton et al., 2009), although contribution of AQP4 has not yet been characterized. Chronic pain is a debilitating condition in SCI patients (Yezierski 1996; Finnerup et al., 2001; Siddall et al., 2003; Hulsebosch, 2005). So, what could be the possible mechanism linking AQP4, astrocytic swelling, and hyperexcitability of pain processing neurons, that underlies pain development after SCI? A plausible hypothesis is that swollen astrocytes release excess glutamate (Milligan and MacVicar, 2006) that causes hyperexcitability in pain-processing neurons. It has been well documented that glutamate receptor inhibition attenuates pain and reduces neuronal hyperexcitability after SCI (reviewed in Hulsebosch et al., 2005; 2009). It has also been shown that ammonia-induced astrocytic swelling triggers the release of glutamate and causes neuronal hyperexcitability (Rose, 2007). However, this mechanism has not yet been studied in relation to SCI-dependent neuropathic pain. In human SCI AQP4-overexpressing astrocytes may trigger the above described mechanism if they are localized near pain-processing neurons (e.g. in the vicinity of pain processing neurons in dorsal horns), even in the absence of a large population of AQP4-overexpressing astrocytes. Not all SCI patients develop pain. Published studies have reported divergent incidence of chronic pain among individuals with SCI, ranging from 26 to 96% (Dijkers et al., 2009). This individual variability may be attributed to the unpredictable localization of AQP4-overexpressing astrocytes in injured spinal cords. Our analysis of three human SCI samples showed that abundance and distribution of AQP4-overexpressing astrocytes may vary significantly among individual SCI patients. Consistent with this feature of SCI pain, we also found inter-individual variability in AQP4 levels among equally injured rats in the chronic-post-injury phase, although GFAP levels were indistinguishable (not shown), indicating that differential AQP4 expression may explain astrocytic contribution to pain in SCI rats that develop pain, in agreement with our previous study (Nesic et al., 2005).
V. E) Effect on the size of fluid-filled cavities
Fluid-filled cavities almost always form at the SCI lesion site in human and rat injuries, but not in mouse SCI (Stokes and Jakeman, 2002). Although the fluid-filled cavities are permanent, they do not usually pose a clinical problem for SCI patients, so little attention has been given to the mechanisms underlying their development/maintenance. However, understanding the processes that contribute to permanent filling of cavities with fluid is necessary for a better understanding of cavity expansion into syrinx in some SCI patients. Syringes are longitudinally orientated fluid-filled cavities extending away from the lesion site (Klekamp and Samii, 2002; Stoodley et al., 1999). Formation of syringes in some SCI patients, or posttraumatic syrongomyelia (PTS), is a devastating neurological condition characterized by progressive loss of sensation or strength, pain, profuse sweating, spasticity and autonomic dysreflexia (Biyani and Masry, 1994). Syringes occur in about 28% (Backe et al., 1991; Williams, 1992), although reported incidence varied depending on the study. The mechanisms underlying enlargement of fluid-filled cavities and development of PTS are poorly understood (for review, see Klekamp and Samii, 2001; Klekamp, 2002), and as a result, surgical treatments have long term efficacy in less than 50% of patients with PTS. Without exception, activated astrocytes form the wall of both cavities and syringes in SCI patients and animal models of SCI (Reddy et al., 1989; Inman and Steward, 2003; Seki and Feghlings, 2008), even in cases where syringes are not associated with traumatic injury to the spinal cord (Levin et al., 2004). This suggests a role for astrocytes in fluid-filled cavity formation; however, it is not yet confirmed. We found intense AQP4 labeling in astrocytes around fluid-filled cavities in rat SCI (see Fig. 2B, 2B1), suggesting a possible role for AQP4 in fluid accumulation within cavities.
To shed light on the possible link between AQP4 and fluid-filled cavity formation, we analyzed cavity sizes using the T2-weighted Magnetic Resonance Imaging (MRI) method (Weirich et al., 1990; Bilgen et al., 2000; Narayana et al., 1999), while AQP4 protein levels were measured in the same SCI rats by Western blotting. Fluid accumulation appears in T2 MRI images as a hyperintense (“white”) signal similar to the subarachnoid cerebrospinal fluid signal intensity (marked with red arrow in Fig. 4A). Soon after SCI (7d), we found a diffuse hyperintense signal; e.g. vasogenic edema throughout the gray and white matter (see black arrow) that was especially intense at and around the site of injury (images 1-3). Vasogenic edema demonstrated with T2MRI imaging was additionally confirmed with increased spinal water content early (3d) after SCI (Fig. 4B). Significant decreases in AQP4 early after SCI may contribute to the accumulation of vasogenic edema by slowing the process of fluid removal (Zador et al., 2008), but this remains to be confirmed.
Fig. 4.
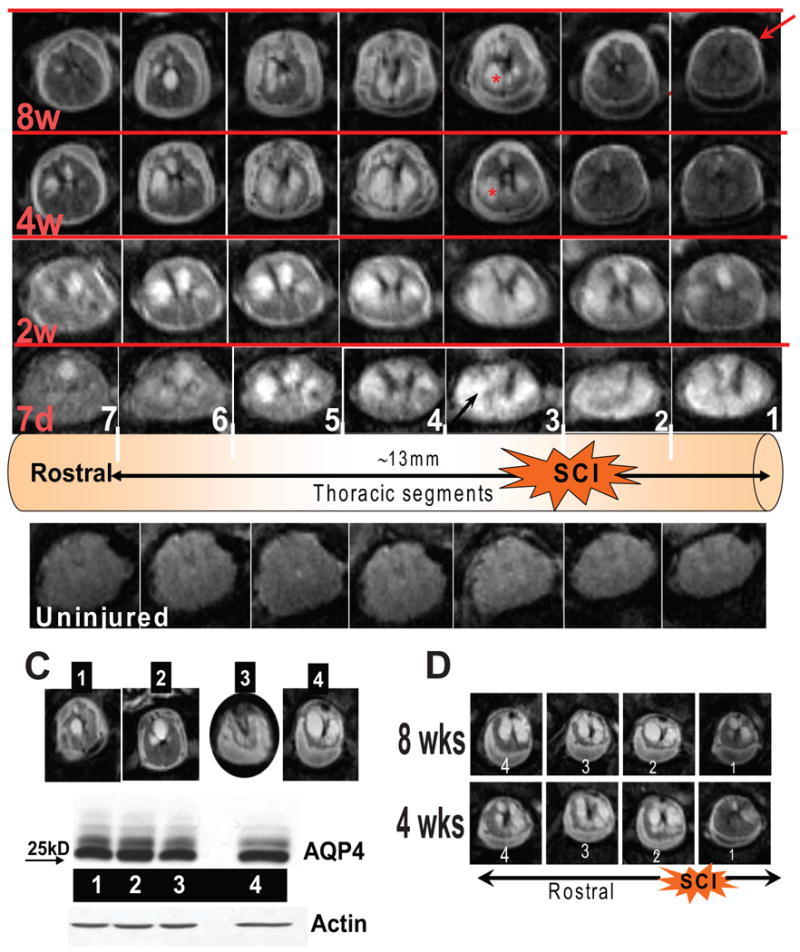

A) The bottom line represents typical T2-MRI images of an uninjured spinal cord imaged in the same thoracic regions as those in the upper rows. Here we show 7 consecutive mages, around the SCI epicenter (images 1-3, right to left). The schematic representation above the uninjured cord shows the epicenter of SCI, and the approximate length of the cord that was imaged and presented here. The upper rows represent a moderately injured rat spinal cord imaged at 7 days, 2 wks, 4 wks and 8 wks after SCI. Numbers in white indicate individual images in columns, and red letters are time points after SCI (in rows). Arrows and asterisks are explained in the text. B) Increased water content was found not only at 3d after SCI (due to vasogenic edema) but also in the thoracic region of injured spinal cords 2 and 5months after SCI (∼20mm long; mean±S.D; p<0.05; reported in Nesic et al., 2006). C) MRI images of four SCI rats (1-4) 8 wks after SCI at the epicenter of SCI. Rat No. 4 had the largest cyst. D) Four T2 MRI images of an SCI rat whose cysts spanned ∼12 mm (about 4 segments) and enlarged from 4wks to 8 wks.
However, considerable fluid accumulation within cavities (round, well defined structures, labeled with a red star in Fig. 4A) was also visible in injured cords, as early as 2wks post-SCI (corresponding to the time of AQP4 restoration, Fig. 3B) and persisted up to 8 wks after SCI (corresponding to robust increases in AQP4, Fig. 2F, G). Fluid accumulation within cavities also resulted in significantly increased water content of injured spinal cords 2 or 5 months after SCI, as reported earlier (Fig. 4B, Nesic et al., 2006). Therefore, we asked whether AQP4 overexpression may contribute to the fluid accumulation within cavities.
We measured AQP4 levels in SCI rats (n=10, in two independent experiments) 8wks after SCI where sizes of fluid-filled cavities were quantified at 4, 6 and 8 wks after SCI. However, we did not find any correlation between AQP4 protein levels (8 wks post-SCI) and cavity sizes (8 wks post-SCI), including fluid-filled cavities that enlarged over time. A representative example of 4 SCI rats, their fluid-filled cavities and AQP4 levels both measured 8wks post-SCI is presented in Fig. 4C. Although cavities varied significantly in size, AQP4 levels were indistinguishable, suggesting that AQP4 likely does not have an important role in fluid-filled cavity formation or its enlargement after rodent SCI (not previously reported). A fluid-filled cavity in SCI rat No. 4 (Fig. 4C) increased from 4 to 8 wks after SCI (Fig. 4D), in contrast to other SCI rats in this group (not shown, see typical example presented in Fig. 4A), whose cavities stayed the same or decreased from 4 to 8 wks. It thus appears that fluid-filled cavity enlargement was also not associated with significantly altered AQP4 levels. Although AQP4 protein levels were not correlated with the total fluid accumulation in chronically injured SCI rats, the role of specifically altered AQP4 distribution/function in the formation of fluid-filled cavities cannot be ruled out.
VI. AQP4 upregulation and hypoxia
To shed some light on mechanisms that trigger and maintain AQP4 increases in chronically injured rat cords, we analyzed mRNA levels of thousands of genes expressed in chronically injured cords, using DNA microarrays and transcriptional profiling of 8 equally injured rats (indistinguishable BBB scores of those rats are presented in Nesic et al., 2005). Our goal was to identify all mRNAs that were significantly correlated with AQP4 mRNA levels in those 8 SCI rats (coefficient of correlation ≥±0.8; p<0.01), and thus give us an insight into the genes that were possibly co-regulated with the aqp4 gene in chronically injured cords. Out of 3389 genes expressed in all injured spinal cords, we found 112 transcripts positively and 11 mRNAs negatively correlated with AQP4 mRNA levels. Partial results of this analysis are presented in Table 1.
Table 1.
| Name | Accession No. | Correlation Coefficient | Significance |
|---|---|---|---|
| HIF1 | Y09507 | 0.84 | ** (p<0.01) |
| NKCC1 | AF086758 | 0.87 | ** |
| inward rectifier 10 | X83585 | 0.9 | ** |
| inward rectifier 9 | X83581 | 0.83 | ** |
| inwardly rectifying 1 | U27558 | 0.9 | ** |
| K+ channel protein, beta subunit | |||
| 2764 | X70662 | 0.85 | ** |
| CI channel RCI1 | D13985 | 0.86 | ** |
| GFAP | AF028784cds#1_s_ | 0.184 | |
| GFAP | AF028784mRNA#2_a | -0.22 | |
| KCC1 | U55815_at | -0.73 | ** |
Among significantly correlated mRNAs we noted Hypoxia induced factor, HIF-1α (Table 1, r=0.84; p<0.01), a transcription factor that has already been shown to regulate AQP4 expression. It has been found that increased HIF-1α activation triggers AQP4 expression after traumatic brain injury (Ding et al., 2009). As we have already reported; there is a significant 50% increase in protein levels of HIF-1α (Nesic et al., 2008) in chronically injured cords (42d after SCI), suggesting that HIF-1α maybe be one of the upstream regulators of persistent AQP4 upregulation in rat SCI. Conversely, AQP4 upregulation may also be a protective reaction to hypoxia. The promoter region of the aqp4 gene contains a putative antioxidant-responsive element (ARE; Umenishi and Verkman, 1988), which is also activated under hypoxic conditions (Walleh et al., 1997). It has already been shown that activators of ARE can induce AQP4 expression in brain trauma (Zhao et al., 2005). Therefore, AQP4 upregulation may reflect a protective reaction of injured cords to persistently hypoxic conditions. Severe hypoxic conditions did not exist only in the chronically injured cords; they were also present in early post-SCI phases. Acute hypoxia has been well documented, including increases in HIF-1α (Xiaowei et al., 2006). Therefore, the appearance of AQP4-overexpressing astrocytes even early after SCI (Fig. 2C) may also be explained by hypoxia.
It is well established that hypoxia significantly alters ion transport mechanisms as a consequence of the cellular energy depletion and resultant intracellular sodium overload (Malek et al., 2003). Those harmful mechanisms leading to sodium overload are also seen in SCI (Lopachin et al., 1999), where inhibition of sodium channels shows significant neuroprotective and therapeutic potential (Schwartz and Fehlings 2001; Hawryluk et al., 2008). As a compensatory reaction to the sodium overload, potassium and chloride effluxes are stimulated to maintain cellular osmolality and electroneutrality (Malek et al., 2003). Therefore, hypoxic insult to neurons results in increased extracellular potassium and chloride by more than ten-fold (Blank and Kirshner, 1977), which are than taken up by astrocytes via different pathways, including inward-rectifying potassium channels and chloride transporters (Kahle et al., 2009). Rapid removal of extracellular K+ and Cl- by astrocytes must be accompanied by obligatory water influx, presumably through AQP4 channels. Consistent with hypoxia-induced alterations in ion transport mechanisms involving astrocytic uptake of potassium and chloride, we found transcript levels of several K+ channels and K+/Cl- transporters to be significantly correlated with AQP4 mRNA levels (Table 1). For example, we found a significant correlation between AQP4 mRNA and mRNA for the Na-K-2Cl cotransporter (NKCC1; r=0.87; p<0.01), whose protein levels are increased after SCI (Cramer et al., 2008). It has been shown that hypoxia increases NKCC1-mediated ion influx in astrocytes, and that deletion or blocking of NKCC1 in astrocytes exposed to hypoxia prevents astrocytic water uptake (Lenart et al., 2004). Therefore, it is possible that hypoxia after SCI increases the expression of NKCC1 and AQP4, as a compensatory, homeostatic reaction to protect neurons.
Ions taken up by astrocytes (including NKCC1-mediated uptake) during normal neuronal activity cause transient astrocytic swelling (MacVicar et al., 2002) and extracellular space shrinkage (Ostby et al., 2009), without damaging to the surrounding tissue, because excess water and ions are removed from the CNS parenchyma via the astrocytic syncytium (Walz, 2000). However, constant hypoxia-driven accumulation of extracellular ions likely exceeds removal capacity of astrocytic network, in addition to already disturbed communication within the astrocytic syncytium after SCI. It has been shown that proteins essential for the communication within astrocytic network, such as gap junction protein, connexin (Cx43) change after SCI (Theriault et al., 1997; Lee et al., 2005), indicating functional rearrangements in astroglial syncytyium in injured spinal cords. Consequences of the damaged functions of the panglial syncytium in neuropathological conditions are eloquently described in the article by J. Rash in this issue of Neuroscience. As a result of the ineffective removal of excess water via the impaired astrocytic syncytium after SCI, chronically swollen astrocytes may negatively impact neuronal excitability and axonal conductivity (Sykova, 2004; Mulligan and MacVicar, 2006).
However, the overall effect of AQP4-driven astrocytic swelling on functional recovery after SCI will depend on the balance between protective astrocytic roles in removing extracellular ions and harmful effects of astrocytic swelling. To test this hypothesis we used NKCC1 inhibitor bumetanide (0.1 mM), known to inhibit NKCC1-mediated ion uptake and astrocytic swelling caused by hypoxia (Lenart et al., 2004). Moderately injured rats received bumetanide intrathecally via osmotic minipumps for 4 wks, while motor recovery was assessed using the BBB test. Bumetanide administration significantly impaired motor recovery (Fig. 5A), indicating that NKCC1- driven ion uptake by astrocytes has a more protective role in hypoxic condition in injured spinal cords. Bumetanide also decreased GFAP levels (Fig. 5B), consistent with its effect on reducing astrocytic swelling (Sykova et al., 1999). Therefore, the reduction of astrocytic swelling resulting from the inhibition of NKCC1-mediated hypoxia-driven uptake of extracellular ions did not lead to improved motor recovery after SCI.
Interestingly, like the aqp4 gene, the gene for NKCC1 is also affected by the transcriptional activation of ARE (Cho et al., 2005), consistent with our hypothesis that AQP4 and NKCC1 are co-regulated in chronically injured rat cords as part of the protective reaction to hypoxia. The possible transcriptional regulator of both genes may be transcriptional factor Nuclear Factor E2 (Nrf2), which activates ARE, but this hypothesis remains to be tested in future experiments. In general, activation of Nrf2 in astrocytes is neuroprotective against a wide array of insults, including hypoxia (Vargas and Johnson, 2009), and a possible involvement of Nrf2 in AQP4 upregulation would support our hypothesis that AQP4 over-expression after SCI is a protective astrocytic reaction.
We also found an inverse correlation between AQP4 mRNA and another chloride cotransporter, KCC1 (r= - 0.73, p<0.01), which has an opposing role to NKCC1, since it extrudes chloride ions from astrocytes (Kettenmann and Ransom, 2004), and is downregulated after SCI (Cramer et al., 2008). The inverse correlation between AQP4 and KCC1 may suggest that increases in AQP4 are driven by ion influxes, rather than effluxes, at least in hypoxic conditions after SCI. Interestingly, AQP4 levels were NOT correlated with NKCC1 or KCC1 in uninjured cords (r= 0.24 and 0.55 respectively; n=5).
As expected, GFAP mRNA levels (detected with two gene-specific probes on Affymterix microarrays) were not correlated with AQP4 mRNA levels (Table 1, r=0.18 and -0.22), consistent with the existence of various populations of astrocytes that differ in expression levels of AQP4 (e.g. AQP4-positive and AQP4-negative) and further validating the results of our correlation analysis.
VII. AQP4-negative astrocytes – where do they come from?
The astrocytic population undergoes significant (∼60%) depletion at the epicenter of injury after moderate rodent SCI (Grossman et al., 2005). That is consistent with our finding that 3d after SCI there were significant decreases in both GFAP and AQP4 (Fig. 6A), and that a cell-sparing agent, the potent anti-inflammatory interleukin 1 receptor antagonist (IL1ra; Nesic et al., 2001) could reverse some of those SCI-induced depletions, indicating that loss of astrocytes contributes to the decrease in AQP4 levels early after SCI.
Fig. 6.
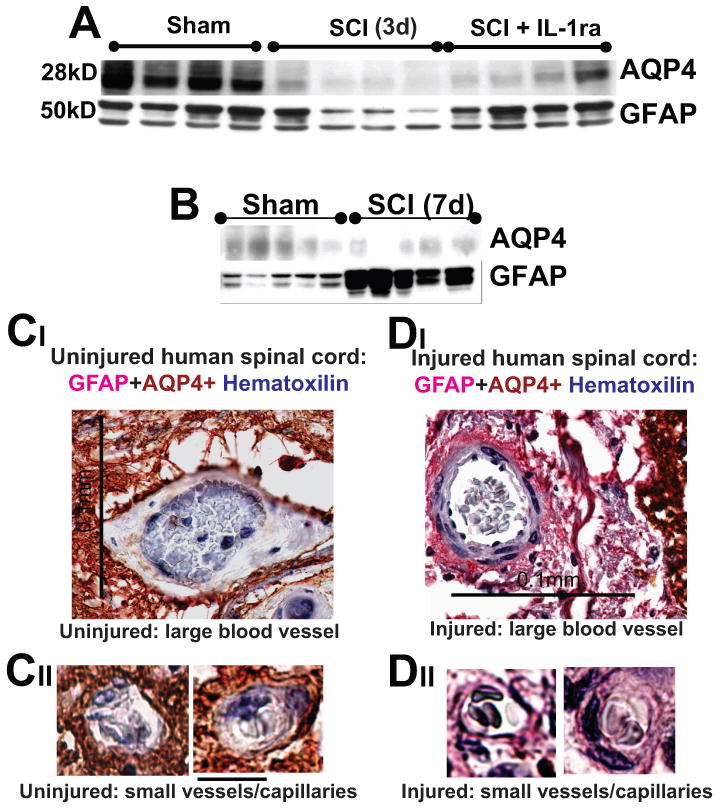
A) AQP4 and GFAP Western blots 3d after SCI at the lesion site (T10). IL1ra (750ng/ml) was administered intrathecally for 3d in a group of SCI rats (n=5), while a second group of SCI rats received vehicle intrathecally for 7d (n=4). Rats were sacrificed at 3d post-SCI and Western blots performed. IL1ra was administered via osmotic minipumps and i.th. cathaters. We have also shown that IL1ra exerted expected effects: it significantly decreased Cox-2 upregulation after SCI, or reduced IL-6 mRNAs (not shown; both Cox-2 and IL-6 are known IL-1 transcriptional targets – Touzani et al., 1999; Igwe et al., 2001). Therefore, the IL-1ra effects we observed in injured cords were mediated via specific blocking of IL-1R activation. B) AQP4 and GFAP Western blots showed that 7d after SCI, AQP4 levels were still reduced at the lesion site in moderate SCI, while GFAP levels showed significantly increased levels compared to sham-treated rats (n=5 per group), indicating replacement and activation of astrocytes early after SCI (already reported in Nesic et al., 2006). C, D) Large and small blood vessels in uninjured and injured human spinal cord (C2, 1 year after SCI, SCI patient No. 29; Guest et al., 2005). Calibration line in CI :0.1 mm; and in CIII: 10 μm. GFAP-labeled astrocytic processes devoid of AQP4 (pink) surrounded blood vessel in chronically injured cords. Erythrocytes in blood vessel were stained blue with hematoxilline.
However, SCI also triggers processes that attempt to replace all lost astrocytes. Lytlle and Wrathall, (2007) showed that lost astrocytes are all replenished with GFAP-labeled cells from 7 until 28d after SCI. Zai and Wrathall, (2005) reported that ∼20% of all BrdU–labeled cells undergoing proliferation 3d after SCI are GFAP-positive, indicating that a substantial fraction of surviving astrocytes undergoes proliferation. In addition, activation of glial progenitor cells and astrogliogenesis also contribute to the replacement of astrocytes in injured cords. Glial progenitor cells in injured cords are the largest population of cells that undergoes proliferation and differentiation after SCI (Zai and Wrathall, (2005), and the vast majority of them differentiate into astrocytes (Horner et al., 2000; Zai and Wrathall, 2005; Yang et al., 2006). However, the phenotypes of GFAP-positive cells generated via processes of astrocytic proliferation and astrogliogenesis after SCI are poorly understood, and to what extent those newly generated GFAP-positive cells resemble mature, resting astrocytes in uninjured cords.
At 7d post-SCI, GFAP levels were significantly increased (58%; Fig. 6B), in contrast to GFAP levels at 3d post-SCI (Fig.6A), suggesting that astrocytic replacement was taking place between 3d and 7d post-SCI, and that those “new” astrocytes are in the activated form (reflected in the elevated GFAP levels, compared to sham-controls). However, Western blot analysis showed that AQP4 levels remained low at 7d post-SCI (Fig. 6B; Nesic et al., 2006), indicating suppression of AQP4 in newly generated astrocytes, and perhaps in surviving astrocytes as well. Consistent with this, we found that IL-1ra completely reversed GFAP levels, but only partially restored AQP4 levels (Fig. 6A), suggesting that surviving astrocytes may also have reduced AQP4 expression levels early after SCI.
Our data also indicate that some of those “new” astrocytes are immature cells (will be reported elsewhere), resembling transitional cells in the normal developmental astrogliogenesis, and have suppressed AQP4; similar to embryonic astrocytes. Astrocytic AQP4 expression becomes apparent only during postnatal development (Wen et al., 1999; Ferrari et al., submitted). Therefore, AQP4 decreases early after SCI (Figs. 3B; 7A, B) may result from suppressed AQP4 expression both in surviving and newly generated astrocytes. What could be the possible consequences of the presence of AQP4-negative astrocytes in injured spinal cord parenchyma is an intriguing question. Given the intimate contact between blood vessels and AQP4-expressing astrocytes (Amiry-Moghaddam et al., 2003), the first question is whether AQP4-negative astrocytes also establish close contact with blood vessels. Here we show one rather large blood vessel in an uninjured spinal cord surrounded by dense astrocytic processes, all expressing AQP4 (Fig. 6C). However, AQP4- negative astrocytic processes also establish equally close contact with larger blood vessels (Fig. 6DI) and small blood vessels/cappilaries (Fig. 6DII) in human SCI, but the ultrastructural organization of this contact and molecular interaction between blood vessels and AQP4-negative astrocytes, and whether those astrocytes affect the function of the blood vessels in injured spinal cords remain to be investigated.
VIII. Summary and conclusions
Several AQPs have been found in rodent spinal cords, but only one was identified in human spinal cords – AQP4, the subject of this report. More detailed studies of other AQPs in human SCI, and their possible roles in pathophysiological outcomes described here, will undoubtedly add to the complexity of AQPs' role in SCI. For example, we have shown that AQP1 levels significantly and persistently increase in rat SCI, in part due to AQP1 induction in astrocytes (Nesic et al., 2008). Therefore, the role of AQP4 in the pathophysiology after SCI will have to be studied in concert with AQP1 or other AQPs that may also prove to be relevant players in deregulation of ion/water homeostasis in human SCI.
The summary and conclusion of our data presented here are depicted in Fig. 7 (next page).
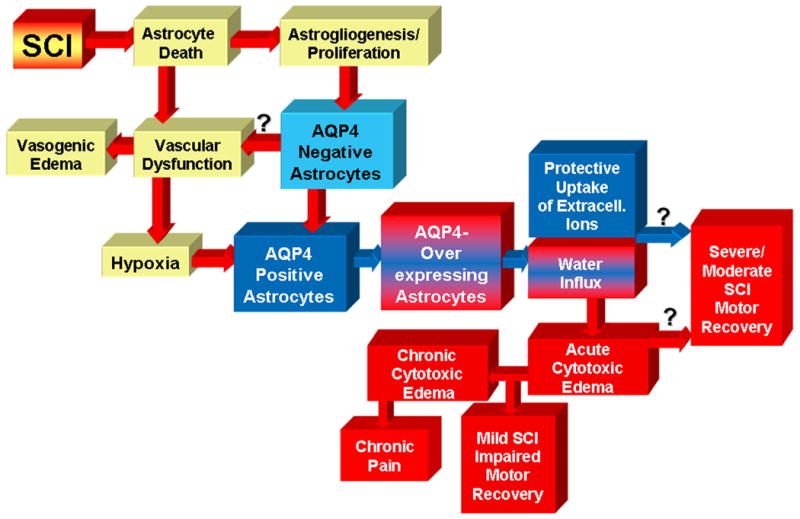
Analysis of AQP4 expression in three chronically injured SCI patients showed the presence of two populations of astrocytes: AQP4-overexpressing and AQP4-negative astroglia (labeled with GFAP; Fig. 7A, B).
AQP4-negative astrocytes are likely generated during the process of SCI-induced replacement of lost astrocytes, and can be rescued with anti-inflammatory cell sparing interventions. AQP4-negative astrocytes are immature cells since they share some similarities with transitional astroglial cells during normal developmental astrogliogenesis (their description will be reported elsewhere). AQP4-negative astrocytes establish close contact with blood vessels in injured cords (rat and human), similar to normal AQP4-expressing astrocytes in uninjured cords. It is thus possible that immature AQP4-negative astrocytes contribute to the chronic dysfunction of blood vessels, described in animal models of SCI that includes vascular hyperpermeability, consequent vasogenic edema, or chronic hypoxia – a possibility that remains to be tested (Fig. 7A).
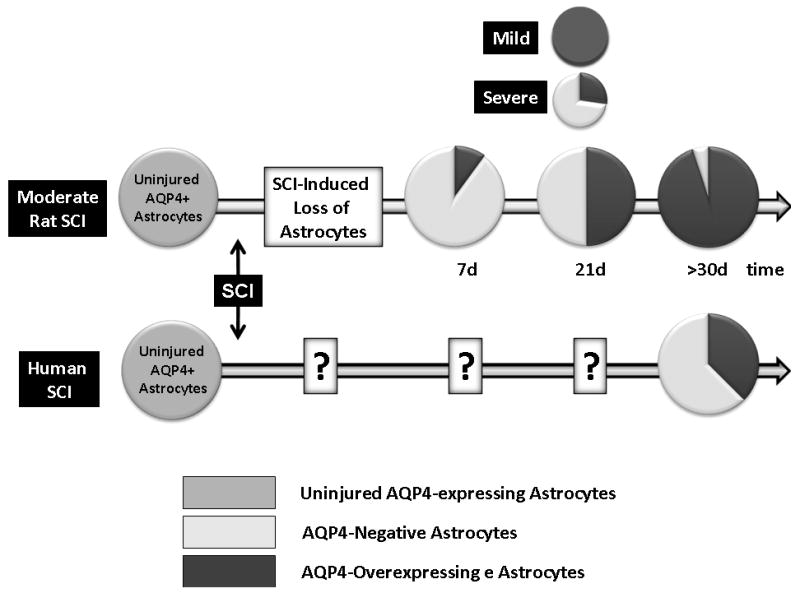
Those two populations of astrocytes also exist in a rat model of contusion SCI. However, the abundance of AQP4-negative astrocytes changes over time: it is higher at early time points after rat SCI (Fig. 7B). Gradually, AQP4-negative astrocytes become replaced with, or transformed into AQP4-expressing astrocytes that predominate in chronic post-injury phase (Fig. 7A, B). The AQP4/GFAP ratio (measured with Western blots in rat SCI; Fig. 7B) varies not only with time post-injury, but also with the severity of injury. More severe injuries have lower AQP4/GFAP ratios, indicating a higher number of AQP4-negative astrocytes, and thus are more similar to human SCI (Fig. 7B). Therefore, it appears that more severe rat contusion injury better mimics astrocytic changes observed in human SCI. However, the time course of astrocytic changes in human SCI remains to be determined.
Although AQP4 overexpression implies negative impact of cytotoxic edema on neuronal/axonal excitability, better motor recovery of SCI rats was not associated with lower AQP4 levels. This likely results from a low abundance of AQP4 over-expressing astrocytes during the time post-injury when gross motor recovery takes place in moderate and severe SCI (Fig. 7B). Therefore, we predict that therapeutic inhibition of AQP4 will have a positive effect on the gross motor recovery only in mild SCI, but will not be beneficial in moderate or severe SCI (Fig. 7B).
AQP4 overexpression- mediated astrocytic swelling may be more relevant to sensory recovery, since mechanisms underlying sensory dysfunction such as pain development take place in the chronic post-injury phase in a rat SCI (Fig. 7A), when AQP4-overexpressing astrocytes predominate. AQP4 levels are increased in SCI rats that develop neuropathic pain-like behavior. We propose a mechanism that links astrocytic swelling and pain: swelling-induced release of glutamate that can induce hyperexcitability of pain-processing neurons, and chronic pain.
In contrast, formation of fluid-filled cavities in chronically injured cords does not appear to be affected by the extent of AQP4 increases. Although AQP4 protein levels were not correlated with the total fluid accumulation within cavities, the role of specifically altered AQP4 distribution in the formation of fluid-filled cavities cannot be ruled out.
Our analysis of transcriptional profiles of chronically injured rat spinal cords suggests that persistent hypoxia may trigger and maintain AQP4 overexpression in AQP4-expressing astrocytes. This analysis also suggested that AQP4 increases may be coupled to a K+/Cl- uptake mechanism, as a protective reaction to hypoxia. Bumetanide, NKCC1 inhibitor decreased astrocytic swelling and significantly impaired motor recovery of moderately injured rats, suggesting that hypoxia-driven mechanisms, although leading to astrocytic swelling may also be protective in moderately (or severely) injured rats. Therefore, the effect of AQP4 inhibition on functional recovery after SCI will depend not only on the time interval after SCI or animal models used in the study, but also on the balance between protective role of increased AQP4 in hypoxia and deleterious effects of astrocytic swelling.
It is also possible that AQP4-overexpression is maladaptive reaction to hypoxia that leads to excessive astrocytic swelling in chronically injured cords, affecting primarily sensory recovery after SCI. The antioxidants acetazolamide (Tanimura et al., 2009) and edaravone (Kikuchi et al., 2009) have recently been shown to be neuroprotective by inhibiting/decreasing AQP4 in brain ischemia. Although those antioxidants are not strictly selective for AQP4, they are promising therapeutic tools in manipulating AQP4 functions in SCI.
Taken together our data and the reports of others that attempt to shed light on the role of AQP4 in human SCI raise more questions than provide answers. A critical contribution to the field however will be the discovery of selective, specific and safe pharmacological agents to inhibit AQP4 functions, which can be then be used to directly test the hypotheses proposed in this report.
XIX. Methods
Rat model of contusion SCI
We used Sprague Dawley male rats. Rats received a partial laminectomy at T10. Moderate SCI was then inflicted using an Infinite Horizon device (LLC, Lexington, KY) and 150kD force, 1s dwell time. These forces produce substantial motor deficits, although SCI rats exhibit spontaneous recovery of weight-supported plantar stepping with occasional coordination (The average final BBB score for rat SCI is 11.5.) The surgical procedures are described in Nesic et al., 2008; all procedures comply with the recommendations in the NIH Guide for the Care and Use of Laboratory Animals, and are approved by UTMB's Institutional Animal Care and Use Committee. We use the minimal number of animals necessary for each experiment, and the suffering of animals is minimized.
Locomotor assessments
Gross motor recovery of SCI rats was assessed using open field tests with the Basso Beattie and Bresnahan scale (BBB rank scores; Basso et al., 1995) assigned as measures of hind limb activity to document plantar placement with weight-bearing stance.
Water content was determined according to the protocol described in Li and Tator (1999). The percent water content was calculated as follows: water content (%) = [(wet weight-dry weight)/wet weight] ×100.
Histological analysis
Preparation of injured human spinal cords for histological analysis is described in Guest et al., 2005.
Immunohistochemistry
Uninjured and injured human spinal cord sections (5-6 microns thickness) were subjected to a deparaffinization/rehydration procedure by xylene treatment. Several steps of incubations in decreasing ethanol concentration from 100% to 70% were followed with a final incubation in Phosphate-buffered saline (PBS). Antigen retrieval was performed at high temperature in the presence of 10mM sodium citrate. Immunohystochemistry was performed by incubating with two primary antibodies, AQP4 (Chemicon; 1:400) and GFAP (Chemicon; 1:800 dilution). AQP4 was detected with LSAB2 system-horse radish peroxidase (DakoCytomation) and a DAB substrate that gave a brown end-product at the site of the target antigen. GFAP-bound antibody was detected with an alkaline phosphatase secondary detection system and a substrate that gave a pink precipitate from fast red chromogen. Counterstaining with Harris hematoxylin was used to detect the cell nuclei (dark blue).
Immunofluorescence
Immunofluorescent detection of AQP4 and GFAP in human spinal cord sections was performed using the same antibodies as in immunohistchemistry. After deparafinization/rehydration and antigen retrieval procedures, AQP4 immunolabeling was determined with the secondary antibody (Alexa Fluor goat anti-rabbit IgG; 1:500).
Immunofluorescent detection of AQP4 in rat spinal cord sections is described in detail in Nesic et al., (2008). Uninjured and injured rats were intracardially perfused with 300ml of 0.1M PBS, followed by 500 mL of 4% paraformaldehyde in 0.1 M phosphate buffer. The spinal cords were removed and postfixed in 4% paraformaldehyde for 2 h at 4°C, then rinsed and cryoprotected in 30% sucrose in phosphate buffer for 48 h at 4°C. We used primary antibodies: Rabbit AQP-4 (Chemicon; 1: 10,000); mouse monoclonal GFAP (Chemicon; 1:1000).
Labeled spinal cord sections were studied with a confocal laser scanning system (BIO-RAD Radiance 2100, K-2 system). For double immunofluorescence staining, to avoid “bleed through” of fluorescense signals between adjacent color channels, the data were collected one channel at a time by sequential scanning. Images were collected using the 488 and 568 nm excitation lines of an Argon-Krypton laser. The co-localization of the two antigens was shown by yellow color in the images obtained after overlaying the green and red channels. Digital images were saved and processed with Adobe Photoshop for final editing. For quantitative analysis of the intensity of immunolabeling, sections were imaged using the same exposure times, and the average density was measured with MetaMorph software (Molecular Devices, Downingtown, PA). Five randomly selected sections from the thoracic spinal cord segment in each animal were analyzed and the relative optical densities were averaged to provide a mean number for each rat.
Electrophoresis and Western Blotting
As described in Nesic et al., 2006. We used primary antibodies: Rabbit AQP-4 (Chemicon; 1: 1000); Mouse monoclonal GFAP (Chemicon; 1:71000).
MRI imaging
MRI that includes dual-echo and diffusion-weighted images was performed in the MRI facility at UT Houston (http://www.uth.tmc.edu/radiology/narayana/pub index.htm) headed by Dr. P. Narayana. The method is described in Dr. Narayana's publications: Weirich et al. (1990); Bilgen et al. (2000) and Narayana et al. (1999). To quantify the hyperintense regions in rapid acquisition relaxation enhancement (RARE) images we used Image Pro-Plus 5 software. Intensity thresholds levels were determined for the hyperintense lesions by comparison with the normal uninjured spinal cord tissue. The thresholds were then applied to areas of interest in SCI images. The volume of injury was calculated by multiplying each lesion area by slice thickness (1mm), then summing the volumes for all slices to obtain the total lesion volume.
DNA microarrays. (detailed description in Nesic et al., 2003,2005)
Total RNA was prepared from frozen spinal cord segments using TRI-Reagent (Molecular Research Center, Cincinnati, OH, USA). Spinal segments were homogenized in TRI-Reagent, and total RNA extracted in chloroform, ethanol-precipitated, and stored at −80°C. Total RNA was assayed for integrity on 1% denaturing agarose gels. Approximately 15μg of total RNA was used for each target. The rat RGA microarray from Affymetrix (Santa Clara, CA, USA) was used for all hybridizations; the results were analyzed with Affymetrix GeneChip Analysis Suite 5.0 software. Finding genes with significantly changed expression levels in any group being compared was done using the Statistical Analysis of Microarrays (SAM), a robust statistical method devised specifically for the analysis of microarray data (Tusher et al., 2001), which was used on the dataset that resulted from filtering out (a) genes absent in all samples and (b) all ESTs (Expressed Sequence Tags). We accepted only those mRNA values with a fold change that was higher than 1.5 for upregulated genes and lower than 0.66 for downregulated genes. This pre-filtering procedure decreased the number of genes being analyzed, and thus reduced the number of false positives from the SAM analysis. Pearson correlation analysis was done using the SPSS statistical package.
Statistical Analysis
All statistical tests were evaluated at the α level of significance of 0.05, two-tailed. We use parametric methods (t-test) for our analyses. For multiple-group comparisons, data wee analyzed using one way analysis of variance (one way ANOVA). The LSD multiple comparisons post-hoc test was used to determine p values. p values less than 0.05 are considered significant.
Acknowledgments
Supported by NIH NS05417 and Mission Connect, a project of TIRR Foundation. I would like to thank Dr. David Konkel for critically editing the manuscript. Human spinal cord sections were donated from the University of Miami spinal cord collection.
List of Abbreviations
- ALS
amyotrophic lateral sclerosis
- AQPs
Aquaporins
- AQP4
Aquaporin 4
- BBB
Basso, Beattie, Bresnahan motor recovery score
- BSCB
blood-spinal cord barrier
- CNS
Central Nervous System
- FDA
Food and Drug Administration
- GFAP
Glial Fibrillary Acidic Protein
- IL-1ra
Interleukin 1 receptor antagonist
- MS
multiple sclerosis
- NMO
Neuromyelitis optica
- PTS
posttraumatic syringomyelia
- SCI
spinal cord injury
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdullaev IF, Rudkouskaya A, Schools GP, Kimelberg HK, Mongin AA. Pharmacological comparison of swelling-activated excitatory amino acid release and Cl- currents in cultured rat astrocytes. J Physiol. 2006 May 1;572(Pt 3):677–89. doi: 10.1113/jphysiol.2005.103820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amiry-Moghaddam M, Otsuka T, Hurn PD, Traystman RJ, Haug FM, Froehner SC, Adams ME, Neely JD, Agre P, Ottersen OP, Bhardwaj A. An alpha-syntrophin-dependent pool of AQP4 in astroglial end-feet confers bidirectional water flow between blood and brain. Proc Natl Acad Sci U S A. 2003 Feb 18;100(4):2106–11. doi: 10.1073/pnas.0437946100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backe HA, Betz RR, Mesgarzadeh M, Beck T, Clancy M. Post-traumatic spinal cord cysts evaluated by magnetic resonance imaging. Paraplegia. 1991;29:607–612. doi: 10.1038/sc.1991.89. [DOI] [PubMed] [Google Scholar]
- Badaut J. Aquaglyceroporin 9 in brain pathologies. Neuroscience. 2009 doi: 10.1016/j.neuroscience.2009.10.030. [DOI] [PubMed] [Google Scholar]
- Barry D, McDermott K. Differentiation of radial glia from radial precursor cells and transformation into astrocytes in the developing rat spinal cord. Glia. 2005 May;50(3):187–97. doi: 10.1002/glia.20166. [DOI] [PubMed] [Google Scholar]
- Basso DM, Beattie MS, Bresnahan JC. A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma. 1995;12:1–21. doi: 10.1089/neu.1995.12.1. [DOI] [PubMed] [Google Scholar]
- Beattie MS, Li Q, Bresnahan JC. Cell death and plasticity after experimental spinal cord injury. Prog Brain Res. 2000;128:9–21. doi: 10.1016/S0079-6123(00)28003-5. [DOI] [PubMed] [Google Scholar]
- Benton RL, Maddie MA, Dincman TA, Hagg T, Whittemore SR. Transcriptional activation of endothelial cells by TGFbeta coincides with acute microvascular plasticity following focal spinal cord ischaemia/reperfusion injury. ASN Neuro. 2009 Aug 26;1(3):pii, e00015. doi: 10.1042/AN20090008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton RL, Maddie MA, Minnillo DR, Hagg T, Whittemore SR. Griffonia simplicifolia isolectin B4 identifies a specific subpopulation of angiogenic blood vessels following contusive spinal cord injury in the adult mouse. J Comp Neurol. 2008 Mar 1;507(1):1031–52. doi: 10.1002/cne.21570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilgen MB, Abbe R, Narayana PA. Dynamic contrast enhanced magnetic resonance imaging of experimental spinal cord injury: in vivo serial studies. Magn Reson Med. 2001;45:614–622. doi: 10.1002/mrm.1083. [DOI] [PubMed] [Google Scholar]; cord injury. Neurochem Res. 28:1693–1703. doi: 10.1023/a:1026013106016. [DOI] [PubMed] [Google Scholar]
- Biyani A, Masry WS. Posttraumatic syringomyelia. A review of the literature. Paraplegia. 1994:32723–731. doi: 10.1038/sc.1994.117. [DOI] [PubMed] [Google Scholar]
- Blank WF, Jr, Kirshner HS. The kinetics of extracellular potassium changes during hypoxia and anoxia in the cat cerebral cortex. Brain Res. 1977 Mar 4;123(1):113–24. doi: 10.1016/0006-8993(77)90646-1. [DOI] [PubMed] [Google Scholar]
- Carlton SM, Du J, Tan HY, Nesic O, Hargett GL, Bopp AC, Yamani A, Lin Q, Willis WD, Hulsebosch CE. Peripheral and central sensitization in remote spinal cord regions contribute to central neuropathic pain after spinal cord injury. Pain. 2009 Oct 21; doi: 10.1016/j.pain.2009.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayli SR, Kocak A, Yilmaz U, Tekiner A, Erbil M, Ozturk C, Batcioglu K, Saim Yologlu. Effect of combined treatment with melatonin and methylprednisolone on neurological recovery after experimental spinal cord injury. European Spine Journal. 2004;13:724–732. doi: 10.1007/s00586-003-0550-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hye-Youn Cho, Reddy Sekhar P, DeBiase Andrea, Yamamoto Masayuki, Kleeberger Steven R. Gene expression profiling of NRF2-mediated protection against oxidative injury. Free Radical Biology and Medicine. 2005 February 1;38(3):325–343. doi: 10.1016/j.freeradbiomed.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Cramer SW, Baggott C, Cain J, Tilghman J, Allcock B, Miranpuri G, Rajpal S, Sun D, Resnick D. The role of cation-dependent chloride transporters in neuropathic pain following spinal cord injury. Mol Pain. 2008 Sep 17;4:36. doi: 10.1186/1744-8069-4-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkers M, Bryce T, Zanca J. Prevalence of chronic pain after traumatic spinal cord injury: a systematic review. J Rehabil Res Dev. 2009;46:13–29. [PubMed] [Google Scholar]
- Ding JY, Kreipke CW, Speirs SL, Schafer P, Schafer S, Rafols JA. Hypoxia-inducible factor-1alpha signaling in aquaporin upregulation after traumatic brain injury. Neurosci Lett. 2009 Mar 27;453(1):68–72. doi: 10.1016/j.neulet.2009.01.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellingson BM, Ulmer JL, Schmit BD. Morphology and morphometry of human chronic spinal cord injury using diffusion tensor imaging and fuzzy logic. Ann Biomed Eng. 2008 Feb;36(2):224–36. doi: 10.1007/s10439-007-9415-6. Epub 2007 Dec 8. [DOI] [PubMed] [Google Scholar]
- Evans A, Stoodley N, Halpin S. Magnetic resonance imaging of intraspinal cystic lesions: a pictorial review. Curr Probl Diagn Radiol. 2002 May-Jun;31(3):79–94. doi: 10.1067/cdr.2002.125402. Review. [DOI] [PubMed] [Google Scholar]
- Finnerup NB, Johannesen IL, Sindrup SH, Bach FW, Jensen TS. Pain and dysesthesia in patients with spinal cord injury: A postal survey. Spinal Cord. 2001;39:256–262. doi: 10.1038/sj.sc.3101161. [DOI] [PubMed] [Google Scholar]
- Gagnon KB, Adragna NC, Fyffe RE, Lauf PK. Characterization of glial cell K-Cl cotransport. doi: 10.1159/000104160. [DOI] [PubMed] [Google Scholar]; Harsan LA, Poulet P, Guignard B, Parizel N, Skoff RP, Ghandour MS. Astrocytic hypertrophy in dysmyelination influences the diffusion anisotropy of white matter. Neurosci Res. 2007 Apr;85(5):935–44. doi: 10.1002/jnr.21201. [DOI] [PubMed] [Google Scholar]
- Gwak YS, Hulsebosch CE. Remote astrocytic and microglial activation modulates neuronal hyperexcitability and below-level neuropathic pain after spinal injury in rat. Neuroscience. 2009 Jul 7;161(3):895–903. doi: 10.1016/j.neuroscience.2009.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guest JD, Hiester ED, Bunge RP. Demyelination and Schwann cell responses adjacent to injury epicenter cavities following chronic human spinal cord injury. Experimental Neurology. 2005 April;192(2):384–393. doi: 10.1016/j.expneurol.2004.11.033. [DOI] [PubMed] [Google Scholar]
- Harsan LA, Poulet P, Guignard B, Parizel N, Skoff RP, Ghandour MS. Astrocytic hypertrophy in dysmyelination influences the diffusion anisotropy of white matter. J Neurosci Res. 2007 Apr;85(5):935–44. doi: 10.1002/jnr.21201. [DOI] [PubMed] [Google Scholar]
- Hawryluk GW, Rowland J, Kwon BK. Fehlings MGProtection and repair of the injured spinal cord: a review of completed, ongoing, and planned clinical trials for acute spinal cord injury. Neurosurg Focus. 2008;25(5):E14. doi: 10.3171/FOC.2008.25.11.E14. [DOI] [PubMed] [Google Scholar]
- Hinson SR, McKeon A, Lennon VA. Neurological autoimmunity targeting aquaporin-4. Neuroscience. 2009 Oct 19; doi: 10.1016/j.neuroscience.2009.08.032. [DOI] [PubMed] [Google Scholar]
- Horner PJ, Power AE, Kempermann G, Kuhn HG, Palmer TD, Winkler J, Thal LJ, et al. Proliferation and differentiation of progenitor cells throughout the intact adult rat spinal cord. J Neurosci. 2000;20:2218–222. doi: 10.1523/JNEUROSCI.20-06-02218.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulsebosch CE. From discovery to clinical trials: treatment strategies for central neuropathic pain after spinal cord injury. Curr Pharm Des. 2005;11:1411–1420. doi: 10.2174/1381612053507864. [DOI] [PubMed] [Google Scholar]
- Hulsebosch CE, Hains BC, Crown ED, Carlton SM. Mechanisms of chronic central neuropathic pain after spinal cord injury. Brain Res Rev. 2009;60:202–213. doi: 10.1016/j.brainresrev.2008.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igwe OJ, Murray JN, Moolwaney AS. Interleukin 1-induced cyclooxygenase and nitric oxide synthase gene expression in the rat dorsal root ganglia is modulated by antioxidants. Neuroscience. 2001;105(4):971–85. doi: 10.1016/s0306-4522(01)00253-6. [DOI] [PubMed] [Google Scholar]
- Imura T, Tane K, Toyoda N, Fushiki S. Endothelial cell-derived bone morphogenetic proteins regulate glial differentiation of cortical progenitors. Eur J Neurosci. 2008 Apr;27(7):1596–606. doi: 10.1111/j.1460-9568.2008.06134.x. [DOI] [PubMed] [Google Scholar]
- Inman DM, Steward O. Physical size does not determine the unique histopathological response seen in the injured mouse spinal cord. J Neurotrauma. 2003 Jan;20(1):33–42. doi: 10.1089/08977150360517164. [DOI] [PubMed] [Google Scholar]
- Kahle Kristopher T, Simard J Marc, Staley Kevin J, Nahed Brian V, Jones Pamela S, Dandan Sun AJ. Molecular Mechanisms of Ischemic Cerebral Edema: Role of Electroneutral Ion Transport. Physiology. 2009 August;24(4):257–265. doi: 10.1152/physiol.00015.2009. [DOI] [PubMed] [Google Scholar]
- Kalyani K Hobson, Rao MS. Neuroepithelial stem cells from the embryonic spinal cord: isolation, characterization, and clonal analysis. Dev Biol. 1997;186:202–223. doi: 10.1006/dbio.1997.8592. [DOI] [PubMed] [Google Scholar]
- Kettenmann Helmut, Ransom Bruce R., editors. Neuroglia. 2. Oxford University Press; USA: Sep 30, 2004. [Google Scholar]
- Kiyoshi Kikuchi, Tancharoen Salunya, Matsuda Fumiyo, Biswas Kamal Krishna, Ito Takashi, Morimoto Yoko, Oyama Yoko, Takenouchi Kazunori, Miura Naoki, Arimura Noboru, Nawa Yuko, Meng Xiaojie, Shrestha Binita, Arimura Shinichiro, Iwata Masahiro, Mera Kentaro, Sameshima Hisayo, Ohno Yoshiko, Maenosono Ryuichi, Tajima Yutaka, Uchikado Hisaaki, Kuramoto Terukazu, Nakayama Kenji, Shigemori Minoru, Yoshida Yoshihiro, Hashiguchi Teruto, Maruyama Ikuro, Kawahara Ko-ichi. Edaravone attenuates cerebral ischemic injury by suppressing aquaporin-4. Biochemical and Biophysical Research Communications. 2009 doi: 10.1016/j.bbrc.2009.09.015. [DOI] [PubMed] [Google Scholar]
- Klekamp Jörg, Samii Madjid, Matthies C. Syringomyelia: Diagnosis and Treatment. Springer; 2001. [Google Scholar]; Josephson A, Greitz D, Klason T, Olson L, Spenger C. A spinal sac constriction model supports the theory that induced pressure gradients in the cord cause edema and cyst formation. Neurosurgery. 2001;46:636–646. doi: 10.1097/00006123-200103000-00039. [DOI] [PubMed] [Google Scholar]
- Klekamp J. The pathophysiology of syringomyelia - historical overview and current concept. Acta Neurochir. 2002;144:649–664. doi: 10.1007/s00701-002-0944-3. [DOI] [PubMed] [Google Scholar]
- Krenz NR, Weaver LC. Changes in the morphology of sympathetic preganglionic neurons parallel the development of autonomic dysreflexia after spinal cord injury in rats. Neurosci Lett. 1998 Feb 27;243(1-3):61–4. doi: 10.1016/s0304-3940(98)00101-3. [DOI] [PubMed] [Google Scholar]
- Lee IH, Lindqvist E, Kiehn O, Widenfalk J, Olson L. Glial and neuronal connexin expression patterns in the rat spinal cord during development and following injury. J Comp Neurol. 2005 Aug 15;489(1):1–10. doi: 10.1002/cne.20567. [DOI] [PubMed] [Google Scholar]
- Lee SW, Kim WJ, Choi YK, Song HS, Son MJ, Gelman IH, Kim YJ, Kim KW. SSeCKS regulates angiogenesis and tight junction formation in the blood–brain barrier. Nat Med. 2003;9:900–906. doi: 10.1038/nm889. [DOI] [PubMed] [Google Scholar]
- Lee HS, Han J, Bai HJ, Kim KW. Brain angiogenesis in developmental and pathological processes: regulation, molecular and cellular communication at the neurovascular interface. FEBS J. 2009 Sep;276(17):4622–35. doi: 10.1111/j.1742-4658.2009.07174.x. [DOI] [PubMed] [Google Scholar]
- Lenart B, Kintner DB, Shull GE, Sun D. Na-K-Cl cotransporter-mediated intracellular Na+ accumulation affects Ca2+ signaling in astrocytes in an in vitro ischemic model. J Neurosci. 2004 Oct 27;24(43):9585–97. doi: 10.1523/JNEUROSCI.2569-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine David N. The pathogenesis of syringomyelia associated with lesions at the foramen magnum: a critical review of existing theories and proposal of a new hypothesis. Journal of the Neurological Sciences. 2004 May 15;220(1-2):3–21. doi: 10.1016/j.jns.2004.01.014. [DOI] [PubMed] [Google Scholar]
- LoPachin RM, Gaughan CL, Lehning EJ, Kaneko Y, Kelly TM, Blight A. Experimental spinal cord injury: spatiotemporal characterization of elemental concentrations and water contents in axons and neuroglia. J Neurophysiol. 1999 Nov;82(5):2143–53. doi: 10.1152/jn.1999.82.5.2143. [DOI] [PubMed] [Google Scholar]
- Lytle JM, Wrathall JR. Glial cell loss, proliferation and replacement in the contused murine spinal cord. Eur J Neurosci. 2007 Mar;25(6):1711–24. doi: 10.1111/j.1460-9568.2007.05390.x. [DOI] [PubMed] [Google Scholar]
- MacVicar BA, Feighan D, Brown A, Ransom B. Intrinsic optical signals in the rat optic nerve: role for K(+) uptake via NKCC1 and swelling of astrocytes. Glia. 2002 Feb;37(2):114–23. doi: 10.1002/glia.10023. [DOI] [PubMed] [Google Scholar]
- Malek SA, Coderre E, Stys PK. Aberrant chloride transport contributes to anoxic/ischemic white matter injury. J Neurosci. 2003 May 1;23(9):3826–36. doi: 10.1523/JNEUROSCI.23-09-03826.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihai G, Nout YS, Tovar CA, Miller BA, Schmalbrock P, Bresnahan JC, Beattie MS. Longitudinal comparison of two severities of unilateral cervical spinal cord injury using magnetic resonance imaging in rats. J Neurotrauma. 2008 Jan;25(1):1–18. doi: 10.1089/neu.2007.0338. [DOI] [PubMed] [Google Scholar]
- Miller SM. Methylprednisolone in acute spinal cord injury: a tarnished standard. Journal of Neurosurgical Anesthesiology. 2008;20(2):140–142. doi: 10.1097/01.ana.0000314442.40952.0d. [DOI] [PubMed] [Google Scholar]
- Milligan ED, Watkins LR. Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci. 2009 Jan;10(1):23–36. doi: 10.1038/nrn2533. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan SJ, MacVicar BA. VRACs CARVe a path for novel mechanisms of communication in the CNS. Sci STKE. 2006 Oct 17;2006(357):pe42. doi: 10.1126/stke.3572006pe42. Review. [DOI] [PubMed] [Google Scholar]
- Misu T, Fujihara K, Kakita A, Konno H, Nakamura M, Watanabe S, Takahashi T, Nakashima I, Takahashi H, Itoyama Y. Loss of aquaporin 4 in lesions of neuromyelitis optica: distinction from multiple sclerosis. Brain. 2007;130(5):1224–1234. doi: 10.1093/brain/awm047. [DOI] [PubMed] [Google Scholar]
- Bilgen MB, Abbe R, Narayana PA. Dynamic contrast enhanced magnetic resonance imaging of experimental spinal cord injury: in vivo serial studies. Magn Reson Med. 2001;45:614–622. doi: 10.1002/mrm.1083. [DOI] [PubMed] [Google Scholar]; cord injury. Neurochem Res. 28:1693–1703. doi: 10.1023/a:1026013106016. [DOI] [PubMed] [Google Scholar]
- Nesic O, Xu GY, McAdoo D, High KW, Hulsebosch C, Perez-Pol R. IL-1 receptor antagonist prevents apoptosis and caspase-3 activation after spinal cord injury. J Neurotrauma. 2001 Sep;18(9):947–56. doi: 10.1089/089771501750451857. [DOI] [PubMed] [Google Scholar]
- Nesic O, Svrakic NM, Xu GY, McAdoo D, Westlund KN, Hulsebosch CE, Ye Z, Galante A, Soteropoulos P, Tolias P, Young W, Hart RP, Perez-Polo JR. DNA microarray analysis of the contused spinal cord: effect of NMDA receptor inhibition. J Neurosci Res. 2002 May 15;68(4):406–23. doi: 10.1002/jnr.10171. [DOI] [PubMed] [Google Scholar]
- Nesic O, Lee J, Johnson KM, Ye Z, Xu GY, Unabia GC, Wood TG, McAdoo DJ, Westlund KN, Hulsebosch CE, Regino Perez-Polo J. Transcriptional profiling of spinal cord injury-induced central neuropathic pain. J Neurochem. 2005;95:998–1014. doi: 10.1111/j.1471-4159.2005.03462.x. [DOI] [PubMed] [Google Scholar]
- Nesic O, Lee J, Ye Z, Unabia GC, Rafati D, Hulsebosch CE, Perez-Polo JR. Acute and chronic changes in aquaporin 4 expression after spinal cord injury. Neuroscience. 2006;143:779–792. doi: 10.1016/j.neuroscience.2006.08.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesic O, Lee J, Unabia GC, Johnson K, Ye Z, Vergara L, Hulsebosch CE, Perez-Polo JR. Aquaporin 1 - a novel player in spinal cord injury. J Neurochem. 2008;105:628–640. doi: 10.1111/j.1471-4159.2007.05177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicaise C, Soyfoo MS, Authelet M, De Decker R, Bataveljic D, Delporte C, Pochet R. Aquaporin-4 overexpression in rat ALS model. Anat Rec (Hoboken) 2009 Feb;292(2):207–13. doi: 10.1002/ar.20838. [DOI] [PubMed] [Google Scholar]
- Noble LJ, Wrathall JR. Distribution and time course of protein extravasation in the rat spinal cord after contusive injury. Brain Res. 1989 Mar 13;482(1):57–66. doi: 10.1016/0006-8993(89)90542-8. [DOI] [PubMed] [Google Scholar]
- Oshio K, Binder DK, Yang B, Schecter S, Verkman AS, Manley GT. Expression of aquaporin water channels in mouse spinal cord. Neuroscience. 2004;127:685–693. doi: 10.1016/j.neuroscience.2004.03.016. [DOI] [PubMed] [Google Scholar]
- Østby I, Øyehaug L, Einevoll GT, Nagelhus EA, Plahte E, et al. Astrocytic Mechanisms Explaining Neural-Activity-Induced Shrinkage of Extraneuronal Space. PLoS Comput Biol. 2009;5(1):e1000272. doi: 10.1371/journal.pcbi.1000272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pistollato F, Chen HL, Rood BR, Zhang HZ, D'Avella D, Denaro L, Gardiman M, te Kronnie G, Schwartz PH, Favaro E, Indraccolo S, Basso G, Panchision DM. Hypoxia and HIF1alpha repress the differentiative effects of BMPs in high-grade glioma. Stem Cells. 2009 Jan;27(1):7–17. doi: 10.1634/stemcells.2008-0402. [DOI] [PubMed] [Google Scholar]
- Popovich PG, Horner PJ, Mullin BB, Stokes BT. A quantitative spatial analysis of the blood-spinal cord barrier. I. Permeability changes after experimental spinal contusion injury. Exp Neurol. 1996 Dec;142(2):258–75. doi: 10.1006/exnr.1996.0196. [DOI] [PubMed] [Google Scholar]
- Reddy KK, Del Bigio MR, Sutherland GR. Ultrastructure of the human posttraumatic syrinx. J Neurosurg. 1989 Aug;71(2):239–43. doi: 10.3171/jns.1989.71.2.0239. [DOI] [PubMed] [Google Scholar]
- Rose C. Effect of ammonia on astrocytic glutamate uptake/release mechanisms. J Neurochem. 2006 Apr;97 1:11. doi: 10.1111/j.1471-4159.2006.03796.x. [DOI] [PubMed] [Google Scholar]
- Rosenberg LJ, Zai LJ, Wrathall JR. Chronic alterations in the cellular composition of spinal cord white matter following contusion injury. Glia. 2005 Jan 1;49(1):107–20. doi: 10.1002/glia.20096. [DOI] [PubMed] [Google Scholar]
- Rozet I. Methylprednisolone in acute apinal cord injury is there any other ethical choice. Journal Neurosurgical Anesthesiology. 2008;20(2):137–139. doi: 10.1097/01.ana.0000314441.63823.b0. [DOI] [PubMed] [Google Scholar]
- Saadoun S, Bell BA, Verkman AS, Papadopoulos MC. Greatly improved neurological outcome after spinal cord compression injury in AQP4-deficient mice. Brain. 2008 Apr;131(Pt 4):1087–98. doi: 10.1093/brain/awn014. [DOI] [PubMed] [Google Scholar]
- Saadoun S, Tait MJ, Reza A, Davies DC, Bell BA, Verkman AS, Papadopoulos MC. AQP4 gene deletion in mice does not alter blood-brain barrier integrity or brain morphology. Neuroscience. 2009 Jul 7;161(3):764–72. doi: 10.1016/j.neuroscience.2009.03.069. Epub 2009 Apr 5. Review. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Rabchevsky AG, Fugaccia I, Main JA, Lumpp JE., Jr Experimental modeling of spinal cord injury: characterization of a force-defined injury device. J Neurotrauma. 2003 Feb;20(2):179–93. doi: 10.1089/08977150360547099. [DOI] [PubMed] [Google Scholar]
- Schwartz G, Fehlings MG. Evaluation of the neuroprotective effects of sodium channel blockers after spinal cord injury: improved behavioral and neuroanatomical recovery with riluzole. J Neurosurg. 2001;94:245–256. doi: 10.3171/spi.2001.94.2.0245. [DOI] [PubMed] [Google Scholar]
- See J, Mamontov P, Ahn K, Wine-Lee L, Crenshaw EB, 3rd, Grinspan JB. BMP signaling mutant mice exhibit glial cell maturation defects. Mol Cell Neurosci. 2007 May;35(1):171–82. doi: 10.1016/j.mcn.2007.02.012. Epub 2007 Feb 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki T, Fehlings MG. Mechanistic insights into posttraumatic syringomyelia based on a novel in vivo animal model. Laboratory investigation. J Neurosurg Spine. 2008 Apr;8(4):365–75. doi: 10.3171/SPI/2008/8/4/365. [DOI] [PubMed] [Google Scholar]
- Seifert G, Hüttmann K, Binder DK, Hartmann C, Wyczynski A, Neusch C, Steinhäuser C. Analysis of astroglial K+ channel expression in the developing hippocampus reveals a predominant role of the Kir4.1 subunit. J Neurosci. 2009 Jun 10;29(23):7474–88. doi: 10.1523/JNEUROSCI.3790-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. HIF-1: mediator of physiological and pathophysiological responses to hypoxia. J Appl Physiol. 2000 Apr;88(4):1474–80. doi: 10.1152/jappl.2000.88.4.1474. [DOI] [PubMed] [Google Scholar]
- Sharma HS, Olsson Y. Edema formation and cellular alterations following spinal cord injury in the rat and their modification with p-chlorophenylalanine. Acta Neuropathol. 1990;79(6):604–10. doi: 10.1007/BF00294237. [DOI] [PubMed] [Google Scholar]
- Sharma HS. Pathophysiology of blood-spinal cord barrier in traumatic injury and repair. Curr Pharm Des. Shepard MJ, Bracken MB.Magnetic resonance imaging and neurological recovery in acute spinal cord injury: observations from the National Acute Spinal Cord Injury Study 3. Spinal Cord. 1999 Dec;37(12):833–7. doi: 10.1038/sj.sc.3100927. [DOI] [PubMed] [Google Scholar]
- Shibuya S, Hara H, Wakayama Y, Inoue M, Jimi T, Matsuzaki Y. Aquaporin 4 mRNA levels in neuromuscular tissues of wild-type and dystrophin-deficient mice. Tohoku J Exp Med. 2008 Aug;215(4):313–9. doi: 10.1620/tjem.215.313. [DOI] [PubMed] [Google Scholar]
- Shields SD, Mazario J, Skinner K, Basbaum AI. Anatomical and functional analysis of aquaporin 1, a water channel in primary afferent neurons. Pain. 2007 Sep;131(1-2):8–20. doi: 10.1016/j.pain.2006.11.018. [DOI] [PubMed] [Google Scholar]
- Simard Marie, Arcuino Gregory, Takano Takahiro, Liu Qing Song, Nedergaard Maiken. Signaling at the Gliovascular Interface. The Journal of Neuroscience. 2003 October 8;23(27):9254–9262. doi: 10.1523/JNEUROSCI.23-27-09254.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solenov EI, Vetrivel L, Oshio K, Manley GT, Verkman AS. Optical measurement of swelling and water transport in spinal cord slices from aquaporin null mice. J Neurosci Methods. 2002 Jan 15;113(1):85–90. doi: 10.1016/s0165-0270(01)00481-2. [DOI] [PubMed] [Google Scholar]
- Sroga JM, Jones TB, Kigerl KA, McGaughy VM, Popovich PG. Rats and mice exhibit distinct inflammatory reactions after spinal cord injury. J Comp Neurol. 2003 Jul 21;462(2):223–40. doi: 10.1002/cne.10736. [DOI] [PubMed] [Google Scholar]
- Stokes, Jakeman LB. Experimental modelling of human spinal cord injury: a model that crosses the species barrier and mimics the spectrum of human cytopathology. J Comp Neurol. 2003 Jul 21;462(2):223–40. doi: 10.1038/sj.sc.3101254. [DOI] [PubMed] [Google Scholar]
- Syková E. Diffusion properties of the brain in health and disease. Neurochem Int. 2004 Sep;45(4):453–66. doi: 10.1016/j.neuint.2003.11.009. [DOI] [PubMed] [Google Scholar]; Tator CH, Koyanagi I. Vascular mechanisms in the pathophysiology of human spinal cord injury. J Neurosurg. 1997 Nov;87(5):800. doi: 10.3171/jns.1997.86.3.0483. [DOI] [PubMed] [Google Scholar]
- Syková E, Vargová L, Prokopová S, Simonová Z. Glial swelling and astrogliosis produce diffusion barriers in the rat spinal cord. Glia. 1999;25:56–70. doi: 10.1002/(sici)1098-1136(19990101)25:1<56::aid-glia6>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Tanimura Y, Hiroaki Y, Fujiyoshi Y. Acetazolamide reversibly inhibits water conduction by quaporin-4. J Struct Biol. 2009;166:16–21. doi: 10.1016/j.jsb.2008.11.010. [DOI] [PubMed] [Google Scholar]
- Taya K, Marmarou C, Okuno K, Prieto R, Marmarou A. Effect of Secondary Insults upon Aquaporin-4 Water Channels Following Experimental Cortical Contusion in Rats. J Neurotrauma. 2009 Aug 25; doi: 10.1089/neu.2009.0933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thuret S, Moon LDF, Gage Fred H. Therapeutic interventions after spinal cord injury. Nature Reviews Neuroscience. 2006;7(8):628–643. doi: 10.1038/nrn1955. [DOI] [PubMed] [Google Scholar]
- Touzani O, Boutin H, Chuquet J, Rothwell N. Potential mechanisms of interleukin-1 involvement in cerebral ischaemia. J Neuroimmunol. 1999;100:203–215. doi: 10.1016/s0165-5728(99)00202-7. [DOI] [PubMed] [Google Scholar]
- Umenishi F, Verkman AS. Isolation and functional analysis of alternative promoters in the human aquaporin-4 water channel gene. Genomics. 1998 Jun 15;50(3):373–7. doi: 10.1006/geno.1998.5337. [DOI] [PubMed] [Google Scholar]
- Valencia-de Ita S, Lawand NB, Lin Q, Castaneda-Hernandez G, Willis WD. Role of the Na+-K+-2Cl-cotransporter in the development of capsaicin-induced neurogenic inflammation. J Neurophysiol. 2006 Jun;95(6):3553–61. doi: 10.1152/jn.01091.2005. [DOI] [PubMed] [Google Scholar]
- Vargas MR, Johnson JA. The Nrf2-ARE cytoprotective pathway in astrocytes. Expert Rev Mol Med. 2009 Jun 3;11:e17. doi: 10.1017/S1462399409001094. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkman AS, Binder DK, Bloch O, Auguste K, Papadopoulos MC. Three distinct roles of aquaporin-4 in brain function revealed by knockout mice. Biochim Biophys Acta. 2006 Aug;1758(8):1085–93. doi: 10.1016/j.bbamem.2006.02.018. Epub 2006 Mar 10. [DOI] [PubMed] [Google Scholar]
- Walz W. Role of astrocytes in the clearance of excess extracellular potassium. Neurochem Int. 2000 Apr;36(4-5):291–300. doi: 10.1016/s0197-0186(99)00137-0. [DOI] [PubMed] [Google Scholar]
- Waleh NS, Calaoagan J, Murphy BJ, Knapp AM, Sutherland RM, Laderoute KR. The redox-sensitive human antioxidant responsive element induces gene expression under low oxygen conditions arcinogenesis. 19:1333–1337. doi: 10.1093/carcin/19.8.1333. [DOI] [PubMed] [Google Scholar]
- Weirich, et al. Histopathologic correlation of magnetic resonance imaging signal patterns in a spinal cord injury model. Spine. 1990;15(7):630–8. doi: 10.1097/00007632-199007000-00004. [DOI] [PubMed] [Google Scholar]
- Wen H, Nagelhus EA, Amiry-Moghaddam M, Agre P, Ottersen OP, Nielsen S. Ontogeny of water transport in rat brain: postnatal expression of the aquaporin-4 water channel. Eur J Neurosci. 1999 Mar;11(3):935–45. doi: 10.1046/j.1460-9568.1999.00502.x. [DOI] [PubMed] [Google Scholar]
- Whetstone WD, Hsu JY, Eisenberg M, Werb Z, Noble-Haeusslein LJ. Blood-spinal cord barrier after spinal cord injury: relation to revascularization and wound healing. J Neurosci Res. 2003 Oct 15;74(2):227–39. doi: 10.1002/jnr.10759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RE, Jakeman LB. Don't fence me in: harnessing the beneficial roles of astrocytes for spinal cord repair. Restor Neurol Neurosci. 2008;26(2-3):197–214. Review. [PMC free article] [PubMed] [Google Scholar]
- Williams B. Pathogenesis of post-traumatic syringomyelia. Br J Neurosurg. 1992;6:517–520. doi: 10.3109/02688699209002367. [DOI] [PubMed] [Google Scholar]
- Wolburg H, Noell S, Mack A, Wolburg-Buchholz K, Fallier-Becker P. Brain endothelial cells and the gliovascular complex. Cell Tissue Res. 2009 Jan;335(1):75–96. doi: 10.1007/s00441-008-0658-9. Epub 2008 Jul 16. Review. [DOI] [PubMed] [Google Scholar]
- Xiaowei H, Ninghui Z, Wei X, Yiping T, Linfeng X. The experimental study of hypoxia-inducible factor-1alpha and its target genes in spinal cord injury. Spinal Cord. 2006 Jan;44(1):35–43. doi: 10.1038/sj.sc.3101813. [DOI] [PubMed] [Google Scholar]
- Xu WB, Gu YT, Wang YF, Lu XH, Jia LS, Lv G. Bradykinin preconditioning modulates aquaporin-4 expression after spinal cord ischemic injury in rats. Brain Res. 2008 Dec 30;1246:11–8. doi: 10.1016/j.brainres.2008.09.087. [DOI] [PubMed] [Google Scholar]
- Yang H, Lu P, McKay HM, Bernot T, Keirstead H, Steward O, Gage FH, Edgerton VR. Tuszynski MHEndogenous neurogenesis replaces oligodendrocytes and astrocytes after primate spinal cord injury. J Neurosci. 2006 Feb 22;26(8):2157–66. doi: 10.1523/JNEUROSCI.4070-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Zador Z, Verkman AS. Glial cell aquaporin-4 overexpression in transgenic mice accelerates cytotoxic brain swelling. J Biol Chem. 2008 May 30;283(22):15280–6. doi: 10.1074/jbc.M801425200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yezierski RP. Pain following spinal cord injury: the clinical problem and experimental studies. Pain. 1996;68:185–194. doi: 10.1016/s0304-3959(96)03178-8. [DOI] [PubMed] [Google Scholar]
- Yool AJ, Brown EA, Flynn GA. Roles for novel pharmacological blockers of aquaporins in the treatment of brain oedema and cancer. Clin Exp Pharmacol Physiol. 2009 Jun 29; doi: 10.1111/j.1440-1681.2009.05244.x. [DOI] [PubMed] [Google Scholar]
- Zai LJ, Wrathall JR. Cell proliferation and replacement following contusive spinal cord injury. Glia. 2005;50:247–257. doi: 10.1002/glia.20176. [DOI] [PubMed] [Google Scholar]
- Zador Z, Stiver A, Wang V, Manley GT. Role of aquaporin-4 in cerebral edema and stroke. Handb Exp Pharmacol. 2009;(190):159–70. doi: 10.1007/978-3-540-79885-9_7. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerlin M, Goldman JE. Interactions between glial progenitors and blood vessels during early postnatal corticogenesis: blood vessel contact represents an early stage of astrocyte differentiation. J Comp Neurol. 1997 Nov 3;387(4):537–46. doi: 10.1002/(sici)1096-9861(19971103)387:4<537::aid-cne5>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Zhao Jing, Moore Anthony N, Clifton Guy L, Dash Pramod K. *Sulforaphane enhances aquaporin-4 expression and decreases cerebral edema following traumatic brain injury. Journal of Neuroscience Research. 2005;82(4):499–506. doi: 10.1002/jnr.20649. [DOI] [PubMed] [Google Scholar]
- Zhou J, Kong H, Hua X, Xiao M, Ding J, Hu G. Altered blood-brain barrier integrity in adult aquaporin-4 knockout mice. Neuroreport. 2008 Jan 8;19(1):1–5. doi: 10.1097/WNR.0b013e3282f2b4eb. [DOI] [PubMed] [Google Scholar]
- Zhu C, Qiu L, Wang X, Xu F, Nilsson M, Cooper-Kuhn C, Kuhn HG, Blomgren K. Age-dependent regenerative responses in the striatum and cortex after hypoxia-ischemia. J Cereb Blood Flow Metab. 2009 Feb;29(2):342–54. doi: 10.1038/jcbfm.2008.124. Epub 2008 Nov 5. [DOI] [PubMed] [Google Scholar]